TGFα Promotes Chemoresistance of Malignant Pleural Mesothelioma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
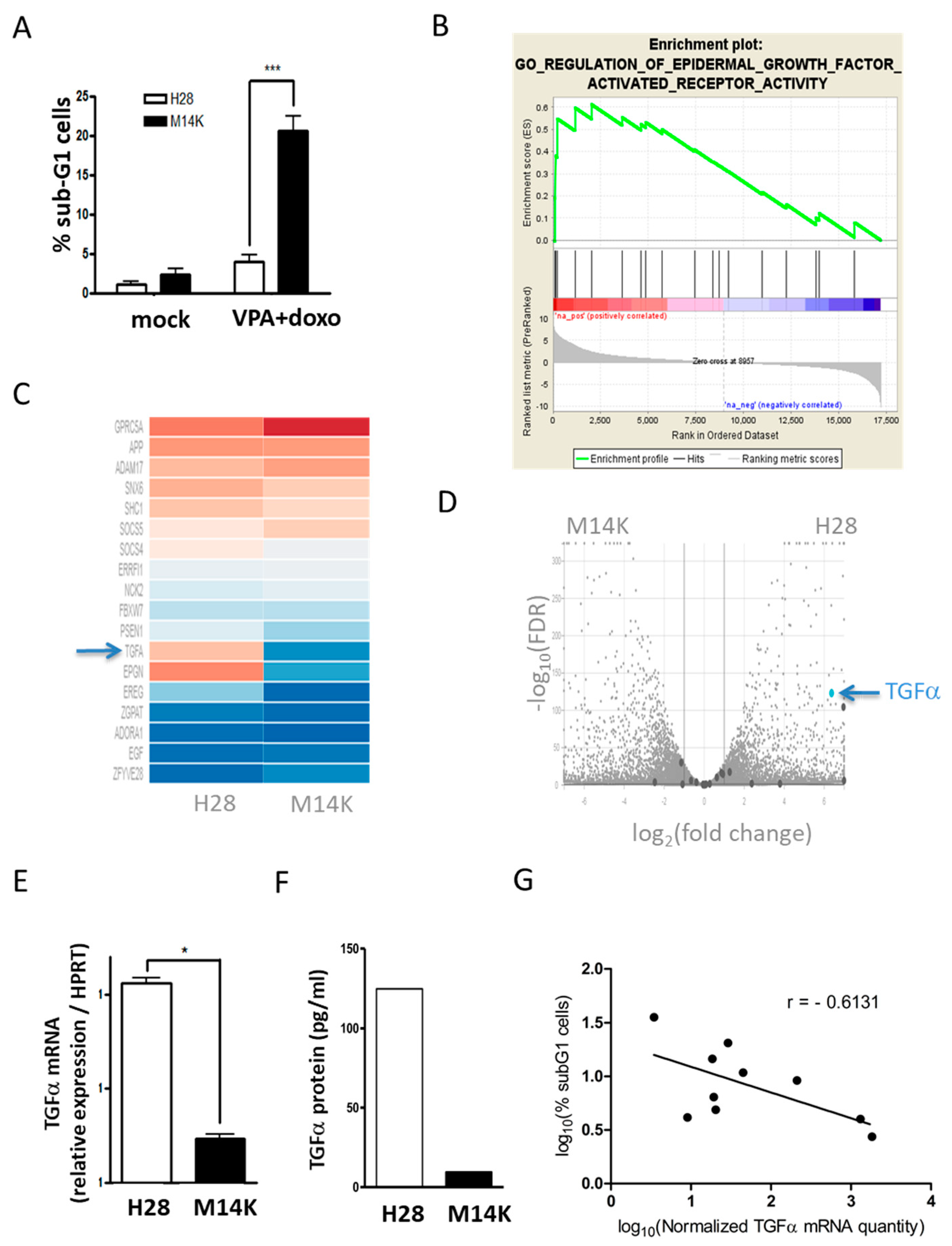
2.1. TGFα is Overexpressed in MPM Cells Resisting VPA-Doxorubicin Induced Apoptosis
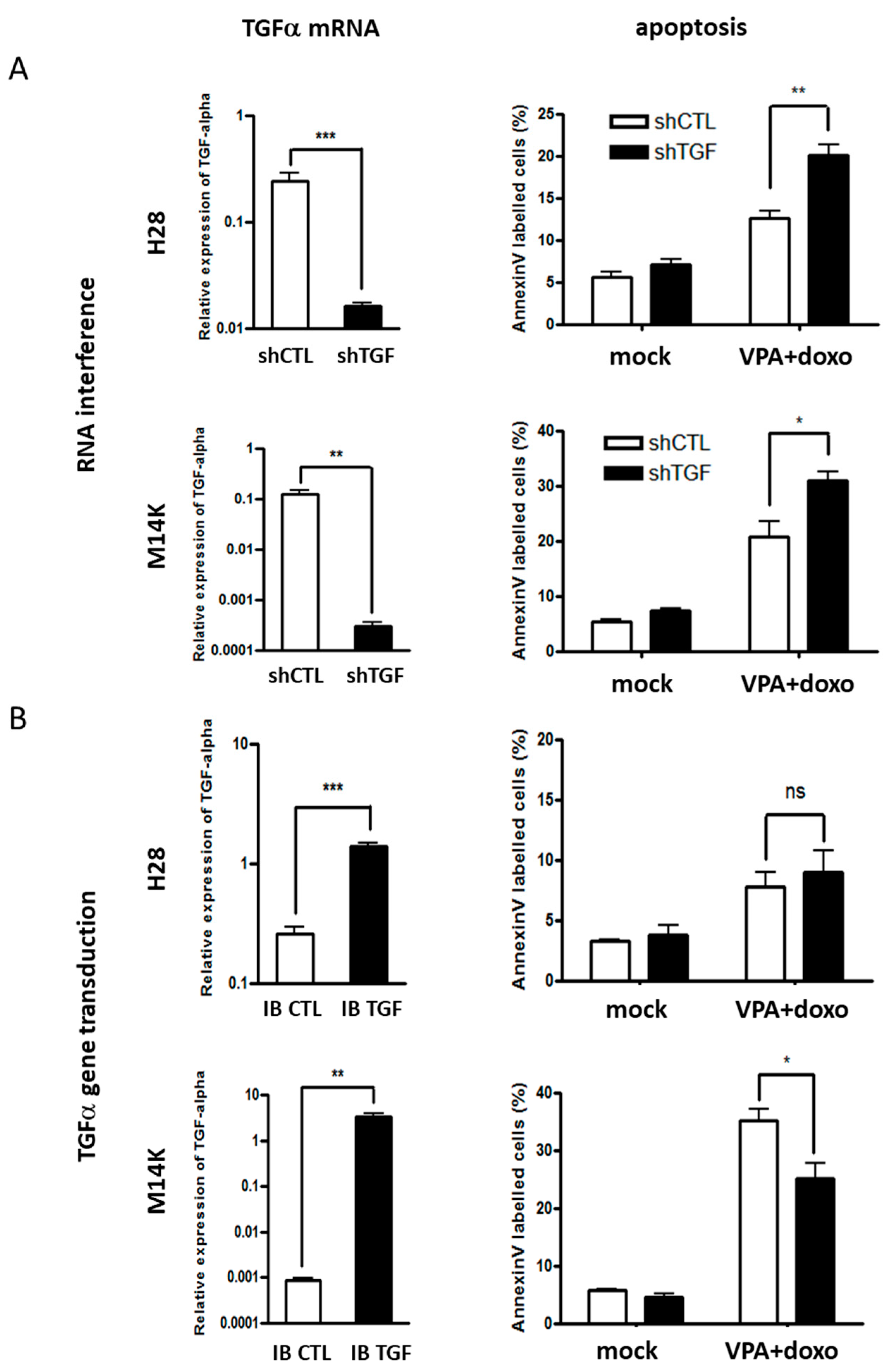
2.2. Modulation of TGFα Expression Influences the Apoptotic Response Induced by Doxorubicin and VPA
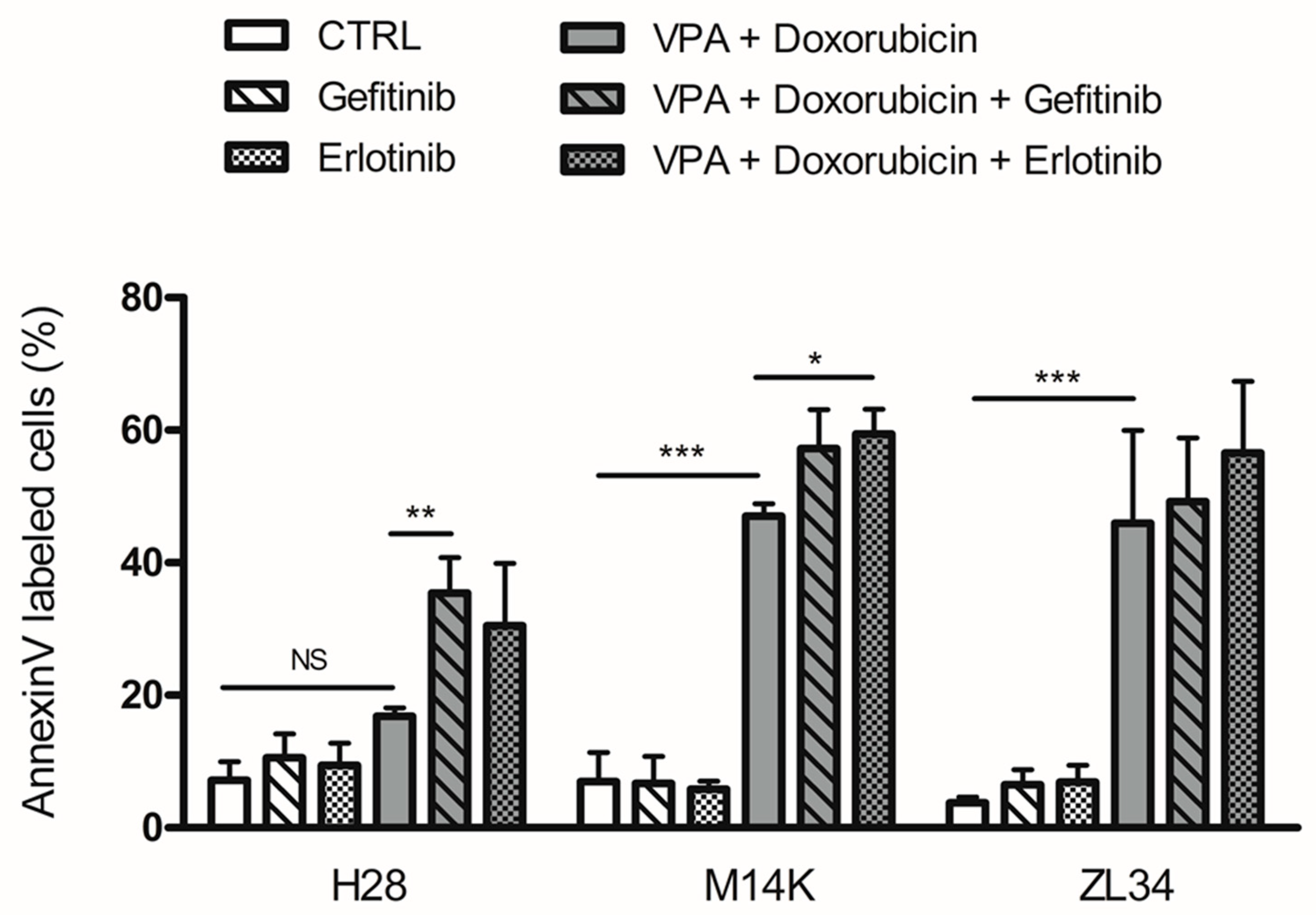
2.3. EGFR Tyrosine Kinase Inhibitors Increase Apoptosis Induced by VPA and Doxorubicin
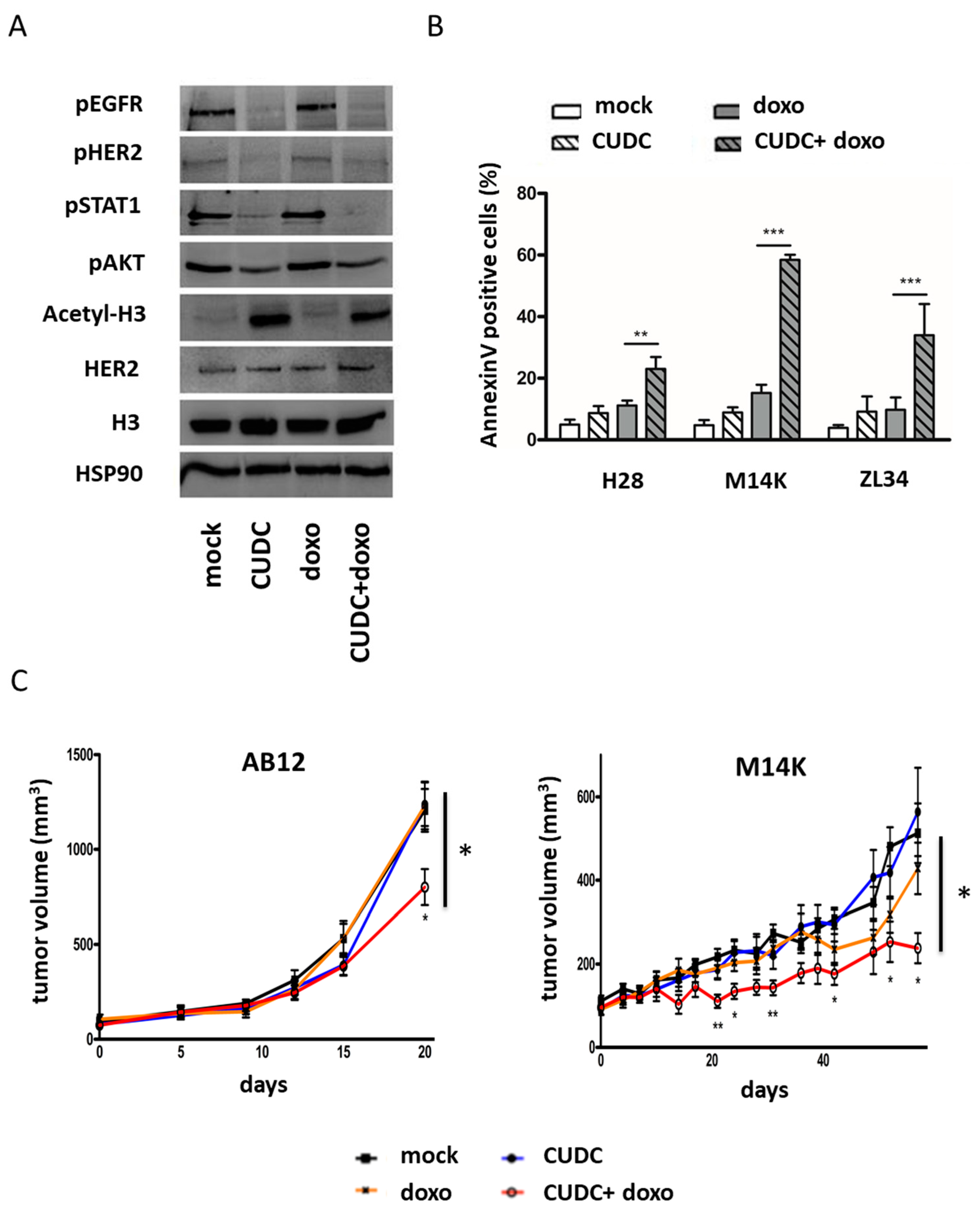
2.4. An Inhibitor Targeting EGFR/HER2 and HDAC Synergizes with Doxorubicin to Induce Apoptosis in MPM Cells and Inhibit Tumor Growth in Two Mesothelioma Mouse Models
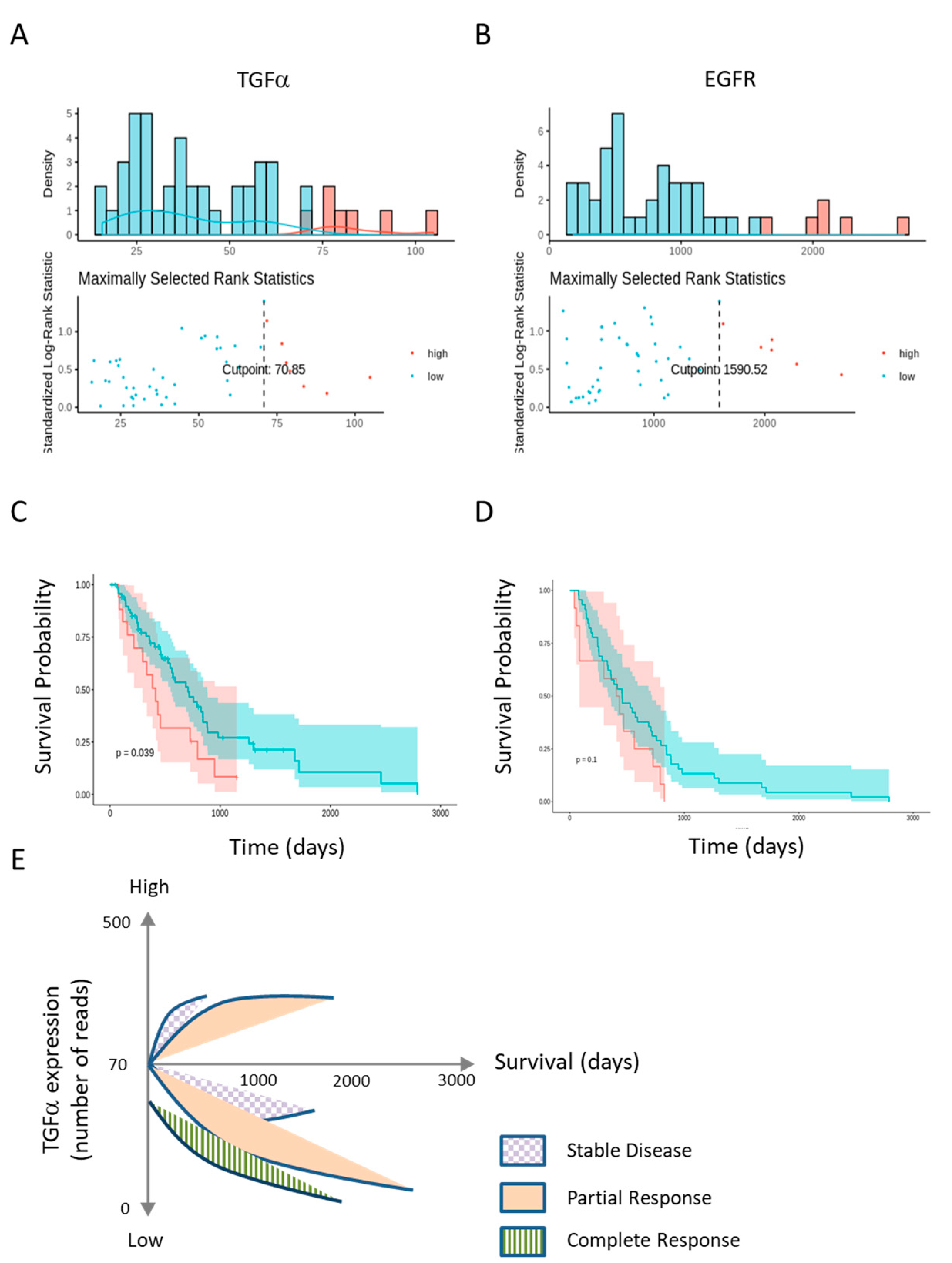
2.5. Low TGFα Expression Predicts Patient’s Survival
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemicals
4.2. RNA Sequencing and Bioinformatics Analyses
4.3. Lentiviral Production and Generation of Stable Cell Lines
4.4. Quantification of TGFα Expression by RT-qPCR and ELISA
4.5. Quantification of Apoptosis
4.6. Cell Lysate Preparation and Immunoblotting
4.7. Mouse Models
4.8. Analysis of the Cancer Genome Atlas Database
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Kanodia, S.; Chao, A.; Miller, A.; Wali, A.; Weissman, D.; Adjei, A.; Baumann, F.; Boffetta, P.; Buck, B.; et al. Consensus report of the 2015 Weinman international conference on mesothelioma. J. Thorac. Oncol. 2016, 11, 1246–1262. [Google Scholar] [CrossRef] [PubMed]
- Frost, G. The latency period of mesothelioma among a cohort of British asbestos workers (1978–2005). Br. J. Cancer 2013, 109, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Van den Borre, L.; Deboosere, P. Asbestos in Belgium: An underestimated health risk. The evolution of mesothelioma mortality rates (1969–2009). Int. J. Occup. Environ. Health 2014, 20, 134–140. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Van Meerbeeck, J.P.; Gaafar, R.; Manegold, C.; Van Klaveren, R.J.; Van Marck, E.A.; Vincent, M.; Legrand, C.; Bottomley, A.; Debruyne, C.; Giaccone, G. Randomized phase III study of cisplatin with or without raltitrexed in patients with malignant pleural mesothelioma: An intergroup study of the European organisation for research and treatment of cancer lung cancer group and the National Cancer Institute of Canada. J. Clin. Oncol. 2005, 23, 6881–6889. [Google Scholar] [CrossRef]
- Ceresoli, G.L.; Zucali, P.A.; Favaretto, A.G.; Grossi, F.; Bidoli, P.; Del Conte, G.; Ceribelli, A.; Bearz, A.; Morenghi, E.; Cavina, R.; et al. Phase II study of pemetrexed plus carboplatin in malignant pleural mesothelioma. J. Clin. Oncol. 2006, 24, 1443–1448. [Google Scholar] [CrossRef]
- Harvey, V.J.; Slevin, M.L.; Ponder, B.A.; Blackshaw, A.J.; Wrigley, P.F. Chemotherapy of diffuse malignant mesothelioma. Phase II trials of single-agent 5-fluorouracil and adriamycin. Cancer 1984, 54, 961–964. [Google Scholar] [CrossRef]
- Sorensen, P.G.; Bach, F.; Bork, E.; Hansen, H.H. Randomized trial of doxorubicin versus cyclophosphamide in diffuse malignant pleural mesothelioma. Cancer Treat. Rep. 1985, 69, 1431–1432. [Google Scholar]
- Scherpereel, A.; Berghmans, T.; Lafitte, J.J.; Colinet, B.; Richez, M.; Bonduelle, Y.; Meert, A.P.; Dhalluin, X.; Leclercq, N.; Paesmans, M.; et al. Valproate-doxorubicin: Promising therapy for progressing mesothelioma. A phase II study. Eur. Respir. J. 2011, 37, 129–135. [Google Scholar] [CrossRef]
- Buikhuisen, W.A.; Hiddinga, B.I.; Baas, P.; van Meerbeeck, J.P. Second line therapy in malignant pleural mesothelioma: A systematic review. Lung Cancer 2015, 89, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L.; Karon, M.; Siegel, S.E.; Avila, F. Effect of Adriamycin on DNA, RNA, and Protein Synthesis in Cell-free Systems and Intact Cells. Cancer Res. 1976, 36, 2891–2895. [Google Scholar] [PubMed]
- Baas, P.; Fennell, D.; Kerr, K.M.; Van Schil, P.E.; Haas, R.L.; Peters, S. Malignant pleural mesothelioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, v31–v39. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Cinatl, J., Jr. Anti-tumor mechanisms of valproate: A novel role for an old drug. Med. Res. Rev. 2002, 22, 492–511. [Google Scholar] [CrossRef] [PubMed]
- Blaheta, R.A.; Nau, H.; Michaelis, M.; Cinatl, J. Valproate and valproate-analogues: Potent tools to fight against cancer. Curr. Med. Chem. 2002, 9, 1417–1433. [Google Scholar] [CrossRef]
- Ghodke-Puranik, Y.; Thorn, C.F.; Lamba, J.K.; Leeder, J.S.; Song, W.; Birnbaum, A.K.; Altman, R.B.; Klein, T.E. Valproic acid pathway: Pharmacokinetics and pharmacodynamics. Pharmacogenet. Genomics 2013, 23, 236–241. [Google Scholar] [CrossRef]
- Vandermeers, F.; Hubert, P.; Delvenne, P.; Mascaux, C.; Grigoriu, B.; Burny, A.; Scherpereel, A.; Willems, L. Valproate, in combination with pemetrexed and cisplatin, provides additional efficacy to the treatment of malignant mesothelioma. Clin. Cancer Res. 2009, 15, 2818–2828. [Google Scholar] [CrossRef]
- Lai, C.-J.; Bao, R.; Tao, X.; Wang, J.; Atoyan, R.; Qu, H.; Wang, D.-G.; Yin, L.; Samson, M.; Forrester, J.; et al. CUDC-101, a multitargeted inhibitor of histone deacetylase, epidermal growth factor receptor, and human epidermal growth factor receptor 2, exerts potent anticancer activity. Cancer Res. 2010, 70, 3647–3656. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Mehta, A.; Boufraqech, M.; Davis, S.; Wang, J.; Tian, Z.; Yu, Z.; Boxer, M.B.; Kiefer, J.A.; et al. Dual inhibition of HDAC and EGFR signaling with CUDC-101 induces potent suppression of tumor growth and metastasis in anaplastic thyroid cancer. Oncotarget 2015, 6, 9073–9085. [Google Scholar] [CrossRef]
- Vandermeers, F.; Sriramareddy, S.N.; Costa, C.; Hubaux, R.; Cosse, J.P.; Willems, L. The role of epigenetics in malignant pleural mesothelioma. Lung Cancer 2013, 81, 311–318. [Google Scholar] [CrossRef]
- Sasaki, T.; Nakamura, T.; Rebhun, R.B.; Cheng, H.; Hale, K.S.; Tsan, R.Z.; Fidler, I.J.; Langley, R.R. Modification of the primary tumor microenvironment by transforming growth factor alpha-epidermal growth factor receptor signaling promotes metastasis in an orthotopic colon cancer model. Am. J. Pathol. 2008, 173, 205–216. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Karki, P.; Smith, K.; Johnson, J.; Lee, E. Astrocyte-derived growth factors and estrogen neuroprotection: Role of transforming growth factor-α in estrogen-induced upregulation of glutamate transporters in astrocytes. Mol. Cell. Endocrinol. 2014, 389, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Nemo, R.; Murcia, N.; Dell, K.M. Transforming growth factor alpha (TGF-alpha) and other targets of tumor necrosis factor-alpha converting enzyme (TACE) in murine polycystic kidney disease. Pediatr. Res. 2005, 57, 732–737. [Google Scholar] [CrossRef] [PubMed]
- Rossini, M.; Rizzo, P.; Bononi, I.; Clementz, A.; Ferrari, R.; Martini, F.; Tognon, M.G. New perspectives on diagnosis and therapy of malignant pleural mesothelioma. Front. Oncol. 2018, 8, 91. [Google Scholar] [CrossRef]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar] [PubMed]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef]
- Kurai, J.; Chikumi, H.; Hashimoto, K.; Takata, M.; Sako, T.; Yamaguchi, K.; Kinoshita, N.; Watanabe, M.; Touge, H.; Makino, H.; et al. Therapeutic antitumor efficacy of anti-epidermal growth factor receptor antibody, cetuximab, against malignant pleural mesothelioma. Int. J. Oncol. 2012, 41, 1610–1618. [Google Scholar] [CrossRef]
- Agarwal, V.; Lind, M.J.; Cawkwell, L. Targeted epidermal growth factor receptor therapy in malignant pleural mesothelioma: Where do we stand? Cancer Treat. Rev. 2011, 37, 533–542. [Google Scholar] [CrossRef]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.P.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II study of erlotinib in patients with malignant pleural mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E.; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L. Cancer and Leukemia Group B (CALGB 30101) Gefitinib in patients with malignant mesothelioma: A phase II study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef]
- Kryeziu, K.; Jungwirth, U.; Hoda, M.A.; Ferk, F.; Knasmüller, S.; Karnthaler-Benbakka, C.; Kowol, C.R.; Berger, W.; Heffeter, P. Synergistic anticancer activity of arsenic trioxide with erlotinib is based on inhibition of EGFR-mediated DNA double-strand break repair. Mol. Cancer Ther. 2013, 12, 1073–1084. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, K.; Murakami, H.; Taniguchi, T.; Fujii, M.; Kawata, S.; Fukui, T.; Kondo, Y.; Osada, H.; Usami, N.; Yokoi, K.; et al. Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis 2009, 30, 1097–1105. [Google Scholar] [CrossRef]
- Stoppoloni, D.; Canino, C.; Cardillo, I.; Verdina, A.; Baldi, A.; Sacchi, A.; Galati, R. Synergistic effect of gefitinib and rofecoxib in mesothelioma cells. Mol. Cancer 2010, 9, 27. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Zhai, H.X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.J.; Bao, R.; Qian, C. Discovery of CUDC-101 as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; LoRusso, P.M.; Papadopoulos, K.P.; Patnaik, A.; Beeram, M.; Smith, L.S.; Rasco, D.W.; Mays, T.A.; Chambers, G.; Ma, A.; et al. Phase I first-in-human study of CUDC-101, a multitargeted inhibitor of HDACs, EGFR, and HER2 in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 5032–5040. [Google Scholar] [CrossRef]
- Trendowski, M.; Zoino, J.N.; Christen, T.D.; Acquafondata, C.; Fondy, T.P. Preparation, In Vivo Administration, Dose-Limiting Toxicities, and Antineoplastic Activity of Cytochalasin B. Transl. Oncol. 2015, 8, 308–317. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Mönkkönen, H.; Jones, M.; Lefley, D.V.; Coleman, R.E.; Holen, I. Antitumor effects of doxorubicin followed by zoledronic acid in a mouse model of breast cancer. J. Natl. Cancer Inst. 2008, 100, 1167–1178. [Google Scholar] [CrossRef]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Singh, B.; Coffey, R.J. From wavy hair to naked proteins: The role of transforming growth factor alpha in health and disease. Semin. Cell Dev. Biol. 2014, 28, 12–21. [Google Scholar] [CrossRef]
- Scherpereel, A.; Wallyn, F.; Albelda, S.M.; Munck, C. Novel therapies for malignant pleural mesothelioma. Lancet Oncol. 2018, 19, e161–e172. [Google Scholar] [CrossRef]
- Kim, R.Y.; Sterman, D.H.; Haas, A.R. Malignant Mesothelioma: Has Anything Changed? Semin. Respir. Crit. Care Med. 2019, 40, 347–360. [Google Scholar] [CrossRef]
- Lazzari, C.; Karachaliou, N.; Bulotta, A.; Viganó, M.; Mirabile, A.; Brioschi, E.; Santarpia, M.; Gianni, L.; Rosell, R.; Gregorc, V. Combination of immunotherapy with chemotherapy and radiotherapy in lung cancer: Is this the beginning of the end for cancer? Adv. Med. Oncol. 2018, 10, 1758835918762094. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, B.L.; Anderson, R. Realizing the clinical potential of immunogenic cell death in cancer chemotherapy and radiotherapy. Int. J. Mol. Sci. 2019, 20, 959. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Gingeras, T.R. Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinformatics 2015, 51, 11–14. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA. 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staumont, B.; Jamakhani, M.; Costa, C.; Vandermeers, F.; Sriramareddy, S.N.; Redouté, G.; Mascaux, C.; Delvenne, P.; Hubert, P.; Safari, R.; et al. TGFα Promotes Chemoresistance of Malignant Pleural Mesothelioma. Cancers 2020, 12, 1484. https://doi.org/10.3390/cancers12061484
Staumont B, Jamakhani M, Costa C, Vandermeers F, Sriramareddy SN, Redouté G, Mascaux C, Delvenne P, Hubert P, Safari R, et al. TGFα Promotes Chemoresistance of Malignant Pleural Mesothelioma. Cancers. 2020; 12(6):1484. https://doi.org/10.3390/cancers12061484
Chicago/Turabian StyleStaumont, Bernard, Majeed Jamakhani, Chrisostome Costa, Fabian Vandermeers, Sathya Neelature Sriramareddy, Gaëlle Redouté, Céline Mascaux, Philippe Delvenne, Pascale Hubert, Roghaiyeh Safari, and et al. 2020. "TGFα Promotes Chemoresistance of Malignant Pleural Mesothelioma" Cancers 12, no. 6: 1484. https://doi.org/10.3390/cancers12061484
APA StyleStaumont, B., Jamakhani, M., Costa, C., Vandermeers, F., Sriramareddy, S. N., Redouté, G., Mascaux, C., Delvenne, P., Hubert, P., Safari, R., & Willems, L. (2020). TGFα Promotes Chemoresistance of Malignant Pleural Mesothelioma. Cancers, 12(6), 1484. https://doi.org/10.3390/cancers12061484