Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma

 , , , ,
, , , , 
 add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
2.1. PDOX Models Derived from Primary and Recurrent Meningiomas
2.2. PDOX Models Recapitulate the Histopathologic Features of the Patient Tumors
2.3. PDOX Models Can also be Generated from Cryopreserved Cells
2.4. Gene Expression Profiles Were Maintained in the PDOX Tumors
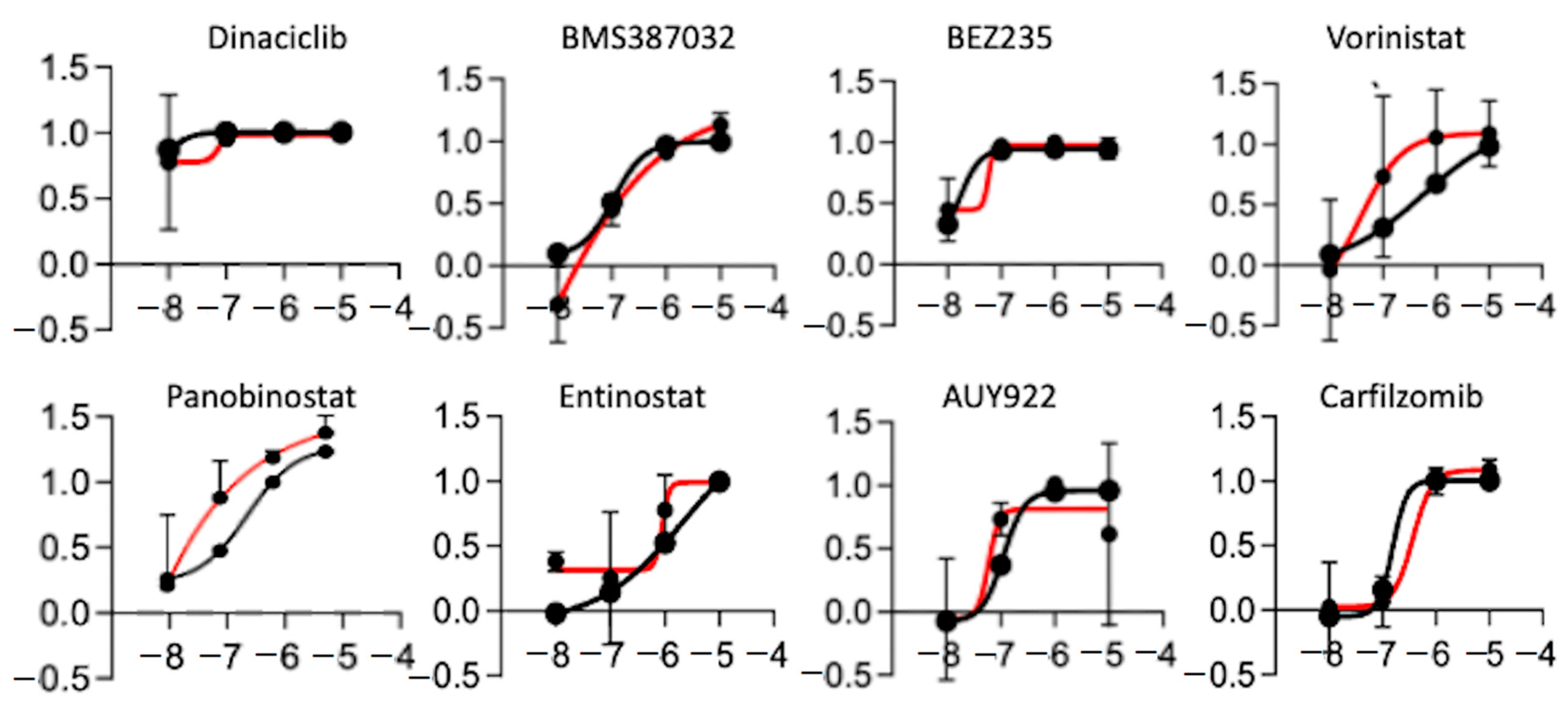
2.5. High-Throughput in Vitro Chemical Screen Identify HDAC Inhibitors
2.6. Panobinostat Significantly Prolonged Animal Survival in the Recurrent Tumor Model
3. Discussion
4. Materials and Methods
4.1. Patient Tumor Tissue Specimen
4.2. Long-Term Storage of Xenograft Cells in Liquid Nitrogen
4.3. Direct Hetero-Transplantation of Primary Tumor Cells into Mouse Brain
4.4. Immunohistochemical (IHC) Staining and Western Blot Analysis
4.5. RNA-Seq Analysis
4.6. Small-Animal Magnetic Resonance Imaging
4.7. Primary Cell Culture Establishment and Propagation
4.8. High-Throughput Drug Screening
4.9. Testing Panobinostat Therapy in Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Angus, S.P.; Oblinger, J.L.; Stuhlmiller, T.J.; DeSouza, P.A.; Beauchamp, R.L.; Witt, L.; Chen, X.; Jordan, J.T.; Gilbert, T.S.K.; Stemmer-Rachamimov, A.; et al. EPH receptor signaling as a novel therapeutic target in NF2-deficient meningioma. Neuro Oncol. 2018, 20, 1185–1196. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Truitt, G.; Boscia, A.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011–2015. Neuro Oncol. 2018, 20, iv1–iv86. [Google Scholar] [CrossRef]
- Patel, A.J.; Wan, Y.-W.; Al-Ouran, R.; Revelli, J.-P.; Cardenas, M.F.; Oneissi, M.; Xi, L.; Jalali, A.; Magnotti, J.F.; Muzny, D.M.; et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc. Natl. Acad. Sci. USA 2019, 116, 21715–21726. [Google Scholar] [CrossRef]
- Vasudevan, H.N.; Braunstein, S.E.; Phillips, J.J.; Pekmezci, M.; Tomlin, B.A.; Wu, A.; Reis, G.F.; Magill, S.T.; Zhang, J.; Feng, F.Y.; et al. Comprehensive Molecular Profiling Identifies FOXM1 as a Key Transcription Factor for Meningioma Proliferation. Cell Rep. 2018, 22, 3672–3683. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Karas, P.J.; Hadley, C.C.; Bayley, V.J.C.; Basit Khan, A.; Jalali, A.; Sweeney, A.D.; Klisch, T.J.; Patel, A.J. The role of merlin/NF2 loss in meningioma biology. Cancers 2019, 11, 1633. [Google Scholar] [CrossRef]
- Aghi, M.K.; Carter, B.S.; Cosgrove, G.R.; Ojemann, R.G.; Amin-Hanjani, S.; Martuza, R.L.; Curry, W.T.; Barker, F.G. Long-term recurrence rates of atypical meningiomas after gross total resection with or without postoperative adjuvant radiation. Neurosurgery 2009, 64, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.H. Characterization of a newly established malignant meningioma cell line of the human brain: IOMM-Lee. Neurosurgery 1990, 27, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Kargiotis, O.; Chetty, C.; Gogineni, V.; Gondi, C.S.; Pulukuri, S.M.; Kyritsis, A.P.; Gujrati, M.; Klopfenstein, J.D.; Dinh, D.H.; Rao, J.S. uPA/uPAR downregulation inhibits radiation-induced migration, invasion and angiogenesis in IOMM-Lee Meningioma cells and decreases tumor growth in vivo. Int. J. Oncol. 2008, 33, 937–947. [Google Scholar] [CrossRef]
- Tummalapalli, P.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. RNA interference-mediated targeting of urokinase plasminogen activator receptor and matrix metalloproteinase-9 gene expression in the IOMM-Lee malignant meningioma cell line inhibits tumor growth, tumor cell invasion and angiogenesis. Int. J. Oncol. 2007, 31, 5–17. [Google Scholar] [CrossRef]
- Cargioli, T.G.; Ugur, H.C.; Ramakrishna, N.; Chan, J.; Black, P.M.; Carroll, R.S. Establishment of an in vivo meningioma model with human telomerase reverse transcriptase. Neurosurgery 2007, 60, 750–760. [Google Scholar] [CrossRef]
- Mei, Y.; Bi, W.L.; Greenwald, N.F.; Agar, N.Y.; Beroukhim, R.; Dunn, G.P.; Dunn, I.F. Genomic profile of human meningioma cell lines. PLoS ONE 2017, 12, e0178322. [Google Scholar] [CrossRef]
- Ragel, B.T.; Couldwell, W.T.; Gillespie, D.L.; Wendland, M.M.; Whang, K.; Jensen, R.L. A comparison of the cell lines used in meningioma research. Surg. Neurol. 2008, 70, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Horowitz, P.M.; Santagata, S.; Jones, R.T.; Mckenna, A.; Getz, G.; Ligon, K.L.; Palescandolo, E.; Van Hummelen, P.; Ducar, M.D.; et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 2013, 45, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Harmanci, A.S.; Youngblood, M.W.; Clark, V.E.; CoÅ Kun, S.; Henegariu, O.; Duran, D.; Erson-Omay, E.Z.; Kaulen, L.D.; Lee, T.I.; Abraham, B.J.; et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Harmancl, A.S.; Bai, H.; Youngblood, M.W.; Lee, T.I.; Baranoski, J.F.; Ercan-Sencicek, A.G.; Abraham, B.J.; Weintraub, A.S.; Hnisz, D.; et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 2016, 48, 1253–1259. [Google Scholar] [CrossRef]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Özduman, K.; Avşar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef]
- Sahm, F.; Schrimpf, D.; Stichel, D.; Jones, D.T.W.; Hielscher, T.; Schefzyk, S.; Okonechnikov, K.; Koelsche, C.; Reuss, D.E.; Capper, D.; et al. DNA methylation-based classification and grading system for meningioma: A multicentre, retrospective analysis. Lancet Oncol. 2017, 18, 682–694. [Google Scholar] [CrossRef]
- Hoffman, R.M. Patient-derived orthotopic xenograft (PDOX) models of melanoma. Int. J. Mol. Sci. 2017, 18, 1875. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, J.; Wang, L. Patient-derived xenografts: A valuable platform for clinical and preclinical research in pancreatic cancer. Chin. Clin. Oncol. 2019, 8, 17. [Google Scholar] [CrossRef]
- Reddavid, R.; Corso, S.; Moya-Rull, D.; Giordano, S.; Degiuli, M. Patient-Derived Orthotopic Xenograft models in gastric cancer: A systematic review. Updates Surg. 2020, 2020, 1–16. [Google Scholar] [CrossRef]
- McCutcheon, I.E.; Friend, K.E.; Gerdes, T.M.; Zhang, B.M.; Wildrick, D.M.; Fuller, G.N. Intracranial injection of human meningioma cells in athymic mice: An orthotopic model for meningioma growth. J. Neurosurg. 2000, 92, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Baia, G.S.; Dinca, E.B.; Ozawa, T.; Kimura, E.T.; McDermott, M.W.; James, C.D.; VandenBerg, S.R.; Lal, A. An orthotopic skull base model of malignant meningioma. Brain Pathol. 2008, 18, 172–179. [Google Scholar] [CrossRef] [PubMed]
- La Cava, F.; Fringuello Mingo, A.; Irrera, P.; Di Vito, A.; Cordaro, A.; Brioschi, C.; Colombo Serra, S.; Cabella, C.; Terreno, E.; Miragoli, L. Orthotopic induction of CH 157 MN convexity and skull base meningiomas into nude mice using stereotactic surgery and MRI characterization. Anim. Model. Exp. Med. 2019, 2, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.L.; Leppla, D.; Rokosz, N.; Wurster, R.D. Matrigel augments xenograft transplantation of meningioma cells into athymic mice. Neurosurgery 1998, 42, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Michelhaugh, S.K.; Guastella, A.R.; Varadarajan, K.; Klinger, N.V.; Parajuli, P.; Ahmad, A.; Sethi, S.; Aboukameel, A.; Kiousis, S.; Zitron, I.M.; et al. Development of patient-derived xenograft models from a spontaneously immortal low-grade meningioma cell line, KCI-MENG1. J. Transl. Med. 2015, 13, 227. [Google Scholar] [CrossRef]
- Tuchen, M.; Wilisch-Neumann, A.; Daniel, E.A.; Baldauf, L.; Pachow, D.; Scholz, J.; Angenstein, F.; Stork, O.; Kirches, E.; Mawrin, C. Receptor tyrosine kinase inhibition by regorafenib/sorafenib inhibits growth and invasion of meningioma cells. Eur. J. Cancer 2017, 73, 9–21. [Google Scholar] [CrossRef]
- Salhia, B.; Rutka, J.T.; Lingwood, C.; Nutikka, A.; Van Furth, W.R. The treatment of malignant meningioma with verotoxin. Neoplasia 2002, 4, 304. [Google Scholar] [CrossRef]
- Nigim, F.; Esaki, S.I.; Hood, M.; Lelic, N.; James, M.F.; Ramesh, V.; Stemmer-Rachamimov, A.; Cahill, D.P.; Brastianos, P.K.; Rabkin, S.D.; et al. A new patient-derived orthotopic malignant meningioma model treated with oncolytic herpes simplex virus. Neuro Oncol. 2016, 18, 1278–1287. [Google Scholar] [CrossRef]
- Yu, L.; Baxter, P.A.; Voicu, H.; Gurusiddappa, S.; Zhao, Y.; Adesina, A.; Man, T.K.; Shu, Q.; Zhang, Y.J.; Zhao, X.M.; et al. A clinically relevant orthotopic xenograft model of ependymoma that maintains the genomic signature of the primary tumor and preserves cancer stem cells in vivo. Neuro Oncol. 2010, 12, 580–594. [Google Scholar] [CrossRef]
- Shu, Q.; Antalffy, B.; Su, J.M.F.; Adesina, A.; Ou, C.N.; Pietsch, T.; Blaney, S.M.; Lau, C.C.; Li, X.N. Valproic acid prolongs survival time of severe combined immunodeficient mice bearing intracerebellar orthotopic medulloblastoma xenografts. Clin. Cancer Res. 2006, 12, 4687–4694. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Serin Harmanci, A.; Harmanci, A.O.; Zhou, X. CaSpER identifies and visualizes CNV events by integrative analysis of single-cell or bulk RNA-sequencing data. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Torigata, M.; Yamakawa, D.; Takakura, N. Elevated expression of Tie1 is accompanied by acquisition of cancer stemness properties in colorectal cancer. Cancer Med. 2017, 6, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Bilican, B.; Goding, C.R. Cell cycle regulation of the T-box transcription factor tbx2. Exp. Cell Res. 2006, 312, 2358–2366. [Google Scholar] [CrossRef]
- Fukushima, H.; Yasumoto, M.; Ogasawara, S.; Akiba, J.; Kitasato, Y.; Nakayama, M.; Naito, Y.; Ishida, Y.; Okabe, Y.; Yasunaga, M.; et al. ARHGEF15 overexpression worsens the prognosis in patients with pancreatic ductal adenocarcinoma through enhancing the motility and proliferative activity of the cancer cells. Mol. Cancer 2016, 15, 32. [Google Scholar] [CrossRef]
- Cheng, S.; Zhang, X.; Huang, N.; Qiu, Q.; Jin, Y.; Jiang, D. Down-regulation of S100A9 inhibits osteosarcoma cell growth through inactivating MAPK and NF-ΚB signaling pathways. BMC Cancer 2016, 16, 253. [Google Scholar] [CrossRef]
- Kano, M.; Seki, N.; Kikkawa, N.; Fujimura, L.; Hoshino, I.; Akutsu, Y.; Chiyomaru, T.; Enokida, H.; Nakagawa, M.; Matsubara, H. MiR-145, miR-133a and miR-133b: Tumor-suppressive miRNAs target FSCN1 in esophageal squamous cell carcinoma. Int. J. Cancer 2010, 127, 2804–2814. [Google Scholar] [CrossRef]
- Chiyomaru, T.; Enokida, H.; Tatarano, S.; Kawahara, K.; Uchida, Y.; Nishiyama, K.; Fujimura, L.; Kikkawa, N.; Seki, N.; Nakagawa, M. MiR-145 and miR-133a function as tumour suppressors and directly regulate FSCN1 expression in bladder cancer. Br. J. Cancer 2010, 102, 883–891. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineagespecific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Cui, S.; Li, C.; Ema, M.; Weinstein, J.; Quaggin, S.E. Rapid isolation of glomeruli coupled with gene expression profiling identifies downstream targets in Pod1 knockout mice. J. Am. Soc. Nephrol. 2005, 16, 3247–3255. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.L.; Mercurio, A.M. VEGF targets the tumour cell. Nat. Rev. Cancer 2013, 13, 871–882. [Google Scholar] [CrossRef]
- Grasso, C.S.; Tang, Y.; Truffaux, N.; Berlow, N.E.; Liu, L.; Debily, M.A.; Quist, M.J.; Davis, L.E.; Huang, E.C.; Woo, P.J.; et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat. Med. 2015, 21, 555. [Google Scholar] [CrossRef] [PubMed]
- Selt, F.; Hohloch, J.; Hielscher, T.; Sahm, F.; Capper, D.; Korshunov, A.; Usta, D.; Brabetz, S.; Ridinger, J.; Ecker, J.; et al. Establishment and application of a novel patient-derived KIAA1549: BRAF-driven pediatric pilocytic astrocytoma model for preclinical drug testing. Oncotarget 2017, 8, 11460. [Google Scholar] [CrossRef] [PubMed]
- Kogiso, M.; Qi, L.; Lindsay, H.; Huang, Y.; Zhao, X.; Liu, Z.; Braun, F.K.; Du, Y.; Zhang, H.; Bae, G.; et al. Xenotransplantation of pediatric low grade gliomas confirms the enrichment of BRAF V600E mutation and preservation of CDKN2A deletion in a novel orthotopic xenograft mouse model of progressive pleomorphic xanthoastrocytoma. Oncotarget 2017, 8, 87455. [Google Scholar] [CrossRef] [PubMed]
- Maletzki, C.; Stier, S.; Gruenert, U.; Gock, M.; Ostwald, C.; Prall, F.; Linnebacher, M. Establishment, Characterization and Chemosensitivity of Three Mismatch Repair Deficient Cell Lines from Sporadic and Inherited Colorectal Carcinomas. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Pryor, R.; Norvaisas, P.; Marinos, G.; Best, L.; Thingholm, L.B.; Quintaneiro, L.M.; De Haes, W.; Esser, D.; Waschina, S.; Lujan, C.; et al. Host-Microbe-Drug-Nutrient Screen Identifies Bacterial Effectors of Metformin Therapy. Cell 2019, 178, 1299–1312. [Google Scholar] [CrossRef]
- Shu, Q.; Wong, K.K.; Su, J.M.; Adesina, A.M.; Yu, L.T.; Tsang, Y.T.M.; Antalffy, B.C.; Baxter, P.; Perlaky, L.; Yang, J.; et al. Direct Orthotopic Transplantation of Fresh Surgical Specimen Preserves CD133 + Tumor Cells in Clinically Relevant Mouse Models of Medulloblastoma and Glioma. Stem Cells 2008, 26, 1414–1424. [Google Scholar] [CrossRef]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Cheek, P.J.; McCullagh, P.; Nelder, J.A. Generalized Linear Models, 2nd ed.; Chapman&Hall/CRC Press: London, UK, 1989. [Google Scholar] [CrossRef]
- Chauvin, C.; Leruste, A.; Tauziede-Espariat, A.; Andrianteranagna, M.; Surdez, D.; Lescure, A.; Han, Z.Y.; Anthony, E.; Richer, W.; Baulande, S.; et al. High-Throughput Drug Screening Identifies Pazopanib and Clofilium Tosylate as Promising Treatments for Malignant Rhabdoid Tumors. Cell Rep. 2017, 21, 1737–1745. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor ID | Age/Gender | WHO Grade | Type | Cell Tumorigenicity | ||
|---|---|---|---|---|---|---|
| Fresh | Cryo | |||||
| K29 | (P) rimary | 72/F | Atypical (II) | C | 7/10 | 3/3 |
| (R) ecurrent | 72/F | Anaplastic (III) | C | 1/4 | 9/10 | |
| K57 | (P) rimary | 65/M | Atypical (II) | C | 0/10 | - |
| K16 | (P) rimary | 71/F | Atypical (II) | C | 0/12 | - |
| (R) ecurrent | 72/F | Atypical (III) * | B | 0/10 | - | |
| K18 | (P) rimary | 27/M | Atypical (II) | C | 0/10 | - |
| K20 | (P) rimary | 38/M | Atypical (II) | A | 0/10 | - |
| K21 | (P) rimary | 59/M | Classic (I) | Not sequenced | 0/10 | - |
| K26 | (P) rimary | 70/F | Classic (I) | A | 0/5 | - |
| K28 | (P) rimary | 33/F | Classic (I) | A | 0/10 | - |
| K30 | (P) rimary | 41/F | Classic (I) | B | 0/5 | - |
| Drug Name | FDA Approved | Drug Name | FDA Approved |
|---|---|---|---|
| Barasertib | Trichostatin | ||
| Bortezomib | yes | Cediranib | |
| Decitabine | yes | Dasatinib | yes |
| Enzastaurin | Panobinostat | ||
| Lapatinib | yes | SAHA | yes |
| MLN8237 | SNS-032 | ||
| NAC | yes | Carfilzomib | |
| Pazopanib | yes | Alvocidib | |
| Ponatinib | AUY922 | ||
| Quinacrine | yes | AZD-8931 | |
| RO4929097 | BEZ 235 | ||
| Ruxolitinib | Cabozantinib | ||
| Selumetinib | Cilengitide | ||
| SJ-172550 | Dinaciclib | ||
| Sodium butyrate | yes | Entinostat | |
| Sorafenib | yes | Fenretinide | |
| SP600125 | Fostamatinib | ||
| Temsirolimus | yes | Lenalidomide | |
| Veliparib | Nilotinib | ||
| Vismodegib | PF 562271 | ||
| Zibotentan | Retinoic acid | ||
| Crizotinib | yes | RO5045337 | |
| GANT61 | Saracatinib | ||
| BMS 754807 | SB-431542 | ||
| OSI-906 | Tamoxifen | ||
| MK-2206 | Vandetanib | ||
| Pelitinib | Vemurafenib | ||
| BIX 01294 | VER-155008 | ||
| Tozasertib | ABT 737 | ||
| Obatoclax | SJ-172550 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Qi, L.; Du, Y.; Huang, L.F.; Braun, F.K.; Kogiso, M.; Zhao, Y.; Li, C.; Lindsay, H.; Zhao, S.; et al. Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma. Cancers 2020, 12, 1478. https://doi.org/10.3390/cancers12061478
Zhang H, Qi L, Du Y, Huang LF, Braun FK, Kogiso M, Zhao Y, Li C, Lindsay H, Zhao S, et al. Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma. Cancers. 2020; 12(6):1478. https://doi.org/10.3390/cancers12061478
Chicago/Turabian StyleZhang, Huiyuan, Lin Qi, Yuchen Du, L. Frank Huang, Frank K. Braun, Mari Kogiso, Yanling Zhao, Can Li, Holly Lindsay, Sibo Zhao, and et al. 2020. "Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma" Cancers 12, no. 6: 1478. https://doi.org/10.3390/cancers12061478
APA StyleZhang, H., Qi, L., Du, Y., Huang, L. F., Braun, F. K., Kogiso, M., Zhao, Y., Li, C., Lindsay, H., Zhao, S., Injac, S. G., Baxter, P. A., Su, J. M., Stephan, C., Keller, C., Heck, K. A., Harmanci, A., Harmanci, A. O., Yang, J., ... Patel, A. J. (2020). Patient-Derived Orthotopic Xenograft (PDOX) Mouse Models of Primary and Recurrent Meningioma. Cancers, 12(6), 1478. https://doi.org/10.3390/cancers12061478