Insights into the Role of Estrogen Receptor β in Triple-Negative Breast Cancer

 , , , , , ,
, , , , , ,  and
and 
Abstract

1. Introduction
2. Pathological Features of TNBC
3. ERβ Structure and Roles in TNBC
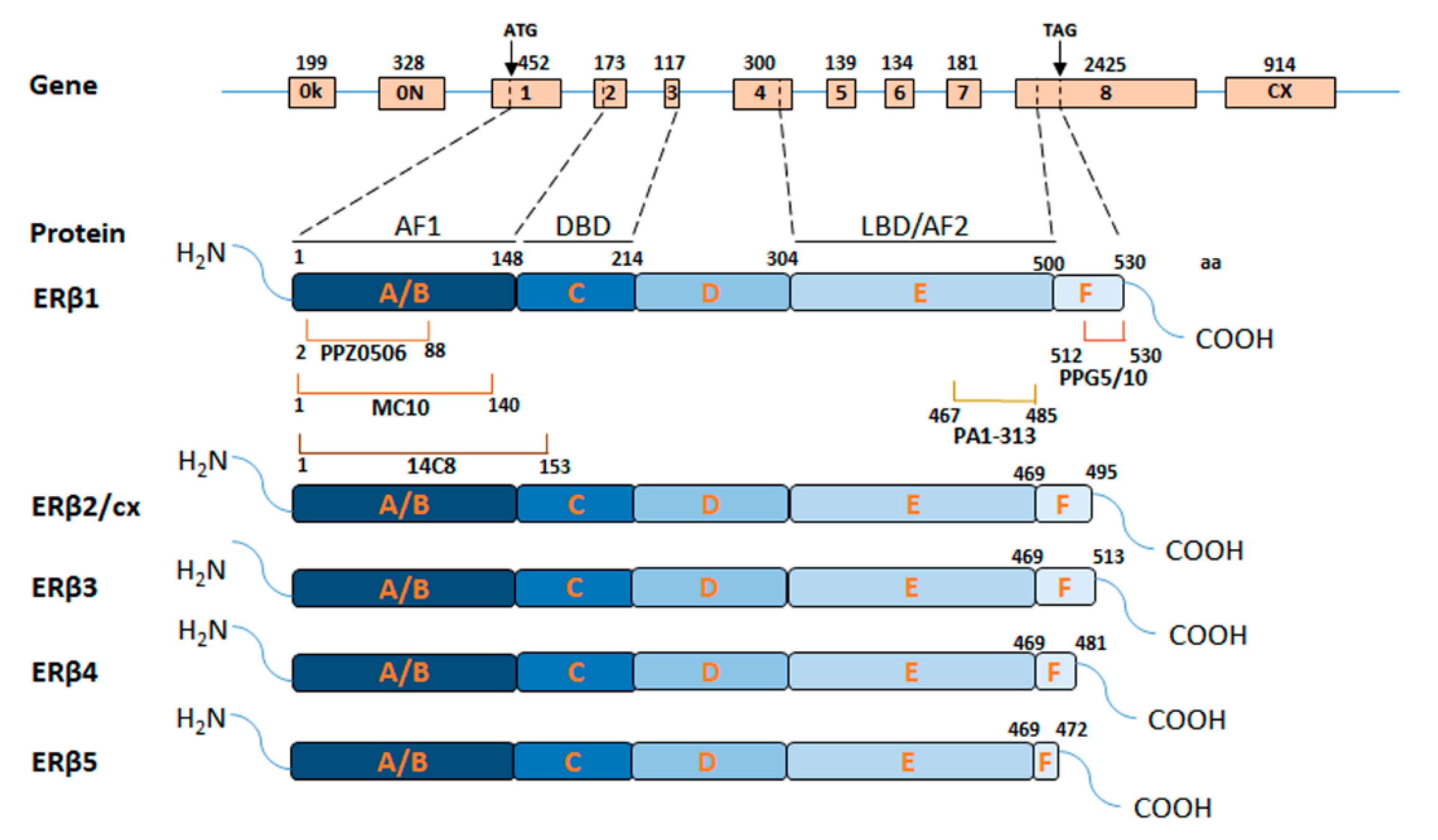
3.1. ERβ Domains and Isoforms
3.2. Issues Raised by Available ERβ Antibodies
3.3. ERβ Ligands and Their Role in TNBC
3.4. ERβ Prognostic Significance in TNBC
4. ERβ Mediated Signaling Pathways in TNBC
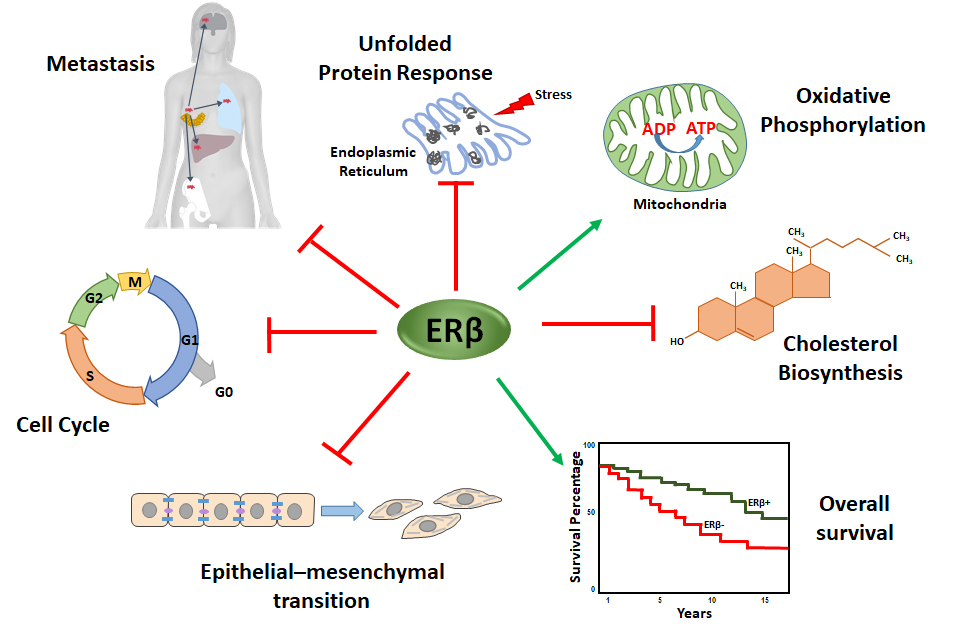
4.1. ERβ Effect on Proliferation and Cell Cycle Progression of TNBC
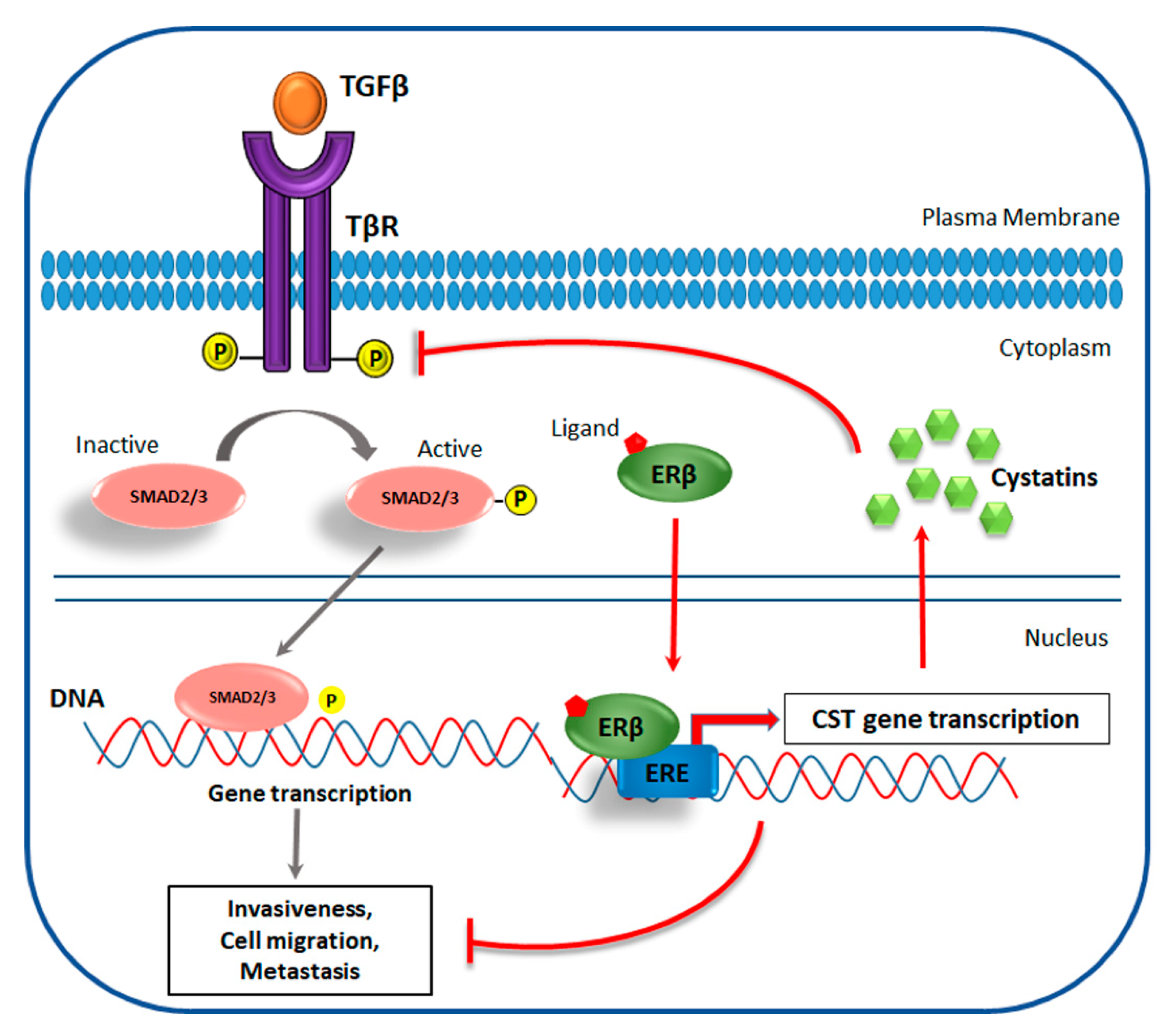
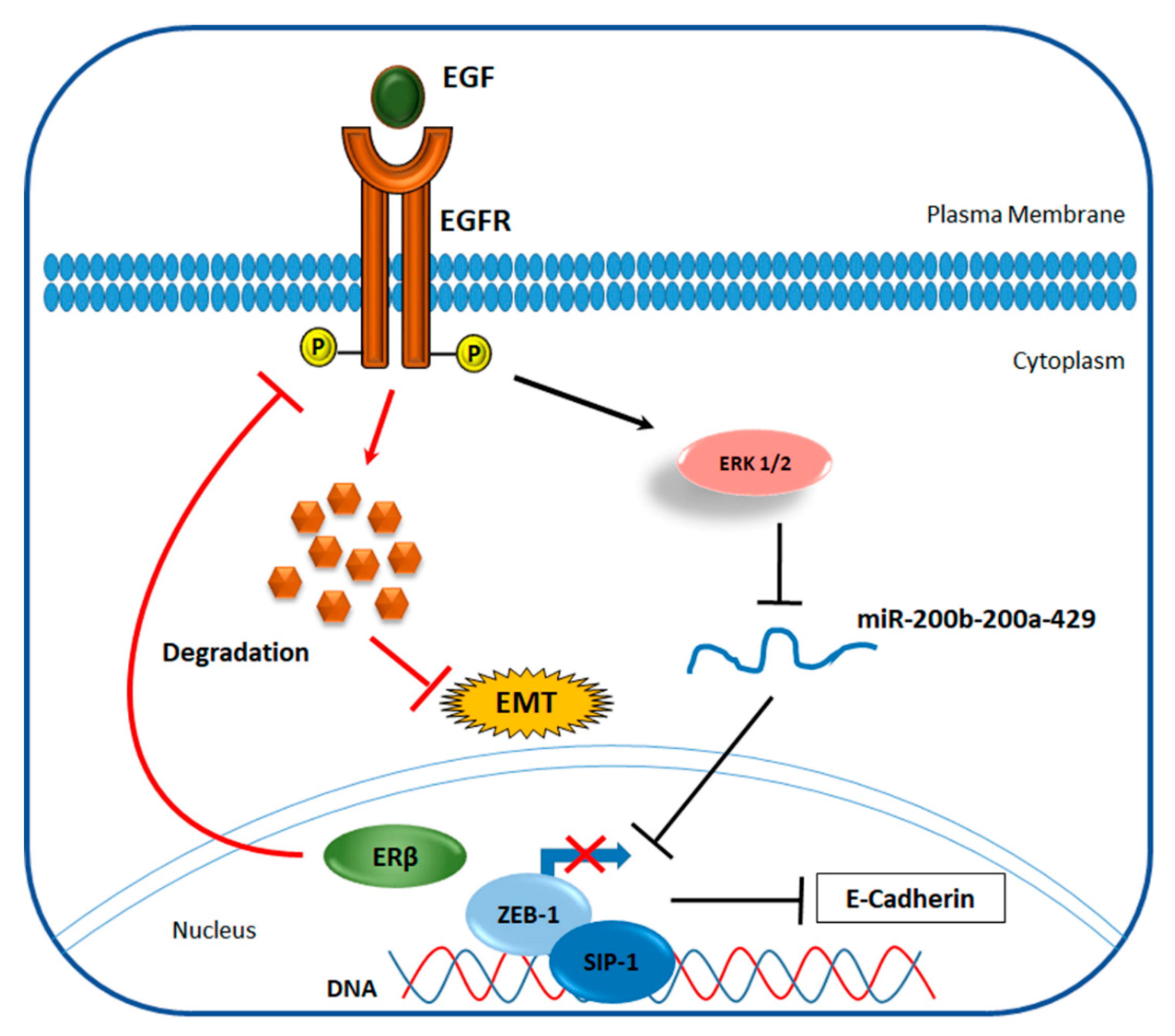
4.2. ERβ Effect on Invasiveness of TNBC
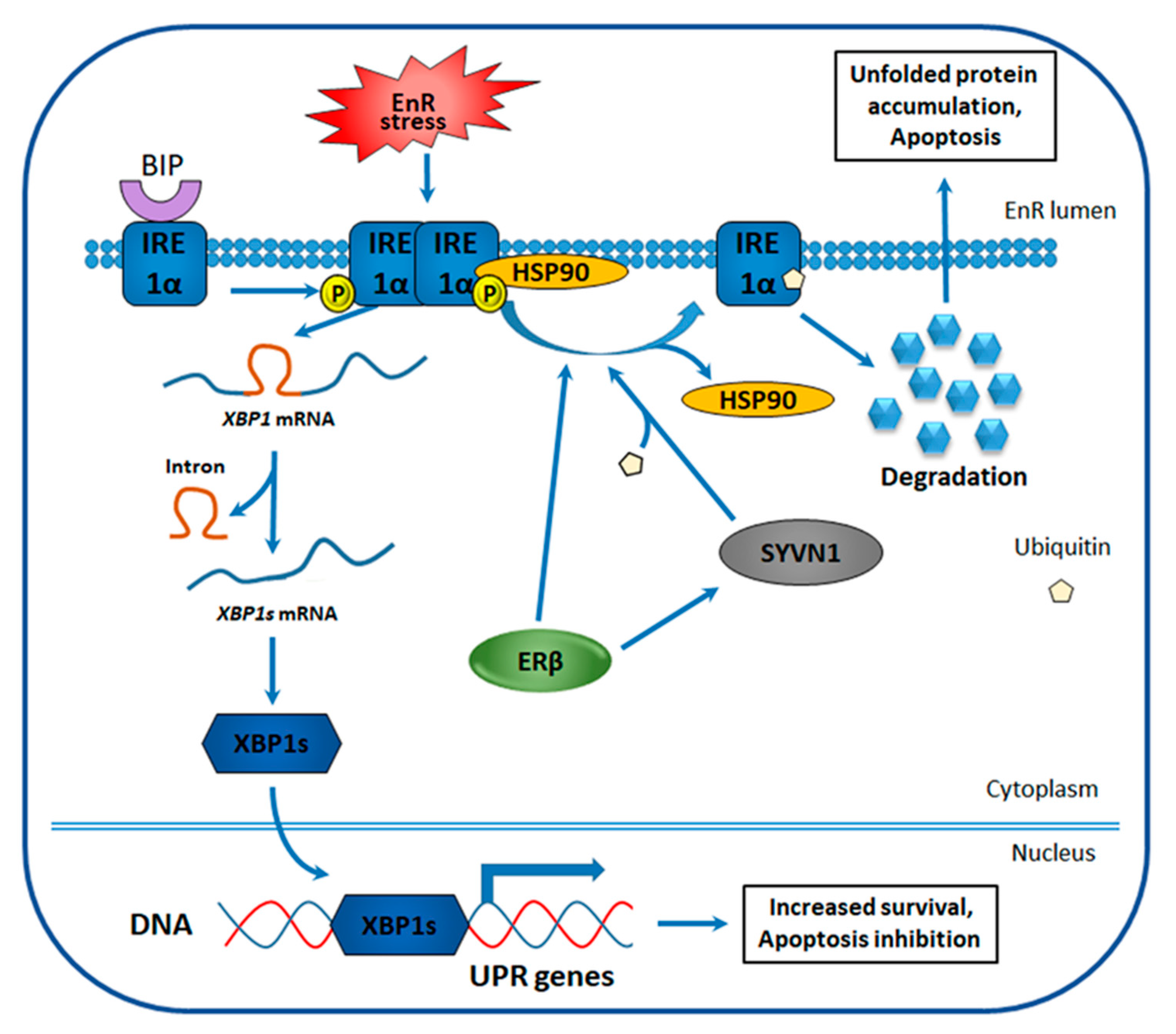
4.3. ERβ Effect on the Unfolded Protein Response in TNBC
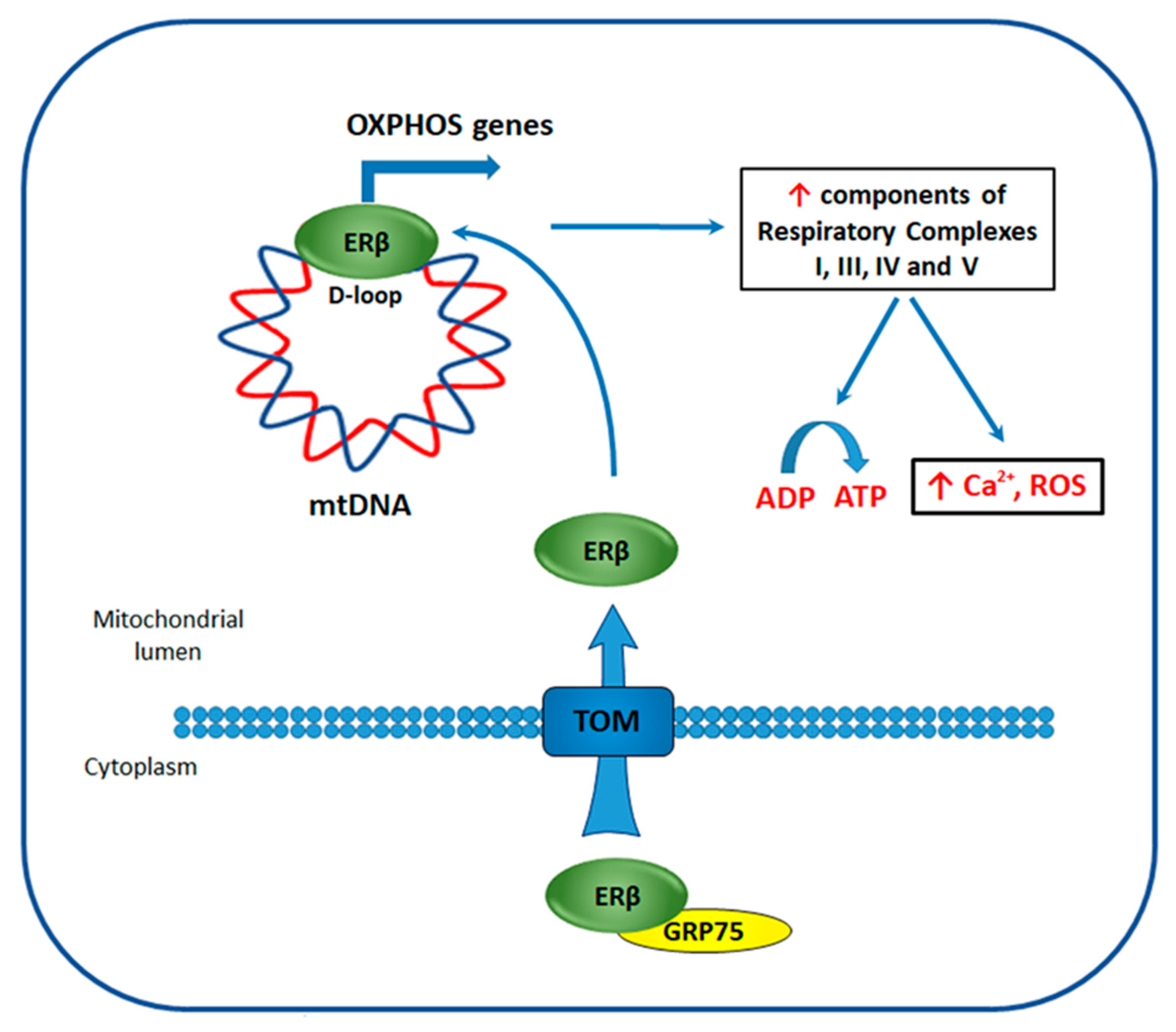
4.4. ERβ Effect on the Bioenergetics of TNBC
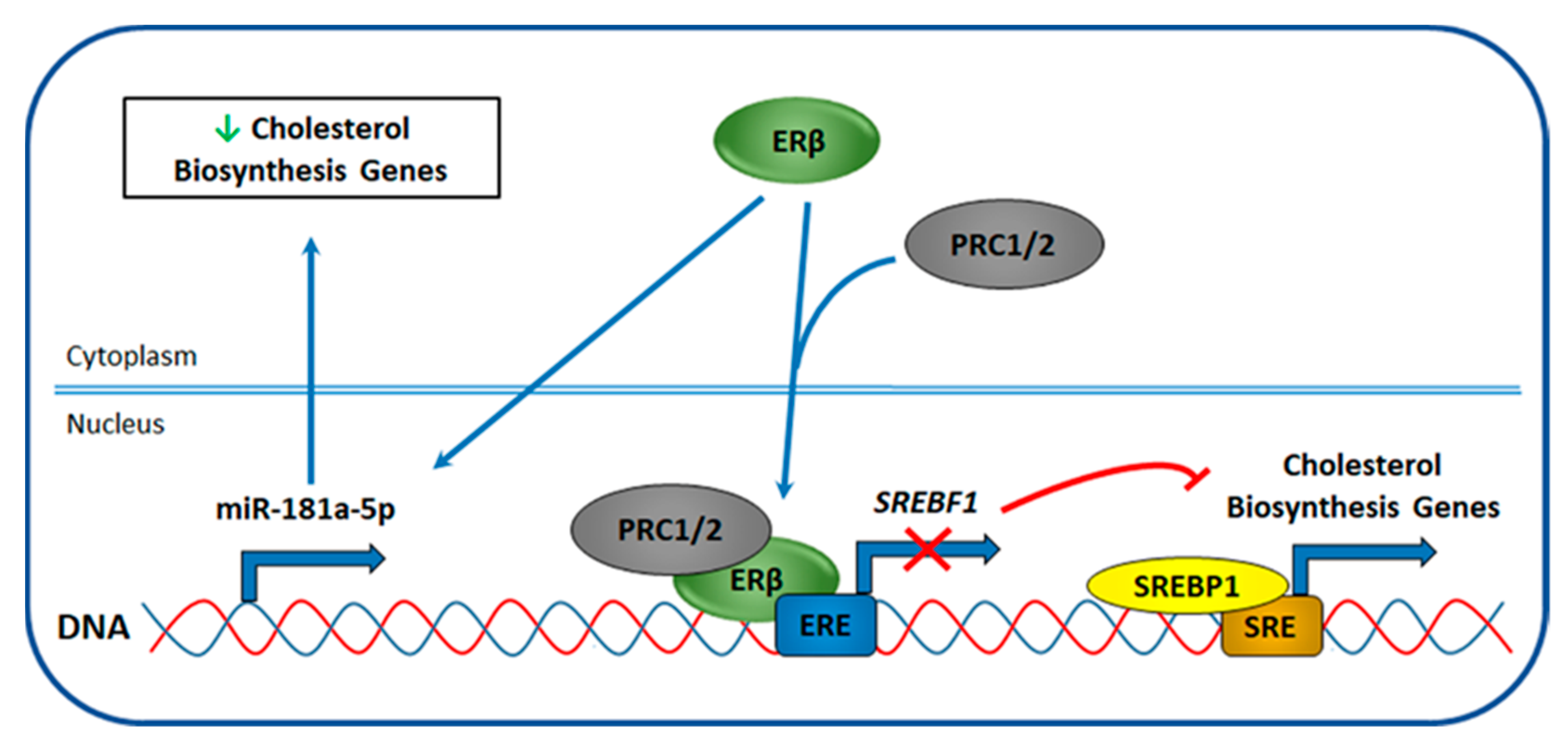
4.5. ERβ Effect on Cholesterol Biosynthesis
4.6. ERβ Effect on AR Signaling Pathways
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 4HT | (Z)-4-hydroxy-tamoxifen |
| ATP | Adenosine Triphosphate |
| AF1/AF2 domain | Activation Function 1/2 domain |
| AKT | Protein Kinase B |
| AR | Androgen Receptor |
| ARE | Androgen-Responsive Element |
| BC | Breast Cancer |
| BIP | Binding Immunoglobulin Protein |
| Cas | CRISPR-associated protein |
| Csc25C | M-phase inducer phosphatase 3 |
| CD24 | CD24 antigen |
| CD44 | CD44 antigen |
| CDH1 | Cadherin-1 |
| CDK | Cyclin-Dependent Kinase |
| CDKN1A | Cyclin-Dependent Kinase Inhibitor 1A |
| ChIP-Seq | Chromatin Immunoprecipitation Sequencing |
| Chk1 | Checkpoint Kinase 1 |
| CO | Cytochrome C Oxidase |
| CRISPR | Clustered Regularly Interspaced Short Palindromic Repeat |
| D-loop | mtDNA Displacement loop |
| DBD | DNA-Binding Domain |
| DFS | Disease-Free Survival |
| DKK1 | Dickkopf-related protein 1 |
| DMFS | Distant Metastasis-Free Survival |
| DPN | 3-bis(4-hydroyphenyl)-propionitrile |
| E2 | 17β-estradiol |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial to Mesenchymal Transition |
| EnR | Endoplasmic Reticulum |
| ERα | Estrogen Receptor α |
| ERβ | Estrogen Receptor β |
| ERE | Estrogen Response Element |
| ERK1/2 | Extracellular Regulated MAP Kinase1/2 |
| GRP75 | Glucose-Regulated Protein 75 |
| HER2/neu | Human Epidermal growth factor Receptor 2 |
| HIF1α | Hypoxia-Inducing Factor 1α |
| HSP90 | Heat Shock Protein 90 |
| IF | Immunofluorescence |
| IHC | Immunohistochemistry |
| IL-1β | Interleukin 1 beta |
| IP | Immunoprecipitation |
| IP-MS | Immunoprecipitation coupled to Mass Spectrometry |
| IRE1α | Inositol-Requiring Enzyme 1α |
| KO | Knockout |
| LAR | Luminal Androgen Receptor |
| LBD domain | Ligand-Binding Domain |
| mAB | Monoclonal Antibody |
| mitoERβ | Mitochondria-targeted ERβ |
| MS | Mass Spectrometry |
| mtDNA | Mitochondrial DNA |
| mTOR | Mammalian Target of Rapamycin |
| NGS | Next Generation Sequencing |
| OS | Overall Survival |
| OXPHOS | Oxidative Phosphorylation |
| p53 | Tumor protein p53 |
| pAB | Polyclonal Antibody |
| PI3K | Phosphatidylinositol 3-Kinase |
| PR | Progesterone Receptor |
| PRC1/2 | Polycomb Repressor Complex 1/2 |
| RAS | Protein belonging to small GTPases superfamily |
| RIME | Rapid Immunoprecipitation Mass spectrometry of Endogenous proteins |
| RORγ | RAR-related Orphan Receptor Gamma |
| ROS | Reactive Oxygen Species |
| RTK | Receptor Tyrosine Kinase |
| SMAD | Homolog of the Caenorhabditis elegans SMA and Drosophila MAD family of genes |
| SERD | Selective Estrogen Receptor Degrader |
| SERM | Selective Estrogen Receptor Modulator |
| SIP1 | Survival of motor neuron protein-Interacting protein 1 |
| SRE | Sterol Regulatory Element |
| SREBF | Sterol Regulatory Element Binding Factor |
| SREBP | Sterol Regulatory Element-Binding Protein |
| SYVN1 | Synoviolin 1 |
| TGFβ | Transforming Growth Factor beta |
| TβR | Receptor of TGFβ |
| TNBC | Triple-Negative Breast Cancer |
| TOM | Translocase of the Outer Membrane of mitochondria |
| TPR | Tetraticopeptide Repeat motifs |
| UPR | Unfolded Protein Response |
| WB | Western Blot |
| WNT4 | Wnt family member 4 |
| XBP1 | X-box-Binding Protein 1 |
| XBP1s | Spliced XBP1 |
| ZEB1 | Zinc finger E-box-binding homeobox 1 |
References
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef]
- Deroo, B.J.; Korach, K.S. Estrogen receptors and human disease. J. Clin. Investig. 2006, 116, 561–570. [Google Scholar] [CrossRef]
- Ali, S.; Coombes, R.C. Estrogen receptor alpha in human breast cancer: Occurrence and significance. J. Mammary Gland Biol. Neoplasia 2000, 5, 271–281. [Google Scholar] [CrossRef]
- Kuiper, G.G.; Enmark, E.; Pelto-Huikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef]
- Speirs, V.; Skliris, G.P.; Burdall, S.E.; Carder, P.J. Distinct expression patterns of ER alpha and ER beta in normal human mammary gland. J. Clin. Pathol. 2002, 55, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E.; Dotzlaw, H.; Watson, P.H.; Murphy, L.C. Altered estrogen receptor α and β messenger RNA expression during human breast tumorigenesis. Cancer Res. 1998, 58, 3197–3201. [Google Scholar] [PubMed]
- Mehta, R.G.; Hawthorne, M.; Mehta, R.R.; Torres, K.E.; Peng, X.; McCormick, D.L.; Kopelovich, L. Differential roles of ERalpha and ERbeta in normal and neoplastic development in the mouse mammary gland. PLoS ONE 2014, 9, e113175. [Google Scholar] [CrossRef] [PubMed]
- Antal, M.C.; Krust, A.; Chambon, P.; Mark, M. Sterility and absence of histopathological defects in nonreproductive organs of a mouse ERbeta-null mutant. Proc. Natl. Acad. Sci. USA 2008, 105, 2433–2438. [Google Scholar] [CrossRef]
- Palmieri, C.; Cheng, G.J.; Saji, S.; Zelada-Hedman, M.; Warri, A.; Weihua, Z.; Van Noorden, S.; Wahlstrom, T.; Coombes, R.C.; Warner, M.; et al. Estrogen receptor beta in breast cancer. Endocr. Relat. Cancer 2002, 9, 1–13. [Google Scholar] [CrossRef]
- Leygue, E.; Murphy, L.C. A bi-faceted role of estrogen receptor beta in breast cancer. Endocr. Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef]
- Roger, P.; Sahla, M.E.; Mäkelä, S.; Gustafsson, J.Å.; Baldet, P.; Rochefort, H. Decreased expression of estrogen receptor β protein in proliferative preinvasive mammary tumors. Cancer Res. 2001, 61, 2537–2541. [Google Scholar] [PubMed]
- Järvinen, T.A.; Pelto-Huikko, M.; Holli, K.; Isola, J. Estrogen receptor β is coexpressed with ERα and PR and associated with nodal status, grade, and proliferation rate in breast cancer. Am. J. Pathol. 2000, 156, 29–35. [Google Scholar] [CrossRef]
- Shaaban, A.M.; O’Neill, P.A.; Davies, M.P.; Sibson, R.; West, C.R.; Smith, P.H.; Foster, C.S. Declining estrogen receptor-β expression defines malignant progression of human breast neoplasia. Am. J. Surg. Pathol. 2003, 27, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Chantzi, Ν.Ι.; Palaiologou, M.; Stylianidou, A.; Goutas, N.; Vassilaros, S.; Kourea, H.P.; Dhimolea, E.; Mitsiou, D.J.; Tiniakos, D.G.; Alexis, Μ.N. Estrogen receptor β2 is inversely correlated with Ki-67 in hyperplastic and noninvasive neoplastic breast lesions. J. Cancer Res. Clin. Oncol. 2014, 140, 1057–1066. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Omoto, Y.; Iwase, H.; Yamashita, H.; Toyama, T.; Coombes, R.C.; Filipovic, A.; Warner, M.; Gustafsson, J.-Å. Differential expression of estrogen receptor α, β1, and β2 in lobular and ductal breast cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 1933–1938. [Google Scholar] [CrossRef]
- Shaaban, A.M.; Jarvis, C.; Moore, F.; West, C.; Dodson, A.; Foster, C.S. Prognostic significance of estrogen receptor Beta in epithelial hyperplasia of usual type with known outcome. Am. J. Surg. Pathol. 2005, 29, 1593–1599. [Google Scholar] [CrossRef]
- Prins, G.S.; Korach, K.S. The role of estrogens and estrogen receptors in normal prostate growth and disease. Steroids 2008, 73, 233–244. [Google Scholar] [CrossRef]
- Bardin, A.; Boulle, N.; Lazennec, G.; Vignon, F.; Pujol, P. Loss of ERβ expression as a common step in estrogen-dependent tumor progression. Endocr. Relat. Cancer 2004, 11, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Lam, E.W.; Sunters, A.; Enmark, E.; De Bella, M.T.; Coombes, R.C.; Gustafsson, J.-Å.; Dahlman-Wright, K. Expression of estrogen receptor β isoforms in normal breast epithelial cells and breast cancer: regulation by methylation. Oncogene 2003, 22, 7600–7606. [Google Scholar] [CrossRef]
- Warner, M.; Wu, W.-F.; Montanholi, L.; Nalvarte, I.; Antonson, P.; Gustafsson, J.-A. Ventral prostate and mammary gland phenotype in mice with complete deletion of the ERβ gene. Proc. Natl. Acad. Sci. USA 2020, 117, 4902–4909. [Google Scholar] [CrossRef]
- Gustafsson, J.A.; Strom, A.; Warner, M. Update on ERbeta. J. Steroid Biochem. Mol. Biol. 2019, 191, 105312. [Google Scholar] [CrossRef] [PubMed]
- Paris, O.; Ferraro, L.; Grober, O.M.; Ravo, M.; De Filippo, M.R.; Giurato, G.; Nassa, G.; Tarallo, R.; Cantarella, C.; Rizzo, F.; et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor beta in hormone-responsive breast cancer. Oncogene 2012, 31, 4196–4206. [Google Scholar] [CrossRef]
- Gruvberger-Saal, S.K.; Bendahl, P.O.; Saal, L.H.; Laakso, M.; Hegardt, C.; Eden, P.; Peterson, C.; Malmstrom, P.; Isola, J.; Borg, A.; et al. Estrogen receptor beta expression is associated with tamoxifen response in ERalpha-negative breast carcinoma. Clin. Cancer Res. 2007, 13, 1987–1994. [Google Scholar] [CrossRef]
- Honma, N.; Horii, R.; Iwase, T.; Saji, S.; Younes, M.; Takubo, K.; Matsuura, M.; Ito, Y.; Akiyama, F.; Sakamoto, G. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J. Clin. Oncol. 2008, 26, 3727–3734. [Google Scholar] [CrossRef] [PubMed]
- Fox, E.M.; Davis, R.J.; Shupnik, M.A. ERβ in breast cancer—Onlooker, passive player, or active protector? Steroids 2008, 73, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E.; Murphy, L. Comparative evaluation of ERalpha and ERbeta significance in breast cancer: State of the art. Expert Rev. Endocrinol. Metab. 2011, 6, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Gustafsson, J.A. The different roles of ER subtypes in cancer biology and therapy. Nat. Rev. Cancer 2011, 11, 597–608. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.-K.; Mak, P.; Hassan, S.; Ho, S.-M. Estrogen receptor (ER)-β isoforms: A key to understanding ER-β signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef]
- Murphy, L.C.; Leygue, E. The role of estrogen receptor-β in breast cancer. Semin. Reprod. Med. 2012, 30, 5–13. [Google Scholar] [CrossRef]
- Skliris, G.P.; Leygue, E.; Curtis-Snell, L.; Watson, P.H.; Murphy, L.C. Expression of oestrogen receptor-beta in oestrogen receptor-alpha negative human breast tumours. Br. J. Cancer 2006, 95, 616–626. [Google Scholar] [CrossRef]
- Novelli, F.; Milella, M.; Melucci, E.; Di Benedetto, A.; Sperduti, I.; Perrone-Donnorso, R.; Perracchio, L.; Venturo, I.; Nistico, C.; Fabi, A.; et al. A divergent role for estrogen receptor-beta in node-positive and node-negative breast cancer classified according to molecular subtypes: An observational prospective study. Breast Cancer Res. 2008, 10, R74. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.; Giurato, G.; Saggese, P.; Pecoraro, G.; Lamberti, J.; Ravo, M.; Rizzo, F.; Rocco, D.; Tarallo, R.; Nyman, T.A.; et al. Interaction Proteomics Identifies ERbeta Association with Chromatin Repressive Complexes to Inhibit Cholesterol Biosynthesis and Exert An Oncosuppressive Role in Triple-negative Breast Cancer. Mol. Cell Proteom. 2020, 19, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Austin, D.; Hamilton, N.; Elshimali, Y.; Pietras, R.; Wu, Y.; Vadgama, J. Estrogen receptor-beta is a potential target for triple negative breast cancer treatment. Oncotarget 2018, 9, 33912–33930. [Google Scholar] [CrossRef] [PubMed]
- Skliris, G.P.; Leygue, E.; Watson, P.H.; Murphy, L.C. Estrogen receptor alpha negative breast cancer patients: Estrogen receptor beta as a therapeutic target. J. Steroid Biochem. Mol. Biol. 2008, 109, 1–10. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Reis-Filho, J.S.; Pusztai, L. Gene expression profiling in breast cancer: Classification, prognostication, and prediction. Lancet 2011, 378, 1812–1823. [Google Scholar] [CrossRef]
- Rakha, E.A.; Reis-Filho, J.S.; Ellis, I.O. Combinatorial biomarker expression in breast cancer. Breast Cancer Res. Treat 2010, 120, 293–308. [Google Scholar] [CrossRef]
- Tang, P.; Tse, G.M. Immunohistochemical Surrogates for Molecular Classification of Breast Carcinoma: A 2015 Update. Arch. Pathol. Lab. Med. 2016, 140, 806–814. [Google Scholar] [CrossRef]
- Perou, C.M. Molecular stratification of triple-negative breast cancers. Oncologist 2011, 16, 61–70. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.J.; Naidu, S.; Topham, A.K.; Guiles, F.; Xu, Y.; McCue, P.; Schwartz, G.F.; Park, P.K.; Rosenberg, A.L.; Brill, K.; et al. Differences in breast carcinoma characteristics in newly diagnosed African-American and Caucasian patients: A single-institution compilation compared with the National Cancer Institute’s Surveillance, Epidemiology, and End Results database. Cancer 2007, 110, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Rakha, E.A.; El-Sayed, M.E.; Green, A.R.; Lee, A.H.; Robertson, J.F.; Ellis, I.O. Prognostic markers in triple-negative breast cancer. Cancer 2007, 109, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Oakman, C.; Viale, G.; Di Leo, A. Management of triple negative breast cancer. Breast 2010, 19, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Penault-Llorca, F.; Viale, G. Pathological and molecular diagnosis of triple-negative breast cancer: A clinical perspective. Ann. Oncol. 2012, 23, vi19–vi22. [Google Scholar] [CrossRef]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef]
- Anders, C.K.; Abramson, V.; Tan, T.; Dent, R. The Evolution of Triple-Negative Breast Cancer: From Biology to Novel Therapeutics. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 34–42. [Google Scholar] [CrossRef]
- da Silva, J.L.; Cardoso Nunes, N.C.; Izetti, P.; de Mesquita, G.G.; de Melo, A.C. Triple negative breast cancer: A thorough review of biomarkers. Crit. Rev. Oncol. Hematol. 2020, 145, 102855. [Google Scholar] [CrossRef]
- Mosselman, S.; Polman, J.; Dijkema, R. ER beta: Identification and characterization of a novel human estrogen receptor. FEBS Lett. 1996, 392, 49–53. [Google Scholar] [CrossRef]
- Nilsson, S.; Gustafsson, J.A. Estrogen receptors: Therapies targeted to receptor subtypes. Clin. Pharmacol. Ther. 2011, 89, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dahlman-Wright, K.; Gustafsson, J.A. Estrogen receptor beta: An overview and update. Nucl. Recept. Signal 2008, 6, e003. [Google Scholar] [CrossRef] [PubMed]
- Pike, A.C.; Brzozowski, A.M.; Hubbard, R.E.; Bonn, T.; Thorsell, A.G.; Engstrom, O.; Ljunggren, J.; Gustafsson, J.A.; Carlquist, M. Structure of the ligand-binding domain of oestrogen receptor beta in the presence of a partial agonist and a full antagonist. EMBO J. 1999, 18, 4608–4618. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Li, X.; Blanchard, A.; Bramwell, V.H.; Pritchard, K.I.; Tu, D.; Shepherd, L.; Myal, Y.; Penner, C.; Watson, P.H.; et al. Expression of both estrogen receptor-beta 1 (ER-beta1) and its co-regulator steroid receptor RNA activator protein (SRAP) are predictive for benefit from tamoxifen therapy in patients with estrogen receptor-alpha (ER-alpha)-negative early breast cancer (EBC). Ann. Oncol. 2013, 24, 1986–1993. [Google Scholar] [CrossRef]
- Leung, Y.K.; Lee, M.T.; Lam, H.M.; Tarapore, P.; Ho, S.M. Estrogen receptor-beta and breast cancer: Translating biology into clinical practice. Steroids 2012, 77, 727–737. [Google Scholar] [CrossRef]
- Haldosen, L.A.; Zhao, C.; Dahlman-Wright, K. Estrogen receptor beta in breast cancer. Mol. Cell. Endocrinol. 2014, 382, 665–672. [Google Scholar] [CrossRef]
- Zhao, C.; Matthews, J.; Tujague, M.; Wan, J.; Strom, A.; Toresson, G.; Lam, E.W.; Cheng, G.; Gustafsson, J.A.; Dahlman-Wright, K. Estrogen receptor β2 negatively regulates the transactivation of estrogen receptor alpha in human breast cancer cells. Cancer Res. 2007, 67, 3955–3962. [Google Scholar] [CrossRef]
- Bozkurt, K.K.; Kapucuoglu, N. Investigation of immunohistochemical ERalpha, ERbeta and ERbetacx expressions in normal and neoplastic breast tissues. Pathol. Res. Pract. 2012, 208, 133–139. [Google Scholar] [CrossRef]
- Girgert, R.; Emons, G.; Grundker, C. Estrogen Signaling in ERalpha-Negative Breast Cancer: ERbeta and GPER. Front. Endocrinol. 2018, 9, 781. [Google Scholar] [CrossRef]
- Tong, D.; Schuster, E.; Seifert, M.; Czerwenka, K.; Leodolte, S.; Zeillinger, R. Expression of estrogen receptor beta isoforms in human breast cancer tissues and cell lines. Breast Cancer Res. Treat 2002, 71, 249–255. [Google Scholar] [CrossRef]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and characterization of human estrogen receptor beta isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Chantzi, N.I.; Tiniakos, D.G.; Palaiologou, M.; Goutas, N.; Filippidis, T.; Vassilaros, S.D.; Dhimolea, E.; Mitsiou, D.J.; Alexis, M.N. Estrogen receptor β2 is associated with poor prognosis in estrogen receptor alpha-negative breast carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 1489–1498. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Wimberly, H.; Han, G.; Pinnaduwage, D.; Murphy, L.C.; Yang, X.R.; Andrulis, I.L.; Sherman, M.; Figueroa, J.; Rimm, D.L. ERbeta splice variant expression in four large cohorts of human breast cancer patient tumors. Breast Cancer Res. Treat 2014, 146, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Subramaniam, M.; Negron, V.; Cicek, M.; Reynolds, C.; Lingle, W.L.; Goetz, M.P.; Ingle, J.N.; Spelsberg, T.C.; Hawse, J.R. Development, characterization, and applications of a novel estrogen receptor beta monoclonal antibody. J. Cell. Biochem. 2012, 113, 711–723. [Google Scholar] [CrossRef]
- Shanle, E.K.; Onitilo, A.A.; Huang, W.; Kim, K.; Zang, C.; Engel, J.M.; Xu, W.; Wisinski, K.B. Prognostic significance of full-length estrogen receptor beta expression in stage I-III triple negative breast cancer. Am. J. Transl. Res. 2015, 7, 1246–1259. [Google Scholar]
- Andersson, S.; Sundberg, M.; Pristovsek, N.; Ibrahim, A.; Jonsson, P.; Katona, B.; Clausson, C.M.; Zieba, A.; Ramstrom, M.; Soderberg, O.; et al. Insufficient antibody validation challenges oestrogen receptor beta research. Nat. Commun. 2017, 8, 15840. [Google Scholar] [CrossRef]
- Nelson, A.W.; Groen, A.J.; Miller, J.L.; Warren, A.Y.; Holmes, K.A.; Tarulli, G.A.; Tilley, W.D.; Katzenellenbogen, B.S.; Hawse, J.R.; Gnanapragasam, V.J.; et al. Comprehensive assessment of estrogen receptor beta antibodies in cancer cell line models and tissue reveals critical limitations in reagent specificity. Mol. Cell. Endocrinol. 2017, 440, 138–150. [Google Scholar] [CrossRef]
- Tremblay, A.; Tremblay, G.B.; Labrie, F.; Giguère, V. Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol. Cell 1999, 3, 513–519. [Google Scholar] [CrossRef]
- Ruff, M.; Gangloff, M.; Wurtz, J.M.; Moras, D. Estrogen receptor transcription and transactivation: Structure-function relationship in DNA- and ligand-binding domains of estrogen receptors. Breast Cancer Res. 2000, 2, 353–359. [Google Scholar] [CrossRef]
- Patel, H.K.; Bihani, T. Selective estrogen receptor modulators (SERMs) and selective estrogen receptor degraders (SERDs) in cancer treatment. Pharmacol. Ther. 2018, 186, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Jordan, V.C. Tamoxifen: Catalyst for the change to targeted therapy. Eur. J. Cancer 2008, 44, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Kuiper, G.G.; Lemmen, J.G.; Carlsson, B.; Corton, J.C.; Safe, S.H.; van der Saag, P.T.; van der Burg, B.; Gustafsson, J.A. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology 1998, 139, 4252–4263. [Google Scholar] [CrossRef] [PubMed]
- Esslimani-Sahla, M.; Simony-Lafontaine, J.; Kramar, A.; Lavaill, R.; Mollevi, C.; Warner, M.; Gustafsson, J.-Å.; Rochefort, H. Estrogen receptor β (ERβ) level but not its ERβcx variant helps to predict tamoxifen resistance in breast cancer. Clin. Cancer Res. 2004, 10, 5769–5776. [Google Scholar] [CrossRef] [PubMed]
- Hopp, T.A.; Weiss, H.L.; Parra, I.S.; Cui, Y.; Osborne, C.K.; Fuqua, S.A. Low levels of estrogen receptor beta protein predict resistance to tamoxifen therapy in breast cancer. Clin. Cancer Res. 2004, 10, 7490–7499. [Google Scholar] [CrossRef] [PubMed]
- Barkhem, T.; Carlsson, B.; Nilsson, Y.; Enmark, E.; Gustafsson, J.; Nilsson, S. Differential response of estrogen receptor alpha and estrogen receptor beta to partial estrogen agonists/antagonists. Mol. Pharmacol. 1998, 54, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.K.; Wakeling, A.; Nicholson, R.I. Fulvestrant: An oestrogen receptor antagonist with a novel mechanism of action. Br. J. Cancer 2004, 90, S2–S6. [Google Scholar] [CrossRef]
- Howell, A.; Osborne, C.K.; Morris, C.; Wakeling, A.E. ICI 182,780 (Faslodex): Development of a novel, “pure” antiestrogen. Cancer 2000, 89, 817–825. [Google Scholar] [CrossRef]
- Mishra, A.K.; Abrahamsson, A.; Dabrosin, C. Fulvestrant inhibits growth of triple negative breast cancer and synergizes with tamoxifen in ERα positive breast cancer by up-regulation of ERβ. Oncotarget 2016, 7, 56876–56888. [Google Scholar] [CrossRef]
- Minutolo, F.; Macchia, M.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A. Estrogen receptor β ligands: Recent advances and biomedical applications. Med. Res. Rev. 2011, 31, 364–442. [Google Scholar] [CrossRef]
- Song, P.; Li, Y.; Dong, Y.; Liang, Y.; Qu, H.; Qi, D.; Lu, Y.; Jin, X.; Guo, Y.; Jia, Y.; et al. Estrogen receptor β inhibits breast cancer cells migration and invasion through CLDN6-mediated autophagy. J. Exp. Clin. Cancer Res. 2019, 38, 354. [Google Scholar] [CrossRef] [PubMed]
- Schüler-Toprak, S.; Häring, J.; Inwald, E.C.; Moehle, C.; Ortmann, O.; Treeck, O. Agonists and knockdown of estrogen receptor β differentially affect invasion of triple-negative breast cancer cells in vitro. BMC Cancer 2016, 16, 951. [Google Scholar] [CrossRef] [PubMed]
- Reese, J.M.; Bruinsma, E.S.; Nelson, A.W.; Chernukhin, I.; Carroll, J.S.; Li, Y.; Subramaniam, M.; Suman, V.J.; Negron, V.; Monroe, D.G. ERβ-mediated induction of cystatins results in suppression of TGFβ signaling and inhibition of triple-negative breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9580–E9589. [Google Scholar] [CrossRef] [PubMed]
- Sareddy, G.R.; Li, X.; Liu, J.; Viswanadhapalli, S.; Garcia, L.; Gruslova, A.; Cavazos, D.; Garcia, M.; Strom, A.M.; Gustafsson, J.A.; et al. Selective Estrogen Receptor β Agonist LY500307 as a Novel Therapeutic Agent for Glioblastoma. Sci. Rep. 2016, 6, 24185. [Google Scholar] [CrossRef]
- Zhao, L.; Huang, S.; Mei, S.; Yang, Z.; Xu, L.; Zhou, N.; Yang, Q.; Shen, Q.; Wang, W.; Le, X.; et al. Pharmacological activation of estrogen receptor beta augments innate immunity to suppress cancer metastasis. Proc. Natl. Acad. Sci. USA 2018, 115, E3673–E3681. [Google Scholar] [CrossRef]
- Sun, J.; Ma, X.; Chen, Y.X.; Rayner, K.; Hibbert, B.; McNulty, M.; Dhaliwal, B.; Simard, T.; Ramirez, D.; O’Brien, E. Attenuation of atherogenesis via the anti-inflammatory effects of the selective estrogen receptor beta modulator 8β-VE2. J. Cardiovasc. Pharmacol. 2011, 58, 399–405. [Google Scholar] [CrossRef]
- Sareddy, G.R.; Vadlamudi, R.K. Cancer therapy using natural ligands that target estrogen receptor beta. Chin. J. Nat. Med. 2015, 13, 801–807. [Google Scholar] [CrossRef]
- Mersereau, J.E.; Levy, N.; Staub, R.E.; Baggett, S.; Zogovic, T.; Zogric, T.; Chow, S.; Ricke, W.A.; Tagliaferri, M.; Cohen, I.; et al. Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol. Cell. Endocrinol. 2008, 283, 49–57. [Google Scholar] [CrossRef]
- Middleton, E.; Kandaswami, C.; Theoharides, T.C. The effects of plant flavonoids on mammalian cells: Implications for inflammation, heart disease, and cancer. Pharmacol. Rev. 2000, 52, 673–751. [Google Scholar]
- Hinsche, O.; Girgert, R.; Emons, G.; Gründker, C. Estrogen receptor β selective agonists reduce invasiveness of triple-negative breast cancer cells. Int. J. Oncol. 2015, 46, 878–884. [Google Scholar] [CrossRef]
- Pan, H.; Zhou, W.; He, W.; Liu, X.; Ding, Q.; Ling, L.; Zha, X.; Wang, S. Genistein inhibits MDA-MB-231 triple-negative breast cancer cell growth by inhibiting NF-kappaB activity via the Notch-1 pathway. Int. J. Mol. Med. 2012, 30, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.G.; Selmin, O.I.; Doetschman, T.C.; Romagnolo, D.F. Epigenetic Activation of BRCA1 by Genistein In Vivo and Triple Negative Breast Cancer Cells Linked to Antagonism toward Aryl Hydrocarbon Receptor. Nutrients 2019, 11, 2559. [Google Scholar] [CrossRef] [PubMed]
- Setchell, K.D.; Clerici, C.; Lephart, E.D.; Cole, S.J.; Heenan, C.; Castellani, D.; Wolfe, B.E.; Nechemias-Zimmer, L.; Brown, N.M.; Lund, T.D.; et al. S-equol, a potent ligand for estrogen receptor beta, is the exclusive enantiomeric form of the soy isoflavone metabolite produced by human intestinal bacterial flora. Am. J. Clin. Nutr. 2005, 81, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Coriano, C.G.; Liu, F.; Sievers, C.K.; Liang, M.; Wang, Y.; Lim, Y.; Yu, M.; Xu, W. A Computational-Based Approach to Identify Estrogen Receptor alpha/beta Heterodimer Selective Ligands. Mol. Pharmacol. 2018, 93, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Park, B.-W.; Kim, K.-S.; Heo, M.-K.; Ko, S.-S.; Lee, K.S.; Hong, S.W.; Yang, W.-I.; Kim, J.-H.; Kim, G.E. Expression of estrogen receptor-β in normal mammary and tumor tissues: Is it protective in breast carcinogenesis? Breast Cancer Res. Treat 2003, 80, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Speirs, V.; Malone, C.; Walton, D.S.; Kerin, M.J.; Atkin, S.L. Increased expression of estrogen receptor β mRNA in tamoxifen-resistant breast cancer patients. Cancer Res. 1999, 59, 5421–5424. [Google Scholar]
- Skliris, G.P.; Carder, P.J.; Lansdown, M.R.; Speirs, V. Immunohistochemical detection of ERbeta in breast cancer: Towards more detailed receptor profiling? Br. J. Cancer 2001, 84, 1095–1098. [Google Scholar] [CrossRef]
- Tan, W.; Li, Q.; Chen, K.; Su, F.; Song, E.; Gong, C. Estrogen receptor beta as a prognostic factor in breast cancer patients: A systematic review and meta-analysis. Oncotarget 2016, 7, 10373–10385. [Google Scholar] [CrossRef]
- Duong, B.N.; Elliott, S.; Frigo, D.E.; Melnik, L.I.; Vanhoy, L.; Tomchuck, S.; Lebeau, H.P.; David, O.; Beckman, B.S.; Alam, J.; et al. AKT regulation of estrogen receptor beta transcriptional activity in breast cancer. Cancer Res. 2006, 66, 8373–8381. [Google Scholar] [CrossRef]
- Baek, J.M.; Chae, B.J.; Song, B.J.; Jung, S.S. The potential role of estrogen receptor beta2 in breast cancer. Int. J. Surg. 2015, 14, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.R.; Anderson, T.J.; Dixon, J.M.; Saunders, P.T. Oestrogen receptor beta and neoadjuvant therapy with tamoxifen: Prediction of response and effects of treatment. Br. J. Cancer 2006, 94, 1333–1338. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, J.; Zhang, C.; Chen, K.; Tang, H.; Tang, J.; Song, C.; Xie, X. ERβ1 inversely correlates with PTEN/PI3K/AKT pathway and predicts a favorable prognosis in triple-negative breast cancer. Breast Cancer Res. Treat 2015, 152, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Feitelson, M.A.; Arzumanyan, A.; Kulathinal, R.J.; Blain, S.W.; Holcombe, R.F.; Mahajna, J.; Marino, M.; Martinez-Chantar, M.L.; Nawroth, R.; Sanchez-Garcia, I.; et al. Sustained proliferation in cancer: Mechanisms and novel therapeutic targets. Semin. Cancer Biol. 2015, 35, S25–S54. [Google Scholar] [CrossRef] [PubMed]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitman, D.C. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef]
- Lin, C.Y.; Strom, A.; Li Kong, S.; Kietz, S.; Thomsen, J.S.; Tee, J.B.; Vega, V.B.; Miller, L.D.; Smeds, J.; Bergh, J.; et al. Inhibitory effects of estrogen receptor beta on specific hormone-responsive gene expression and association with disease outcome in primary breast cancer. Breast Cancer Res. 2007, 9, R25. [Google Scholar] [CrossRef]
- Behrens, D.; Gill, J.H.; Fichtner, I. Loss of tumourigenicity of stably ERbeta-transfected MCF-7 breast cancer cells. Mol. Cell. Endocrinol. 2007, 274, 19–29. [Google Scholar] [CrossRef]
- Shanle, E.K.; Zhao, Z.; Hawse, J.; Wisinski, K.; Keles, S.; Yuan, M.; Xu, W. Research resource: Global identification of estrogen receptor beta target genes in triple negative breast cancer cells. Mol. Endocrinol. 2013, 27, 1762–1775. [Google Scholar] [CrossRef]
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191. [Google Scholar] [CrossRef]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93–115. [Google Scholar] [CrossRef]
- Reese, J.M.; Suman, V.J.; Subramaniam, M.; Wu, X.; Negron, V.; Gingery, A.; Pitel, K.S.; Shah, S.S.; Cunliffe, H.E.; McCullough, A.E.; et al. ERβ1: Characterization, prognosis, and evaluation of treatment strategies in ERα-positive and -negative breast cancer. BMC Cancer 2014, 14, 749. [Google Scholar] [CrossRef]
- Reese, J.M.; Bruinsma, E.S.; Monroe, D.G.; Negron, V.; Suman, V.J.; Ingle, J.N.; Goetz, M.P.; Hawse, J.R. ERbeta inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget 2017, 8, 96506–96521. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.G.; Strom, A.; Lindberg, K.; Gustafsson, J.-A. Estrogen receptor beta decreases survival of p53-defective cancer cells after DNA damage by impairing G 2/M checkpoint signaling. Breast Cancer Res. Treat 2011, 127, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Renoir, J.-M.; Marsaud, V.; Lazennec, G. Estrogen receptor signaling as a target for novel breast cancer therapeutics. Biochem. Pharmacol. 2013, 85, 449–465. [Google Scholar] [CrossRef] [PubMed]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Gustafsson, J.-Å.; Thomas, C. ERβ decreases the invasiveness of triple-negative breast cancer cells by regulating mutant p53 oncogenic function. Oncotarget 2016, 7, 13599. [Google Scholar] [CrossRef]
- Thomas, C.; Rajapaksa, G.; Nikolos, F.; Hao, R.; Katchy, A.; McCollum, C.W.; Bondesson, M.; Quinlan, P.; Thompson, A.; Krishnamurthy, S. ERβ1 represses basal-like breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res. 2012, 14, R148. [Google Scholar] [CrossRef]
- Song, W.; Tang, L.; Xu, Y.; Sun, Q.; Yang, F.; Guan, X. ERβ1 inhibits metastasis of androgen receptor-positive triple-negative breast cancer by suppressing ZEB1. J. Exp. Clin. Cancer Res. 2017, 36, 75. [Google Scholar] [CrossRef]
- Moenner, M.; Pluquet, O.; Bouchecareilh, M.; Chevet, E. Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 2007, 67, 10631–10634. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef]
- Obacz, J.; Avril, T.; Le Reste, P.J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- McGrath, E.P.; Logue, S.E.; Mnich, K.; Deegan, S.; Jäger, R.; Gorman, A.M.; Samali, A. The Unfolded Protein Response in Breast Cancer. Cancers 2018, 10, 344. [Google Scholar] [CrossRef] [PubMed]
- Rajapaksa, G.; Thomas, C.; Gustafsson, J. Estrogen signaling and unfolded protein response in breast cancer. J. Steroid Biochem. Mol. Biol. 2016, 163, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Shajahan, A.N.; Wang, Y.; Tyson, J.J.; Riggins, R.B.; Weiner, L.M.; Bauman, W.T.; Xuan, J.; Zhang, B.; Facey, C.; et al. Endoplasmic reticulum stress, the unfolded protein response, and gene network modeling in antiestrogen resistant breast cancer. Horm. Mol. Biol. Clin. Investig. 2011, 5, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Nichols, P.; Groshen, S.; Spicer, D.; Lee, A.S. GRP78 as potential predictor for breast cancer response to adjuvant taxane therapy. Int. J. Cancer 2011, 128, 726–731. [Google Scholar] [CrossRef]
- Avril, T.; Vauléon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef]
- Kimata, Y.; Kohno, K. Endoplasmic reticulum stress-sensing mechanisms in yeast and mammalian cells. Curr. Opin. Cell Biol. 2011, 23, 135–142. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Turner, B.; Song, J.; Li, R.; Diehn, M.; Le, Q.T.; Khatri, P.; Koong, A.C. Comprehensive Analysis of the Unfolded Protein Response in Breast Cancer Subtypes. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Idowu, M.O.; Kmieciak, M.; Dumur, C.; Burton, R.S.; Grimes, M.M.; Powers, C.N.; Manjili, M.H. CD44(+)/CD24(−/low) cancer stem/progenitor cells are more abundant in triple-negative invasive breast carcinoma phenotype and are associated with poor outcome. Hum. Pathol. 2012, 43, 364–373. [Google Scholar] [CrossRef]
- Lin, Y.; Zhong, Y.; Guan, H.; Zhang, X.; Sun, Q. CD44+/CD24− phenotype contributes to malignant relapse following surgical resection and chemotherapy in patients with invasive ductal carcinoma. J. Exp. Clin. Cancer Res. 2012, 31, 59. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S.; et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature 2012, 487, 380–384. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Rajapaksa, G.; Nikolos, F.; Bado, I.; Clarke, R.; Gustafsson, J.; Thomas, C. ERβ decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene 2015, 34, 4130–4141. [Google Scholar] [CrossRef]
- Gao, B.; Lee, S.M.; Chen, A.; Zhang, J.; Zhang, D.D.; Kannan, K.; Ortmann, R.A.; Fang, D. Synoviolin promotes IRE1 ubiquitination and degradation in synovial fibroblasts from mice with collagen-induced arthritis. EMBO Rep. 2008, 9, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Marcu, M.G.; Doyle, M.; Bertolotti, A.; Ron, D.; Hendershot, L.; Neckers, L. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol. Cell. Biol. 2002, 22, 8506–8513. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Yeung, S.J.; Pan, J.; Lee, M.H. Roles of p53, MYC and HIF-1 in regulating glycolysis—The seventh hallmark of cancer. Cell. Mol. Life Sci. 2008, 65, 3981–3999. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.H.; Jung, W.H.; Koo, J.S. Metabolic phenotypes in triple-negative breast cancer. Tumour Biol. 2013, 34, 1699–1712. [Google Scholar] [CrossRef]
- Lanning, N.J.; Castle, J.P.; Singh, S.J.; Leon, A.N.; Tovar, E.A.; Sanghera, A.; MacKeigan, J.P.; Filipp, F.V.; Graveel, C.R. Metabolic profiling of triple-negative breast cancer cells reveals metabolic vulnerabilities. Cancer Metab. 2017, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Wahdan-Alaswad, R.; Fan, Z.; Edgerton, S.M.; Liu, B.; Deng, X.S.; Arnadottir, S.S.; Richer, J.K.; Anderson, S.M.; Thor, A.D. Glucose promotes breast cancer aggression and reduces metformin efficacy. Cell Cycle 2013, 12, 3759–3769. [Google Scholar] [CrossRef]
- Palorini, R.; Simonetto, T.; Cirulli, C.; Chiaradonna, F. Mitochondrial complex I inhibitors and forced oxidative phosphorylation synergize in inducing cancer cell death. Int. J. Cell Biol. 2013, 2013, 243876. [Google Scholar] [CrossRef] [PubMed]
- Gründker, C.; Wokoun, U.; Hellriegel, M.; Emons, G. Inhibition of aerobic glycolysis enhances the anti-tumor efficacy of Zoptarelin Doxorubicin in triple-negative breast cancer cells. J Obstet. Gynaecol. Res. 2019, 45, 1334–1342. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Liu, R.; Perez, E.J.; Wen, Y.; Stevens, S.M.; Valencia, T.; Brun-Zinkernagel, A.M.; Prokai, L.; Will, Y.; Dykens, J.; et al. Mitochondrial localization of estrogen receptor beta. Proc. Natl. Acad. Sci. USA 2004, 101, 4130–4135. [Google Scholar] [CrossRef] [PubMed]
- Psarra, A.M.; Sekeris, C.E. Steroid and thyroid hormone receptors in mitochondria. IUBMB Life 2008, 60, 210–223. [Google Scholar] [CrossRef]
- Simpkins, J.W.; Yang, S.H.; Sarkar, S.N.; Pearce, V. Estrogen actions on mitochondria—Physiological and pathological implications. Mol. Cell. Endocrinol. 2008, 290, 51–59. [Google Scholar] [CrossRef]
- Song, I.S.; Jeong, Y.J.; Jeong, S.H.; Kim, J.E.; Han, J.; Kim, T.H.; Jang, S.W. Modulation of Mitochondrial ERβ Expression Inhibits Triple-Negative Breast Cancer Tumor Progression by Activating Mitochondrial Function. Cell Physiol. Biochem. 2019, 52, 468–485. [Google Scholar] [CrossRef]
- Chen, J.Q.; Delannoy, M.; Cooke, C.; Yager, J.D. Mitochondrial localization of ERalpha and ERbeta in human MCF7 cells. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E1011–E1022. [Google Scholar] [CrossRef]
- Grober, O.M.; Mutarelli, M.; Giurato, G.; Ravo, M.; Cicatiello, L.; De Filippo, M.R.; Ferraro, L.; Nassa, G.; Papa, M.F.; Paris, O.; et al. Global analysis of estrogen receptor beta binding to breast cancer cell genome reveals an extensive interplay with estrogen receptor alpha for target gene regulation. BMC Genom. 2011, 12, 36. [Google Scholar] [CrossRef]
- Chen, J.Q.; Yager, J.D. Estrogen’s effects on mitochondrial gene expression: Mechanisms and potential contributions to estrogen carcinogenesis. Ann. N. Y. Acad. Sci. 2004, 1028, 258–272. [Google Scholar] [CrossRef]
- Riscal, R.; Skuli, N.; Simon, M.C. Even Cancer Cells Watch Their Cholesterol! Mol. Cell 2019, 76, 220–231. [Google Scholar] [CrossRef]
- Kuzu, O.F.; Noory, M.A.; Robertson, G.P. The Role of Cholesterol in Cancer. Cancer Res. 2016, 76, 2063–2070. [Google Scholar] [CrossRef] [PubMed]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerød, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Cai, D.; Wang, J.; Gao, B.; Li, J.; Wu, F.; Zou, J.X.; Xu, J.; Jiang, Y.; Zou, H.; Huang, Z.; et al. RORγ is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat. Commun. 2019, 10, 4621. [Google Scholar] [CrossRef]
- Cai, D.; Zhang, X.; Chen, H.W. A master regulator of cholesterol biosynthesis constitutes a therapeutic liability of triple negative breast cancer. Mol. Cell. Oncol. 2020, 7, 1701362. [Google Scholar] [CrossRef]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- Alexandrova, E.; Lamberti, J.; Saggese, P.; Pecoraro, G.; Memoli, D.; Cappa, V.M.; Ravo, M.; Iorio, R.; Tarallo, R.; Rizzo, F.; et al. Small Non-Coding RNA Profiling Identifies miR-181a-5p as a Mediator of Estrogen Receptor Beta-Induced Inhibition of Cholesterol Biosynthesis in Triple-Negative Breast Cancer. Cells 2020, 9, 874. [Google Scholar] [CrossRef] [PubMed]
- Doane, A.S.; Danso, M.; Lal, P.; Donaton, M.; Zhang, L.; Hudis, C.; Gerald, W. An estrogen receptor-negative breast cancer subset characterized by a hormonally regulated transcriptional program and response to androgen. Oncogene 2006, 25, 3994–4008. [Google Scholar] [CrossRef] [PubMed]
- Farmer, P.; Bonnefoi, H.; Becette, V.; Tubiana-Hulin, M.; Fumoleau, P.; Larsimont, D.; MacGrogan, G.; Bergh, J.; Cameron, D.; Goldstein, D. Identification of molecular apocrine breast tumours by microarray analysis. Breast Cancer Res. 2005, 7, 2–11. [Google Scholar] [CrossRef]
- Cochrane, D.R.; Bernales, S.; Jacobsen, B.M.; Cittelly, D.M.; Howe, E.N.; D’Amato, N.C.; Spoelstra, N.S.; Edgerton, S.M.; Jean, A.; Guerrero, J. Role of the androgen receptor in breast cancer and preclinical analysis of enzalutamide. Breast Cancer Res. 2014, 16, R7. [Google Scholar] [CrossRef]
- Gucalp, A.; Tolaney, S.; Isakoff, S.J.; Ingle, J.N.; Liu, M.C.; Carey, L.A.; Blackwell, K.; Rugo, H.; Nabell, L.; Forero, A. Phase II trial of bicalutamide in patients with androgen receptor–positive, estrogen receptor–negative metastatic breast cancer. Clin. Cancer Res. 2013, 19, 5505–5512. [Google Scholar] [CrossRef]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor-Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Anestis, A.; Sarantis, P.; Theocharis, S.; Zoi, I.; Tryfonopoulos, D.; Korogiannos, A.; Koumarianou, A.; Xingi, E.; Thomaidou, D.; Kontos, M.; et al. Estrogen receptor beta increases sensitivity to enzalutamide in androgen receptor-positive triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1221–1233. [Google Scholar] [CrossRef]
- Rampurwala, M.M.; Rocque, G.B.; Burkard, M.E. Update on adjuvant chemotherapy for early breast cancer. Breast Cancer 2014, 8, 125–133. [Google Scholar] [CrossRef]
- Gucalp, A.; Traina, T.A. Targeting the androgen receptor in triple-negative breast cancer. Curr. Probl. Cancer 2016, 40, 141–150. [Google Scholar] [CrossRef]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef]
- Treiman, M.; Caspersen, C.; Christensen, S.B. A tool coming of age: Thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases. Trends Pharmacol. Sci. 1998, 19, 131–135. [Google Scholar] [CrossRef]
- Sato, K.; Rajendra, E.; Ohta, T. The UPS: A promising target for breast cancer treatment. BMC Biochem. 2008, 9, S2. [Google Scholar] [CrossRef] [PubMed]
- Beckwitt, C.H.; Brufsky, A.; Oltvai, Z.N.; Wells, A. Statin drugs to reduce breast cancer recurrence and mortality. Breast Cancer Res. 2018, 20, 144. [Google Scholar] [CrossRef] [PubMed]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. 2010, 86, 484–493. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | ERβ Antibodies | Source | Tested Applications | Performance |
|---|---|---|---|---|
| Wu et al. [65] | pAb AB1410 | Chemicon | IHC in human cell lines, IF | Bad |
| mAb GR40 | Calbiochem | IHC in human cell lines, IF | Bad | |
| mAb MC9 | Homemade | WB, IP | Good | |
| mAb MC10 | Homemade | WB, IP, IHC in human cell lines and tissues, IF | Good | |
| mAb PPG5/10 | Thermo Fisher Scientific | IHC in human cell lines and tissues, IF | Good | |
| pAb sc-6820 | Santa Cruz Biotechnology | IHC in human cell lines, IF | Bad | |
| Shanle et al. [66] | pAb PA1-313 | Thermo Fisher Scientific | WB, IHC in mouse xenograft tissues | Good |
| Nelson et al. [68] | mAb 14C8 | Abcam | WB, RIME | Low specificity for RIME application |
| mAb CWK-F12 | DSHB | WB, RIME, IHC in human cell lines | Good | |
| mAb GeneTex 70182 | GeneTex | WB, RIME | Good | |
| mAb MC10 | provided by Wu et al. 2012 | WB, RIME | Good | |
| pAb Millipore 06-629 | Millipore | WB, RIME | Bad for WB application | |
| mAb NCL-ER-BETA | Leica Biosystems | WB, RIME | Bad | |
| mAb PPG5/10 | Thermo Fisher Scientific | WB, RIME | Bad for WB application | |
| pAb Sc8974 | Santa Cruz Biotechnology | WB, RIME | Good | |
| Andersson et al. [67] | mAb 14C8 | GeneTex | WB, IP, IHC in human cell lines and tissues | Bad for WB, IP and IHC in human tissues applications |
| mAb PPG5/10 | DAKO | WB, IP, IHC in human cell lines and tissues | Bad | |
| mAb PPZ0506 | Invitrogen | WB, IP, IHC in human cell lines and tissues | Good | |
| Alexandrova et al. [32] | pAb PA1-313 | Thermo Fisher Scientific | IP | Good |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sellitto, A.; D’Agostino, Y.; Alexandrova, E.; Lamberti, J.; Pecoraro, G.; Memoli, D.; Rocco, D.; Coviello, E.; Giurato, G.; Nassa, G.; et al. Insights into the Role of Estrogen Receptor β in Triple-Negative Breast Cancer. Cancers 2020, 12, 1477. https://doi.org/10.3390/cancers12061477
Sellitto A, D’Agostino Y, Alexandrova E, Lamberti J, Pecoraro G, Memoli D, Rocco D, Coviello E, Giurato G, Nassa G, et al. Insights into the Role of Estrogen Receptor β in Triple-Negative Breast Cancer. Cancers. 2020; 12(6):1477. https://doi.org/10.3390/cancers12061477
Chicago/Turabian StyleSellitto, Assunta, Ylenia D’Agostino, Elena Alexandrova, Jessica Lamberti, Giovanni Pecoraro, Domenico Memoli, Domenico Rocco, Elena Coviello, Giorgio Giurato, Giovanni Nassa, and et al. 2020. "Insights into the Role of Estrogen Receptor β in Triple-Negative Breast Cancer" Cancers 12, no. 6: 1477. https://doi.org/10.3390/cancers12061477
APA StyleSellitto, A., D’Agostino, Y., Alexandrova, E., Lamberti, J., Pecoraro, G., Memoli, D., Rocco, D., Coviello, E., Giurato, G., Nassa, G., Tarallo, R., Weisz, A., & Rizzo, F. (2020). Insights into the Role of Estrogen Receptor β in Triple-Negative Breast Cancer. Cancers, 12(6), 1477. https://doi.org/10.3390/cancers12061477