An Overview of Candidate Therapeutic Target Genes in Ovarian Cancer

 , , , ,
, , , ,  ,
,  and
and
Abstract

1. Introduction
2. The Molecular Landscape of Ovarian Cancer
3. The Role of Estrogen Receptors in Ovarian Cancer
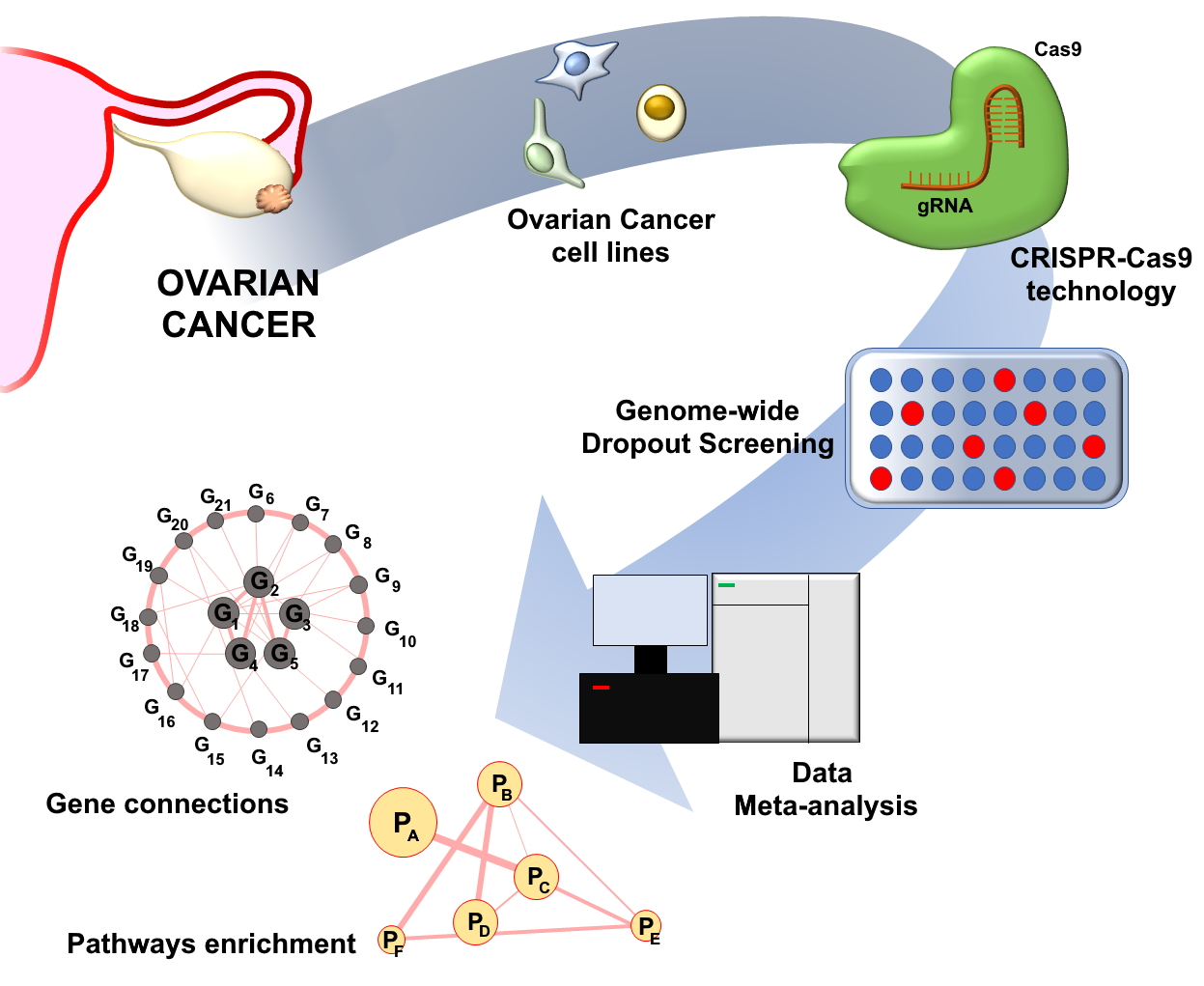
4. Genome-Wide CRISPR-Cas9 Dropout Screening for Identification of Candidate Therapeutic Target Genes in OC
5. Functional Pathways Affected by OC Fitness Genes
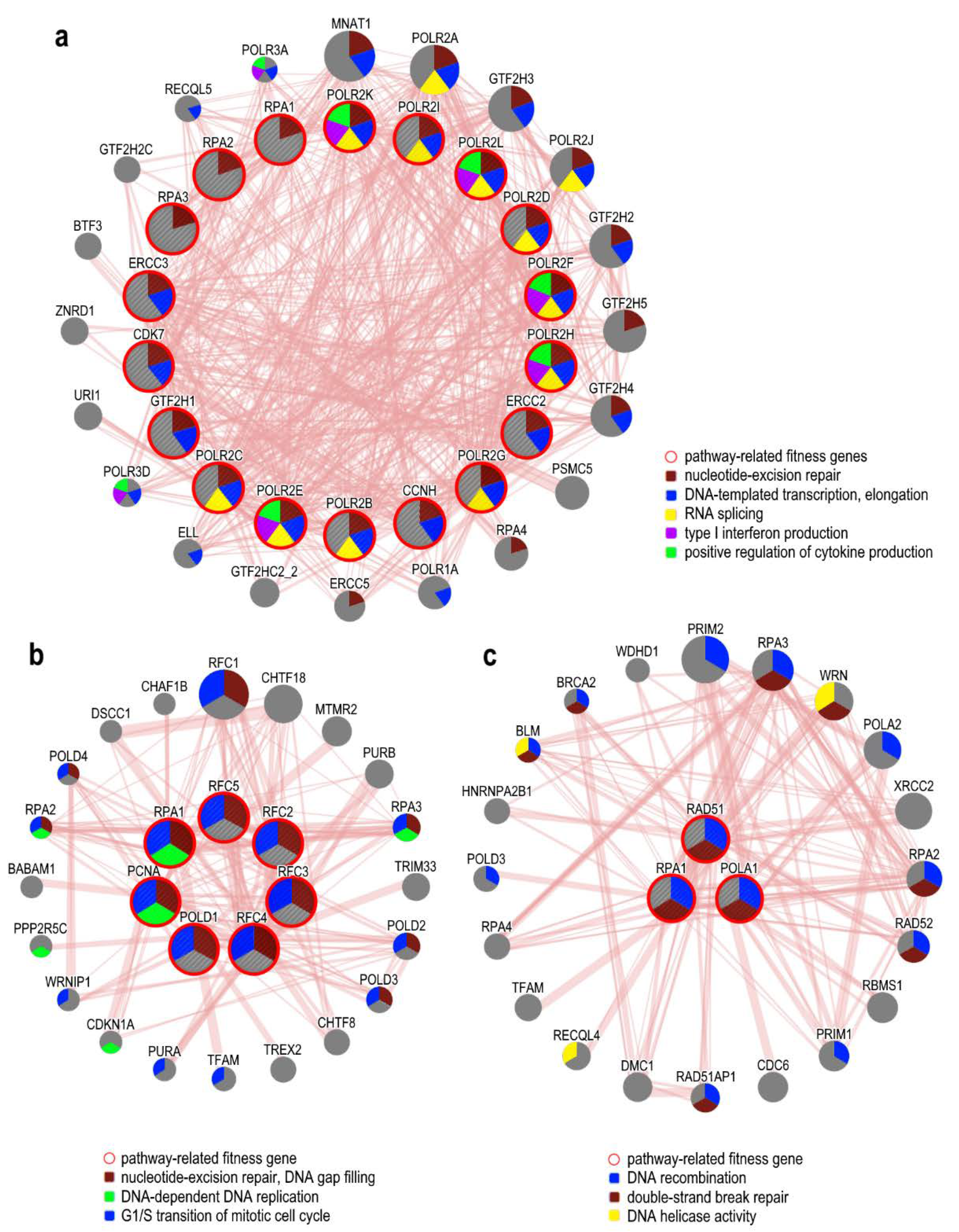
5.1. DNA Damage Response Associated Pathways
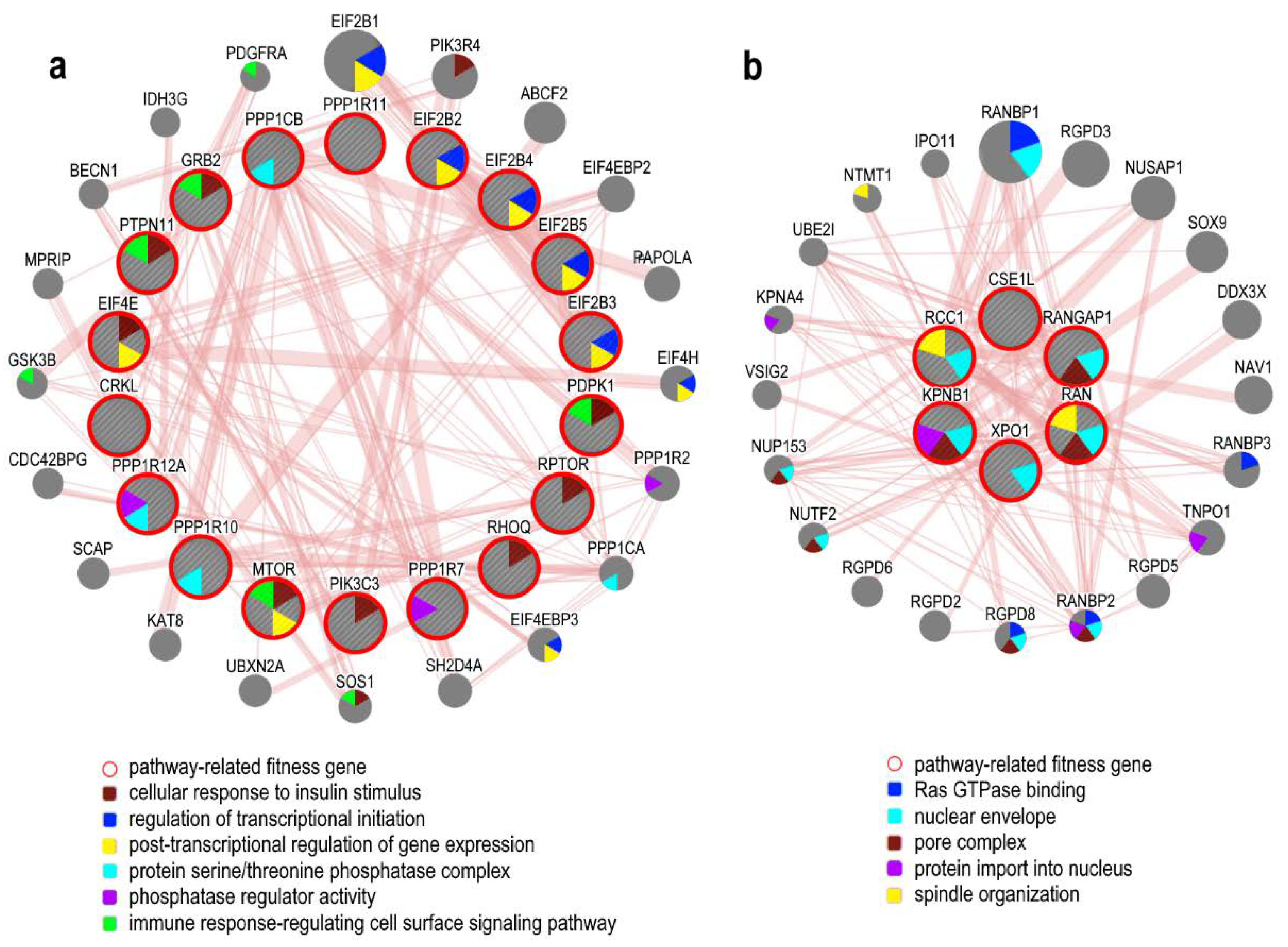
5.2. Hypoxia and Angiogenesis Related Genes
5.3. Proliferative Signals
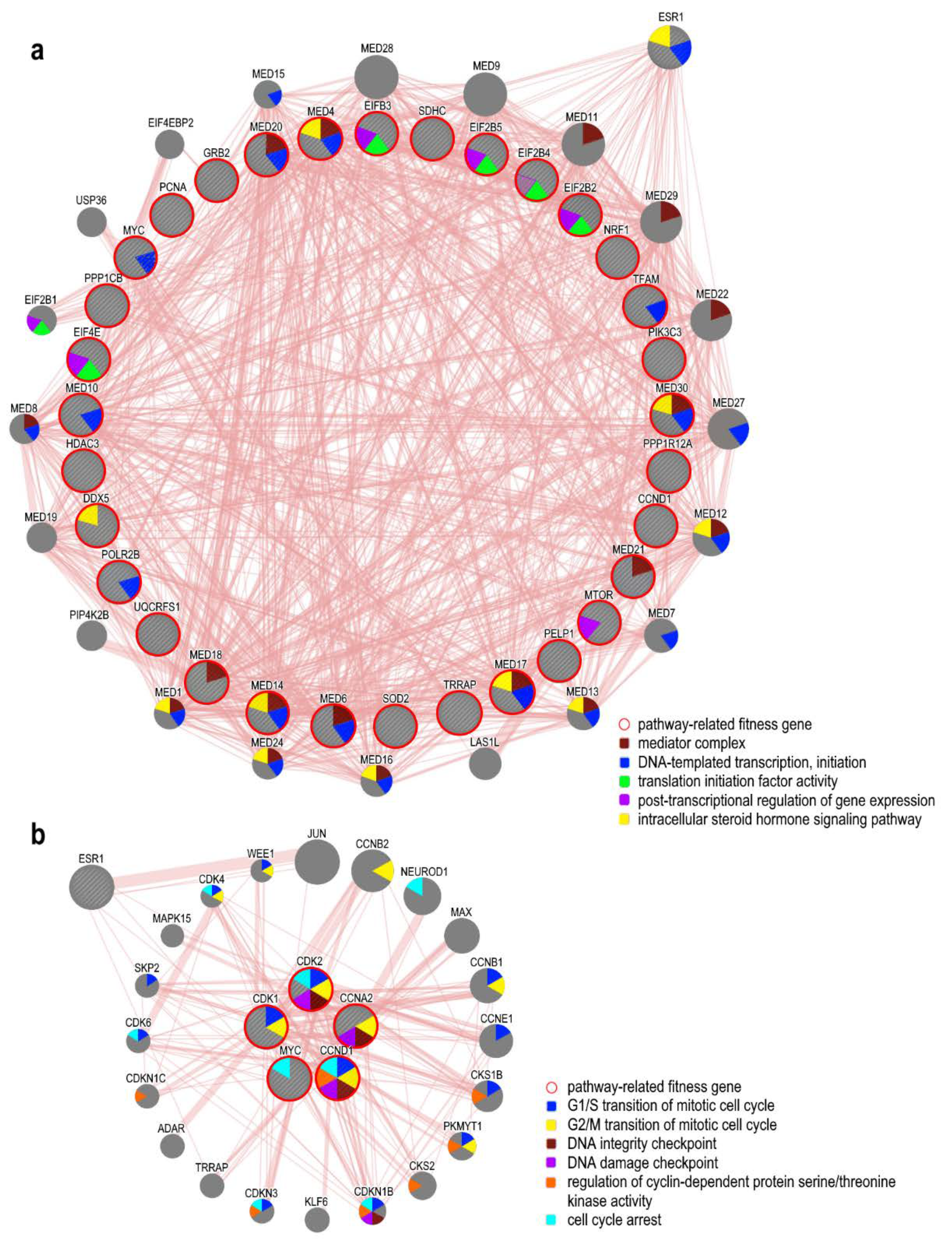
5.4. ER-Related Pathways and Fitness Genes in OC
5.5. Other Suitable Pathways for Targeted Therapies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AKAP12 | A-kinase anchor protein 12 |
| AKT | Protein kinase B |
| APOBEC | Apolipoprotein B mRNA Editing Catalytic Polypeptide-like |
| ARID1A | AT-Rich Interaction Domain 1A |
| ATM | ATM serine/threonine kinase |
| ATR | ATR serine/threonine kinase |
| AURKA | Aurora kinase A |
| BARD1 | BRCA1 associated RING domain 1 |
| BAX | BCL2 associated X, apoptosis regulator |
| BCL2 | BCL2 apoptosis regulator |
| BCL2L1 | BCL2 like 1 |
| BENE | Benzoate transport protein |
| Bit1 | Bcl2-inhibitor of transcription 1 |
| BRAF | B-Raf proto-oncogene, serine/threonine kinase |
| BRCA1 | BReast CAncer gene 1 |
| BRCA2 | BReast CAncer gene 2 |
| BRIP1 | BRCA1 interacting protein C-terminal helicase 1 |
| CASP4 | Caspase 4 |
| CCNA2 | Cyclin-A2 |
| CCNB1 | Cyclin-B1 |
| CCND1 | Cyclin-D1 |
| CCNE1 | Cyclin-E1 |
| CCOC | Clear cell ovarian cancer |
| CDC25 | Cell division cycle 25 homolog A |
| CDH6 | Cadherin-6 |
| CDK | Cyclin-dependent kinases |
| CDK1 | Cyclin-dependent kinases 1 |
| CDK12 | Cyclin-dependent kinases 12 |
| CDK2 | Cyclin-dependent kinases 2 |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A |
| CHEK1 | Checkpoint kinase 1 |
| CHEK2 | Checkpoint kinase 2 |
| COPI | COPI-coated vesicles |
| CREBBP | CREB-binding protein |
| CSE1L | Human homolog of the yeast cse1gene |
| CSMD3 | CUB and Sushi multiple domains 3 |
| CTNNB1 | Catenin beta 1 |
| CTSD | Cathepsin D |
| CXCL11 | C-X-C motif chemokine 11 |
| CXCR7 | C-X-C Chemokine Receptor Type 7 |
| CYR61 | Cysteine rich angiogenic inducer 61 |
| DDR | DNA damage response |
| DES | Desmin |
| DOT1L | Disruptor of telomeric silencing-1-like |
| DSB | Double-strand DNA breaks |
| DYNLL1 | Dynein light chain LC8-type 1 |
| E2 | 17β-estradiol |
| EIF2 | Eukaryotic translation initiation factor 2 |
| EIF2B1 | Eukaryotic translation initiation factor 2B subunit alpha |
| EIF2B2 | Eukaryotic translation initiation factor 2B subunit beta |
| EIF2B3 | Eukaryotic translation initiation factor 2B subunit gamma |
| EIF2B4 | Eukaryotic translation initiation factor 2B subunit delta |
| EIF2B5 | Eukaryotic translation initiation factor 2B subunit epsilon |
| EIF4E | Eukaryotic translation initiation factor 4E |
| EOC | Epithelial ovaria cancer |
| EOVC | Endometrioid ovarian cancers |
| ERBB2 | Erb-b2 receptor tyrosine kinase 2 |
| ERCC | NA excision repair protein ERCC-1-like protein |
| ERCC2 | ERCC excision repair 2 |
| ERCC3 | ERCC excision repair 3 |
| ERE | Estrogen-response element |
| ERK | Extracellular regulated MAP kinase |
| ERα | Estrogen receptor α |
| ERβ | Estrogen receptor β |
| ESR1 | Estrogen receptor 1 |
| ESR2 | Estrogen receptor 2 |
| FAT3 | FAT atypical cadherin 3 |
| FOSL1 | FOS like 1, AP-1 transcription factor subunit |
| FOXM1 | Forkhead box M1 |
| FPP | Farnesyl pyrophosphate |
| GABRA6 | Gamma-aminobutyric acid type A receptor subunit alpha6 |
| GGPP | Geranylgeranyl pyrophosphate |
| GO | Gene Ontology |
| GRSF1 | G-rich RNA sequence binding factor 1 |
| HDAC3 | Histone deacetylase 3 |
| HDI | Human Development Index |
| HER2 | Human epidermal growth factor receptor 2 |
| HGSOC | High-grade serous ovarian cancer |
| HMG-CoA | Hydroxymethyl-glutaryl coenzyme A |
| HMGCR | Hydroxymethyl-glutaryl reductase |
| HR | Homologous recombination |
| HRT | Hormone replacement therapy |
| hTER | Telomerase reverse transcriptase |
| HUS1 | HUS1 checkpoint clamp component |
| ID4 | Inhibitor of DNA binding 4, HLH protein |
| IGFBP3 | Insulin like growth factor binding protein 3 |
| IPA | Ingenuity Pathway Analysis |
| IRF2BP2 | Interferon regulatory factor 2 binding protein 2 |
| KMT2B | Lysine methyltransferase 2B |
| KMT2D | lysine methyltransferase 2D |
| KPNB1 | Karyopherin subunit beta 1 |
| KRAS | KRAS proto-oncogene, GTPase |
| KRT | Keratin |
| LCN2 | Lipocalin-2 |
| LGSOC | Low-grade serous ovarian cancer |
| LOH | Loss of heterozygosity |
| MAPK | Mitogen-Activated Protein Kinase |
| MECOM | MDS1 and EVI1 complex locus |
| MED | Mediator Complex |
| MED14 | Mediator Complex Subunit 14 |
| MED17 | Mediator Complex Subunit 17 |
| MED18 | Mediator Complex Subunit 18 |
| MED20 | Mediator Complex Subunit 20 |
| MED21 | Mediator Complex Subunit 21 |
| MED30 | Mediator Complex Subunit 30 |
| MED4 | Mediator Complex Subunit 4 |
| MED6 | Mediator Complex Subunit 6 |
| MLH1 | MutL homolog 1 |
| MMP | Matrix metalloproteinase |
| MMR | DNA mismatch repair |
| MOC | Mucinous ovarian cancers |
| Mre11 | MRE11 homolog, double strand break repair nuclease |
| MTOR | Mammalian target of rapamycin |
| MTORC1 | Mammalian target of rapamycin complex 1 |
| MUTYH | MutY DNA glycosylase |
| MYC | MYC proto-oncogene |
| NBS1 | Nijmegen Breakage Syndrome 1 |
| NER | Nucleotide excision repair |
| NF1 | Neurofibromin 1 |
| NOTCH4 | Notch Receptor 4 |
| NRAS | NRAS proto-oncogene, GTPase |
| OC | Ovarian cancer |
| P16 | Cyclin-dependent kinase inhibitor 2A |
| P21 | Cyclin-dependent kinase inhibitor 1 |
| P27 | Cyclin-dependent kinase inhibitor 1B |
| PALB2 | Partner and localizer of BRCA2 |
| PARP | Poly ADP ribose polymerase |
| PAX2 | Paired box gene 2 |
| PAX8 | Paired box gene 8 |
| PCNA | Proliferating cell nuclear antigen |
| PERK | Protein kinase-like Endoplasmic Reticulum Kinase |
| PGR | Progesterone receptor |
| PIK3C3 | Phosphatidylinositol 3-Kinase Catalytic Subunit Type 3 |
| PI3K | Phosphatidylinositol 3-kinase |
| PI3KCA | Phosphatidylinositol 3-kinase catalytic alpha polypeptide |
| PLAU | Plasminogen activator, urokinase |
| PLC | Phospholipase C |
| PLK1 | Polo like kinase 1 |
| POLE | DNA polymerase epsilon, catalytic subunit |
| POT1 | Protection of telomeres 1 |
| PP2R1A | Protein phosphatase 2, regulatory subunit A |
| PPP1CB | Protein Phosphatase 1 Catalytic Subunit Beta |
| PPP1R12A | Protein Phosphatase 1 Regulatory Subunit 12A |
| PPP2R1A | Protein Phosphatase 2 Scaffold Subunit Aalpha |
| PTEN | Phosphatase and tensin homolog |
| Rab | Rab Family Small GTPase |
| Rac | Rac Family Small GTPase |
| RAD1 | RAD1 checkpoint DNA exonuclease |
| RAD50 | RAD50 double strand break repair protein |
| RAD51 | RAD51 recombinase |
| RAD51C | RAD51 paralog C |
| RAD51D | RAD51 paralog D |
| RAD9 | Checkpoint Clamp Component A |
| RAN | Ras-related nuclear protein |
| RAP1 | Ras-related protein 1 |
| RASSF1gene | Ras Association Domain Family Member 1 |
| RB1 | Retinoblastoma protein |
| RCF3 | Replication factor C subunit 3 |
| RFC | Replication factor C |
| Rho | Rhodopsin |
| RhoA | Ras homolog family member A |
| RPA | Replication Protein A |
| RPA3 | Replication Protein A3 |
| RRAS2 | Ras-Related Protein R-Ras2 |
| STK11 | Serine/threonine kinase 11 |
| TCGA | The Cancer Genome Atlas |
| TERF1 | Telomeric Repeat Binding Factor 1 |
| TERF2 | Telomeric Repeat Binding Factor 2 |
| TFAP4 | Transcription Factor AP-4 |
| TGFBI | Transforming Growth Factor Beta Induced |
| TIGAR | TP53 induced glycolysis and apoptosis regulator |
| TINF2 | TERF1 Interacting Nuclear Factor 2 |
| TNFSF7 | Tumor Necrosis Factor Ligand Superfamily Member 7 |
| TP53 | Tumor protein p53 |
| TPP1 | Tripeptidyl peptidase 1 |
| TRAM1 | Translocation associated membrane protein 1 |
| TRAP1 | TNF receptor associated protein 1 |
| UBL1 | Ubiquitin-like protein 1 |
| UPR | Unfolded protein response |
| VEGF | Vascular endothelial growth factor |
| VIM | Vimentin |
| WEE1 | WEE1 G2 checkpoint kinase |
| ZMYND8 | Zinc finger MYND-type containing 8 |
| ZNF587B | Zinc finger protein 587B |
References
- World Health Organization. Global Cancer Observatory GLOBOCAN. 2018. Available online: http://gco.iarc.fr/today/data/factsheets/cancers/25-Ovary-fact-sheet.pdf (accessed on 10 May 2020).
- Cancer Tomorrow Powered by GLOBOCAN. 2018. Available online: https://gco.iarc.fr/tomorrow/home (accessed on 28 May 2020).
- Moufarrij, S.; Dandapani, M.; Arthofer, E.; Gomez, S.; Srivastava, A.; Lopez-Acevedo, M.; Villagra, A.; Chiappinelli, K.B. Epigenetic therapy for ovarian cancer: Promise and progress. Clin. Epigenetics 2019, 11, 7. [Google Scholar] [CrossRef] [PubMed]
- Pelucchi, C.; Galeone, C.; Talamini, R.; Bosetti, C.; Montella, M.; Negri, E.; Franceschi, S.; La Vecchia, C. Lifetime ovulatory cycles and ovarian cancer risk in 2 Italian case-control studies. Am. J. Obstet. Gynecol. 2007, 196, 83.e1–83.e7. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, A.S.; Harris, R.; Ltnyre, J. Characteristics Relating to ovarian Cancer Risk: Collaborative Analysis of 12 US case-Control Studies. Am. J. Epidemiol. 1992, 136, 1212–1220. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.-T.; Wu, Q.-J.; Vogtmann, E.; Lin, B.; Wang, Y.-L. Age at menarche and risk of ovarian cancer: A meta-analysis of epidemiological studies. Int. J. Cancer 2012, 132, 2894–2900. [Google Scholar] [CrossRef] [PubMed]
- Tsilidis, K.K.; Allen, N.; Key, T.J.; Dossus, L.; Lukanova, A.; Bakken, K.; Lund, E.; Fournier, A.; Overvad, K.; Hansen, L.; et al. Oral contraceptive use and reproductive factors and risk of ovarian cancer in the European Prospective Investigation into Cancer and Nutrition. Br. J. Cancer 2011, 105, 1436–1442. [Google Scholar] [CrossRef] [PubMed]
- Gates, M.A.; Rosner, B.A.; Hecht, J.L.; Tworoger, S.S. Risk Factors for Epithelial Ovarian Cancer by Histologic Subtype. Am. J. Epidemiol. 2009, 171, 45–53. [Google Scholar] [CrossRef]
- Walker, J.L.; Powell, C.B.; Chen, L.-M.; Carter, J.; Jump, V.L.B.; Parker, L.P.; Borowsky, M.E.; Gibb, R.K. Society of Gynecologic Oncology recommendations for the prevention of ovarian cancer. Cancer 2015, 121, 2108–2120. [Google Scholar] [CrossRef]
- Mallen, A.; Soong, T.R.; Townsend, M.K.; Wenham, R.M.; Crum, C.; Tworoger, S.S. Surgical prevention strategies in ovarian cancer. Gynecol. Oncol. 2018, 151, 166–175. [Google Scholar] [CrossRef]
- Slatnik, C.L.; Duff, E. Ovarian cancer. Nurse Pr. 2015, 40, 47–54. [Google Scholar] [CrossRef]
- Chien, J.; Poole, E.M. Ovarian Cancer Prevention, Screening, and Early Detection: Report from the 11th Biennial Ovarian Cancer Research Symposium. Int. J. Gynecol. Cancer 2017, 27, S20–S22. [Google Scholar] [CrossRef]
- Rasmussen, E.L.K.; Hannibal, C.G.; Dehlendorff, C.; Baandrup, L.; Junge, J.; Vang, R.; Kurman, R.J.; Kjær, S.K. Parity, infertility, oral contraceptives, and hormone replacement therapy and the risk of ovarian serous borderline tumors: A nationwide case-control study. Gynecol. Oncol. 2017, 144, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, L.; Yang, X.; Bie, J.; Li, D.; Sun, C.; Zhang, J.; Meng, Y.; Lin, J. Menopausal Hormone Replacement Therapy and the Risk of Ovarian Cancer: A Meta-Analysis. Front. Endocrinol. 2019, 10, 801. [Google Scholar] [CrossRef] [PubMed]
- Chiaffarino, F.; Pelucchi, C.; Parazzini, F.; Negri, E.; Franceschi, S.; Talamini, R.; Conti, E.; Montella, M.; La Vecchia, C. Reproductive and hormonal factors and ovarian cancer. Ann. Oncol. 2001, 12, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Gaitskell, K.; Green, J.; Pirie, K.; Barnes, I.; Hermon, C.; Reeves, G.K.; Beral, V. Histological subtypes of ovarian cancer associated with parity and breastfeeding in the prospective Million Women Study. Int. J. Cancer 2017, 142, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Koushik, A.; Grundy, A.; Abrahamowicz, M.; Arseneau, J.; Gilbert, L.; Gotlieb, W.H.; Lacaille, J.; Mes-Masson, A.-M.; Parent, M.-E.; Provencher, D.; et al. Hormonal and reproductive factors and the risk of ovarian cancer. Cancer Causes Control 2017, 28, 393–403. [Google Scholar] [CrossRef]
- Luan, N.-N.; Wu, Q.-J.; Gong, T.-T.; Vogtmann, E.; Wang, Y.-L.; Lin, B. Breastfeeding and ovarian cancer risk: A meta-analysis of epidemiologic studies. Am. J. Clin. Nutr. 2013, 98, 1020–1031. [Google Scholar] [CrossRef] [PubMed]
- Riman, T.; Nilsson, S.; Persson, I.R. Review of epidemiological evidence for reproductive and hormonal factors in relation to the risk of epithelial ovarian malignancies. Acta Obstet. Gynecol. Scand. 2004, 83, 783–795. [Google Scholar] [CrossRef]
- Li, D.-P.; Du, C.; Zhang, Z.-M.; Li, G.-X.; Yu, Z.-F.; Wang, X.; Li, P.; Cheng, C.; Liu, Y.-P.; Zhao, Y. Breastfeeding and Ovarian Cancer Risk: A Systematic Review and Meta-analysis of 40 Epidemiological Studies. Asian Pac. J. Cancer Prev. 2014, 15, 4829–4837. [Google Scholar] [CrossRef]
- Risch, H.A.; Marrett, L.D.; Howe, G.R. Parity, Contraception, Infertility, and the Risk of Epithelial Ovarian Cancer. Am. J. Epidemiol. 1994, 140, 585–597. [Google Scholar] [CrossRef]
- Franceschi, S.; Parazzini, F.; Negri, E.; Booth, M.; La Vecchia, C.; Beral, V.; Tzonou, A.; Trichopoulos, D. Pooled analysis of 3 european case-control studies of epithelial ovarian cancer: III. Oral contraceptive use. Int. J. Cancer 1991, 49, 61–65. [Google Scholar] [CrossRef]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer; Beral, V.; Doll, R.; Hermon, C.; Peto, R.; Reeves, G. Ovarian cancer and oral contraceptives: Collaborative reanalysis of data from 45 epidemiological studies including 23 257 women with ovarian cancer and 87 303 controls. Lancet 2008, 371, 303–314. [Google Scholar] [CrossRef]
- Huang, Z.; Gao, Y.; Wen, W.; Li, H.; Zheng, W.; Shu, X.-O.; Beeghly-Fadiel, A. Contraceptive methods and ovarian cancer risk among Chinese women: A report from the Shanghai Women’s Health Study. Int. J. Cancer 2015, 137, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer; Beral, V.; Gaitskell, K.; Hermon, C.; Moser, K.; Reeves, G.; Peto, R. Menopausal hormone use and ovarian cancer risk: Individual participant meta-analysis of 52 epidemiological studies. Lancet 2015, 385, 1835–1842. [Google Scholar] [CrossRef]
- Kurman, R.J.; Carcangiu, M.L.; Herrington, C.S.; Young, R.H. WHO Classification of Tumours of Female Reproductive Organs, 4th ed.; IARC: Lyon, France, 2014.
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian Cancer: An Integrated Review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Lisio, M.-A.; Fu, L.; Goyeneche, A.; Gao, Z.-H.; Telleria, C.M. High-Grade Serous Ovarian Cancer: Basic Sciences, Clinical and Therapeutic Standpoints. Int. J. Mol. Sci. 2019, 20, 952. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer; Beral, V.; Gaitskell, K.; Hermon, C.; Moser, K.; Reeves, G.; Peto, R. Ovarian cancer and smoking: Individual participant meta-analysis including 28 114 women with ovarian cancer from 51 epidemiological studies. Lancet Oncol. 2012, 13, 946–956. [Google Scholar] [CrossRef]
- Faber, M.T.; Kjær, S.K.; Dehlendorff, C.; Chang-Claude, J.; Andersen, K.K.; Høgdall, E.; Webb, P.M.; Jordan, S.J.; Australian Cancer Study (Ovarian Cancer); Australian Ovarian Cancer Study Group; et al. Cigarette smoking and risk of ovarian cancer: A pooled analysis of 21 case-control studies. Cancer Causes Control 2013, 24, 989–1004. [Google Scholar] [CrossRef]
- Merritt, M.A.; Tzoulaki, I.; Brandt, P.A.V.D.; Schouten, L.J.; Tsilidis, K.K.; Weiderpass, E.; Patel, C.J.; Tjønneland, A.; Hansen, L.; Overvad, K.; et al. Nutrient-wide association study of 57 foods/nutrients and epithelial ovarian cancer in the European Prospective Investigation into Cancer and Nutrition study and the Netherlands Cohort Study. Am. J. Clin. Nutr. 2015, 103, 161–167. [Google Scholar] [CrossRef]
- Collaborative Group on Epidemiological Studies of Ovarian Cancer. Ovarian cancer and body size: Individual participant meta-analysis including 25,157 women with ovarian cancer from 47 epidemiological studies. PLoS Med. 2012, 9, e1001200. [Google Scholar] [CrossRef]
- Reid, A.; De Klerk, N.; Musk, A.W. (Bill) Does Exposure to Asbestos Cause Ovarian Cancer? A Systematic Literature Review and Meta-analysis. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 1287–1295. [Google Scholar] [CrossRef]
- Olsen, C.M.; Bain, C.J.; Jordan, S.J.; Nagle, C.; Green, A.C.; Whiteman, D.C.; Webb, P.M. Australian Cancer Study (Ovarian Cancer) and Australian Ovarian Cancer Study Group Recreational Physical Activity and Epithelial Ovarian Cancer: A Case-Control Study, Systematic Review, and Meta-analysis. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2321–2330. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed]
- Toss, A.; Tomasello, C.; Razzaboni, E.; Contu, G.; Grandi, G.; Cagnacci, A.; Schilder, R.J.; Cortesi, L. Hereditary Ovarian Cancer: Not OnlyBRCA1 and 2 Genes. Biomed. Res. Int. 2015, 2015, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Casey, M.J.; Snyder, C.L.; Bewtra, C.; Lynch, J.F.; Butts, M.; Godwin, A.K. Hereditary ovarian carcinoma: Heterogeneity, molecular genetics, pathology, and management. Mol. Oncol. 2009, 3, 97–137. [Google Scholar] [CrossRef]
- American Cancer Association. Available online: https://www.cancer.org/cancer/ovarian-cancer/causes-risks-prevention/what-causes (accessed on 28 May 2020).
- Hart, T.; Chandrashekhar, M.; Aregger, M.; Steinhart, Z.; Brown, K.R.; MacLeod, G.; Mis, M.; Zimmermann, M.; Fradet-Turcotte, A.; Sun, S.; et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 2015, 163, 1515–1526. [Google Scholar] [CrossRef]
- Yu, J.; Yusa, K. Genome-wide CRISPR-Cas9 screening in mammalian cells. Methods 2019, 29–35. [Google Scholar] [CrossRef]
- Behan, F.M.; Iorio, F.; Picco, G.; Gonçalves, E.; Beaver, C.M.; Migliardi, G.; Santos, R.; Rao, Y.; Sassi, F.; Pinnelli, M.; et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 2019, 568, 511–516. [Google Scholar] [CrossRef]
- Matsuo, K.; Sheridan, T.B.; Mabuchi, S.; Yoshino, K.; Hasegawa, K.; Studeman, K.D.; Im, D.D.; Rosenshein, N.B.; Roman, L.D.; Sood, A.K. Estrogen receptor expression and increased risk of lymphovascular space invasion in high-grade serous ovarian carcinoma. Gynecol. Oncol. 2014, 133, 473–479. [Google Scholar] [CrossRef]
- Andersen, C.; Sikora, M.J.; Boisen, M.M.; Ma, T.; Christie, A.; Tseng, G.; Park, Y.S.; Luthra, S.; Chandran, U.; Haluska, P.; et al. Active Estrogen Receptor-alpha Signaling in Ovarian Cancer Models and Clinical Specimens. Clin. Cancer Res. 2017, 23, 3802–3812. [Google Scholar] [CrossRef]
- Paleari, L.; DeCensi, A. Endocrine therapy in ovarian cancer. Curr. Opin. Obstet. Gynecol. 2018, 30, 17–22. [Google Scholar] [CrossRef]
- Koshiyama, M.; Matsumura, N.; Konishi, I. Recent Concepts of Ovarian Carcinogenesis: Type I and Type II. BioMed Res. Int. 2014, 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J.; Shih, I.-M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed]
- Santin, A.D.; Zhan, F.; Bellone, S.; Palmieri, M.; Canè, S.; Bignotti, E.; Anfossi, S.; Gokden, M.; Dunn, D.; Roman, J.J.; et al. Gene expression profiles in primary ovarian serous papillary tumors and normal ovarian epithelium: Identification of candidate molecular markers for ovarian cancer diagnosis and therapy. Int. J. Cancer 2004, 112, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Salani, R.; Kurman, R.J.; Giuntoli, R.; Gardner, G.; Bristow, R.; Wang, T.-L.; Shih, I.-M. Assessment of TP53 mutation using purified tissue samples of ovarian serous carcinomas reveals a higher mutation rate than previously reported and does not correlate with drug resistance. Int. J. Gynecol. Cancer 2008, 18, 487–491. [Google Scholar] [CrossRef]
- Gockley, A.A.; Melamed, A.; Bregar, A.J.; Clemmer, J.T.; Birrer, M.; Schorge, J.O.; Del Carmen, M.G.; Rauh-Hain, J.A. Outcomes of Women With High-Grade and Low-Grade Advanced-Stage Serous Epithelial Ovarian Cancer. Obstet. Gynecol. 2017, 129, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Kurman, R.J. Origin and molecular pathogenesis of ovarian high-grade serous carcinoma. Ann. Oncol. 2013, 24, x16–x21. [Google Scholar] [CrossRef]
- Plaxe, S. Epidemiology of low-grade serous ovarian cancer. Am. J. Obstet. Gynecol. 2008, 198, 459.e1–459.e9. [Google Scholar] [CrossRef]
- Gilks, C.B. Subclassification of ovarian surface epithelial tumors based on correlation of histologic and molecular pathologic data. Int. J. Gynecol. Pathol. 2004, 23, 200–205. [Google Scholar] [CrossRef]
- Ceppi, L.; Birrer, M.J. Translational Advances in Gyneacologic Cancers, 1st ed.; Academic Press: London, UK, 2017. [Google Scholar]
- Hirst, J.; Crow, J.; Godwin, A.K. Ovarian Cancer Genetics: Subtypes and Risk Factors. Ovarian Cancer Pathog. Treat. 2018. [Google Scholar] [CrossRef]
- Cho, K.R.; Shih, I.M. Ovarian cancer. Annu. Rev. Pathol. 2009, 4, 287–313. [Google Scholar] [CrossRef]
- Pierson, W.E.; Peters, P.N.; Chang, M.T.; Chen, L.-M.; Quigley, D.A.; Ashworth, A.; Chapman, J.S. An integrated molecular profile of endometrioid ovarian cancer. Gynecol. Oncol. 2020, 157, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Serebrenik, A.A.; Argyris, P.P.; Jarvis, M.C.; Brown, W.L.; Bazzaro, M.; Vogel, R.I.; Erickson, B.K.; Lee, S.-H.; Goergen, K.M.; Maurer, M.J.; et al. The DNA Cytosine Deaminase APOBEC3B is a Molecular Determinant of Platinum Responsiveness in Clear Cell Ovarian Cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kandalaft, P.L.; Gown, A.M.; Isacson, C. The Lung-Restricted Marker Napsin A Is Highly Expressed in Clear Cell Carcinomas of the Ovary. Am. J. Clin. Pathol. 2014, 142, 830–836. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4, 2126. [Google Scholar] [CrossRef] [PubMed]
- Zorn, K. Gene Expression Profiles of Serous, Endometrioid, and Clear Cell Subtypes of Ovarian and Endometrial Cancer. Clin. Cancer Res. 2005, 11, 6422–6430. [Google Scholar] [CrossRef]
- Schwartz, D.R.; Kardia, S.L.R.; Shedden, K.; Kuick, R.; Michailidis, G.; Taylor, J.M.G.; Misek, D.; Wu, R.; Zhai, Y.; Darrah, D.M.; et al. Gene expression in ovarian cancer reflects both morphology and biological behavior, distinguishing clear cell from other poor-prognosis ovarian carcinomas. Cancer Res. 2002, 62, 4722–4729. [Google Scholar]
- Su, Y.-F.; Tsai, E.-M.; Chen, C.-C.; Wu, C.-C.; Er, T.K. Targeted sequencing of a specific gene panel detects a high frequency of ARID1A and PIK3CA mutations in ovarian clear cell carcinoma. Clin. Chim. Acta 2019, 494, 1–7. [Google Scholar] [CrossRef]
- Babaier, A.; Ghatage, P. Mucinous Cancer of the Ovary: Overview and Current Status. Diagnostics 2020, 10, 52. [Google Scholar] [CrossRef]
- Lenhard, M.; Tereza, L.; Heublein, S.; Ditsch, N.; Himsl, I.; Mayr, D.; Friese, D.M.K.; Jeschke, U. Steroid hormone receptor expression in ovarian cancer: Progesterone receptor B as prognostic marker for patient survival. BMC Cancer 2012, 12, 553. [Google Scholar] [CrossRef]
- Smyth, J.F.; Gourley, C.; Walker, G.; MacKean, M.J.; Stevenson, A.; Williams, A.; Nafussi, A.A.; Rye, T.; Rye, R.; Stewart, M.; et al. Antiestrogen Therapy Is Active in Selected Ovarian Cancer Cases: The Use of Letrozole in Estrogen Receptor-Positive Patients. Clin. Cancer Res. 2007, 13, 3617–3622. [Google Scholar] [CrossRef]
- Argenta, P.A.; Thomas, S.G.; Judson, P.L.; Downs, L.S.; Geller, M.A.; Carson, L.F.; Jonson, A.L.; Ghebre, R. A phase II study of fulvestrant in the treatment of multiply-recurrent epithelial ovarian cancer. Gynecol. Oncol. 2009, 113, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The Nuclear Receptor Superfamily: The Second Decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Charn, T.H.; Liu, E.T.-B.; Chang, E.C.; Lee, Y.K.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Genome-wide dynamics of chromatin binding of estrogen receptors alpha and beta: Mutual restriction and competitive site selection. Mol. Endocrinol. 2009, 24, 47–59. [Google Scholar] [CrossRef]
- Brandenberger, A.W.; Tee, M.K.; Jaffe, R.B. Estrogen Receptor Alpha (ER-?) and Beta (ER-?) mRNAs in Normal Ovary, Ovarian Serous Cystadenocarcinoma and Ovarian Cancer Cell Lines: Down-Regulation of ER-? in Neoplastic Tissues. J. Clin. Endocrinol. Metab. 1998, 83, 1025–1028. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tang, Z.-R.; Zhang, R.; Lian, Z.; Deng, S.-L.; Yu, K. Estrogen-Receptor Expression and Function in Female Reproductive Disease. Cells 2019, 8, 1123. [Google Scholar] [CrossRef]
- Lazennec, G. Estrogen receptor beta, a possible tumor suppressor involved in ovarian carcinogenesis. Cancer Lett. 2006, 231, 151–157. [Google Scholar] [CrossRef]
- Bossard, C.; Busson, M.; Vindrieux, D.; Gaudin, F.; Machelon, V.; Brigitte, M.; Jacquard, C.; Pillon, A.; Balaguer, P.; Balabanian, K.; et al. Potential Role of Estrogen Receptor Beta as a Tumor Suppressor of Epithelial Ovarian Cancer. PLoS ONE 2012, 7, e44787. [Google Scholar] [CrossRef]
- Schlumbrecht, M.P.; Xie, S.S.; Shipley, G.L.; Urbauer, D.L.; Broaddus, R.R. Molecular clustering based on ERα and EIG121 predicts survival in high-grade serous carcinoma of the ovary/peritoneum. Mod. Pathol. 2011, 24, 453–462. [Google Scholar] [CrossRef]
- Halon, A.; Materna, V.; Drag-Zalesinska, M.; Nowak-Markwitz, E.; Gansukh, T.; Donizy, P.; Spaczyński, M.; Zabel, M.; Dietel, M.; Lage, H.; et al. Estrogen Receptor Alpha Expression in Ovarian Cancer Predicts Longer Overall Survival. Pathol. Oncol. Res. 2011, 17, 511–518. [Google Scholar] [CrossRef][Green Version]
- Lannigan, D.A. Estrogen receptor phosphorylation. Steroids 2003, 68, 1–9. [Google Scholar] [CrossRef]
- Matsumura, S.; Ohta, T.; Yamanouchi, K.; Liu, Z.; Sudo, T.; Kojimahara, T.; Seino, M.; Narumi, M.; Tsutsumi, S.; Takahashi, T.; et al. Activation of estrogen receptor α by estradiol and cisplatin induces platinum-resistance in ovarian cancer cells. Cancer Boil. Ther. 2016, 18, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Kimura, A.; Ohmichi, M.; Kawagoe, J.; Kyo, S.; Mabuchi, S.; Takahashi, T.; Ohshima, C.; Arimoto-Ishida, E.; Nishio, Y.; Inoue, M.; et al. Induction of hTERT expression and phosphorylation by estrogen via Akt cascade in human ovarian cancer cell lines. Oncogene 2004, 23, 4505–4515. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, A.; MacLeod, K.; Burns, D.J.; Smyth, J.F.; Langdon, S.P. Estrogen receptor-α mediates gene expression changes and growth response in ovarian cancer cells exposed to estrogen. Endocr. Relat. Cancer 2005, 12, 851–866. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, M.; Ma, H. Paired box gene 2 is associated with estrogen receptor α in ovarian serous tumors: Potential theory basis for targeted therapy. Mol. Clin. Oncol. 2016, 5, 323–326. [Google Scholar] [CrossRef]
- Salvati, A.; Gigantino, V.; Nassa, G.; Giurato, G.; Alexandrova, E.; Rizzo, F.; Tarallo, R.; Weisz, A. The Histone Methyltransferase DOT1L Is a Functional Component of Estrogen Receptor Alpha Signaling in Ovarian Cancer Cells. Cancers 2019, 11, 1720. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-H.; Cheung, L.W.T.; Wong, A.S.T.; Leung, P.C.K. Estrogen Regulates Snail and Slug in the Down-Regulation of E-Cadherin and Induces Metastatic Potential of Ovarian Cancer Cells through Estrogen Receptor α. Mol. Endocrinol. 2008, 22, 2085–2098. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, C.; Mo, Y.; Liu, H.; Zhai, S.; Zhou, H. ERα and ERβ oppositely regulated plexin B1 expression and migration of ovarian cancer SKOV-3 cells. Int. J. Clin. Exp. Med. 2018, 11, 3484–3493. [Google Scholar]
- Benhadjeba, S.; Edjekouane, L.; Sauvé, K.; Carmona, E.; Tremblay, A. Feedback control of the CXCR7/CXCL11 chemokine axis by estrogen receptor α in ovarian cancer. Mol. Oncol. 2018, 12, 1689–1705. [Google Scholar] [CrossRef]
- Zheng, J.; Zhou, J.; Xie, X.; Xie, B.; Lin, J.; Xu, Z.; Zhang, W. Estrogen Decreases Anoikis of Ovarian Cancer Cell Line Caov-3 Through Reducing Release of Bit1. DNA Cell Boil. 2014, 33, 847–853. [Google Scholar] [CrossRef]
- Kodama, M.; Kodama, T.; Newberg, J.Y.; Katayama, H.; Kobayashi, M.; Hanash, S.M.; Yoshihara, K.; Wei, Z.; Tien, J.C.; Rangel, R.; et al. In vivo loss-of-function screens identify KPNB1 as a new druggable oncogene in epithelial ovarian cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E7301–E7310. [Google Scholar] [CrossRef]
- He, Y.; Meghani, K.; Caron, M.-C.; Yang, C.; Ronato, D.A.; Bian, J.; Sharma, A.; Moore, J.; Niraj, J.; Detappe, A.; et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature 2018, 563, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; De Souza, C.; Minn, K.; Chien, J. Genome-scale CRISPR knockout screen identifies TIGAR as a modifier of PARP inhibitor sensitivity. Commun. Boil. 2019, 2, 335. [Google Scholar] [CrossRef]
- Ouyang, Q.; Liu, Y.; Tan, J.; Li, J.; Yang, D.; Zeng, F.; Huang, W.; Kong, Y.; Liu, Z.; Zhou, H.; et al. Loss of ZNF587B and SULF1 contributed to cisplatin resistance in ovarian cancer cell lines based on Genome-scale CRISPR/Cas9 screening. Am. J. Cancer Res. 2019, 9, 988–998. [Google Scholar] [PubMed]
- Stover, E.H.; Baco, M.B.; Cohen, O.; Li, Y.Y.; Christie, E.; Bagul, M.; Goodale, A.; Lee, Y.; Pantel, S.; Rees, M.G.; et al. Pooled Genomic Screens Identify Anti-apoptotic Genes as Targetable Mediators of Chemotherapy Resistance in Ovarian Cancer. Mol. Cancer Res. 2019, 17, 2281–2293. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Dempster, D.J.; Jordan, R.; Mariya, K.; Pan, J.; Guillaume, K.; Root., E.D.; Aviad, T. Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv 2020. [Google Scholar] [CrossRef]
- Broad Institute. DepMap Portal. Available online: https://depmap.org/portal/ (accessed on 10 May 2020).
- Sanger Institute. Cancer Dependency Map Project Score. Available online: https://score.depmap.sanger.ac.uk/ (accessed on 10 May 2020).
- Blayney, J.K.; Davison, T.; McCabe, N.; Walker, S.; Keating, K.; Delaney, T.; Greenan, C.; Williams, A.R.; McCluggage, W.G.; Capes-Davis, A.; et al. Prior knowledge transfer across transcriptional data sets and technologies using compositional statistics yields new mislabelled ovarian cell line. Nucleic Acids Res. 2016, 44, e137. [Google Scholar] [CrossRef]
- Mirza-Aghazadeh-Attari, M.; Ostadian, C.; Saei, A.A.; Mihanfar, A.; Darband, S.G.; Sadighparvar, S.; Kaviani, M.; Kafil, H.S.; Yousefi, B.; Majidinia, M.; et al. DNA damage response and repair in ovarian cancer: Potential targets for therapeutic strategies. DNA Repair 2019, 80, 59–84. [Google Scholar] [CrossRef]
- Nam, E.J.; Kim, Y.T. Alteration of cell-cycle regulation in epithelial ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 1169–1182. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef]
- Sanabria, A.L.A.; Jiménez, C.N.; Sánchez, V.C.; Serrano-Oviedo, L.; Andrés-Pretel, F.; Burgos, M.; Galán-Moya, E.M.; Montero, J.C.; Llopis, J.; Pandiella, A.; et al. Synthetic Lethality Interaction Between Aurora Kinases and CHEK1 Inhibitors in Ovarian Cancer. Mol. Cancer Ther. 2017, 16, 2552–2562. [Google Scholar] [CrossRef] [PubMed]
- Slipicevic, A.; Holth, A.; Hellesylt, E.; Trope, C.G.; Davidson, B.; Flørenes, V.A. Wee1 is a novel independent prognostic marker of poor survival in post-chemotherapy ovarian carcinoma effusions. Gynecol. Oncol. 2014, 135, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Schmid, B.C.; Oehler, M.K. New perspectives in ovarian cancer treatment. Maturitas 2014, 77, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Xu, J.; Zhao, S.; Shi, H.; Yao, S.; Jiang, N. ShRNA-mediated silencing of the RFC3 gene suppress ovarian tumor cells proliferation. Int. J. Clin. Exp. Pathol. 2015, 8, 8968–8975. [Google Scholar] [PubMed]
- Abdel-Fatah, T.M.; Arora, A.; Moseley, P.; Coveney, C.; Perry, C.; Johnson, K.; Kent, C.; Ball, G.; Chan, S.; Madhusudan, S. ATM, ATR and DNA-PKcs expressions correlate to adverse clinical outcomes in epithelial ovarian cancers. BBA Clin. 2014, 2, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; George, E.; Ragland, R.L.; Rafail, S.; Zhang, R.; Krepler, C.; Morgan, M.A.; Herlyn, M.; Brown, E.J.; Simpkins, F.; et al. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin. Cancer Res. 2016, 23, 3097–3108. [Google Scholar] [CrossRef]
- Gralewska, P.; Gajek, A.; Marczak, A.; Rogalska, A. Participation of the ATR/CHK1 pathway in replicative stress targeted therapy of high-grade ovarian cancer. J. Hematol. Oncol. 2020, 13, 1–16. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, J.; Gil-Moreno, A.; García, Á.; Rojo, F.; Xercavins, J.; Salido, E.; Freire, R. Expression of DNA Damage Checkpoint Protein Hus1 in Epithelial Ovarian Tumors Correlates With Prognostic Markers. Int. J. Gynecol. Pathol. 2008, 27, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.L.; Chaudhry, S.; Lopes, G.S.; Levin, N.K.; Tainsky, M. FANCM, RAD1, CHEK1 and TP53I3 act as BRCA-like tumor suppressors and are mutated in hereditary ovarian cancer. Cancer Genet. 2019, 57–64. [Google Scholar] [CrossRef]
- Dudas, A.; Chovanec, M. DNA double-strand break repair by homologous recombination. Mutat. Res. 2004, 566, 131–167. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Dicks, E.; Ramus, S.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef]
- Malisic, E.; Krivokuća, A.; Boljevic, I.; Janković, R. Impact of RAD51 G135C and XRCC1 Arg399Gln polymorphisms on ovarian carcinoma risk in Serbian women. Cancer Biomark. 2015, 15, 685–691. [Google Scholar] [CrossRef] [PubMed]
- Fortier, E.A.; Drobetsky, E.; Wurtele, H. Know your limits: RPA availability and chemoresistance in ovarian cancer. Oncotarget 2019, 10, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Chernikova, S.B.; Game, J.C.; Brown, J.M. Inhibiting homologous recombination for cancer therapy. Cancer Biol. Ther. 2012, 13, 61–68. [Google Scholar] [CrossRef]
- Kopa, P.; Macieja, A.; Galita, G.; Witczak, Z.J.; Poplawski, T. DNA Double Strand Breaks Repair Inhibitors: Relevance as Potential New Anticancer Therapeutics. Curr. Med. Chem. 2019, 26, 1483–1493. [Google Scholar] [CrossRef]
- Zhao, M.; Li, S.; Zhou, L.; Shen, Q.; Zhu, H.; Zhu, X. Prognostic values of excision repair cross-complementing genes mRNA expression in ovarian cancer patients. Life Sci. 2018, 194, 34–39. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, J.; Chen, Y.; Hu, Q.; Shen, H.; Huang, R.; Liu, Q.; Kaur, J.; Long, M.D.; Battaglia, S.; et al. Whole-exome sequencing of ovarian cancer families uncovers putative predisposition genes. Int. J. Cancer 2019, 146, 2147–2155. [Google Scholar] [CrossRef]
- Wu, M.; Sun, Y.; Wu, J.; Liu, G. Identification of Hub Genes in High-Grade Serous Ovarian Cancer Using Weighted Gene Co-Expression Network Analysis. Med. Sci. Monit. 2020, 26, e92210-1. [Google Scholar] [CrossRef]
- Samanta, S.; Tamura, S.; Dubeau, L.; Mhawech-Fauceglia, P.; Miyagi, Y.; Kato, H.; Lieberman, R.; Buckanovich, R.J.; Lin, Y.G.; Neamati, N. Clinicopathological significance of endoplasmic reticulum stress proteins in ovarian carcinoma. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Sevigny, C.; Bhattacharya, P.; Jordan, V.C.; Clarke, R. Estrogen-Induced Apoptosis in Breast Cancers Is Phenocopied by Blocking Dephosphorylation of Eukaryotic Initiation Factor 2 Alpha (eIF2α) Protein. Mol. Cancer Res. 2019, 17, 918–928. [Google Scholar] [CrossRef]
- Weis, S.M.A.; Cheresh, D. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Spannuth, W.A.; Nick, A.M.; Jennings, N.B.; Armaiz-Pena, G.N.; Mangala, L.S.; Danes, C.G.; Lin, Y.G.; Merritt, W.M.; Thaker, P.H.; Kamat, A.A.; et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int. J. Cancer 2009, 124, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Kalli, K.R.; Falowo, O.I.; Bale, L.K.; Zschunke, M.A.; Roche, P.C.; Conover, C.A. Functional Insulin Receptors on Human Epithelial Ovarian Carcinoma Cells: Implications for IGF-II Mitogenic Signaling. Endocrinology 2002, 143, 3259–3267. [Google Scholar] [CrossRef] [PubMed]
- Liefers-Visser, J.; Meijering, R.; Reyners, A.; Van Der Zee, A.; De Jong, S. IGF system targeted therapy: Therapeutic opportunities for ovarian cancer. Cancer Treat. Rev. 2017, 60, 90–99. [Google Scholar] [CrossRef]
- Ghoneum, A.; Said, N. PI3K-AKT-mTOR and NFκB Pathways in Ovarian Cancer: Implications for Targeted Therapeutics. Cancers 2019, 11, 949. [Google Scholar] [CrossRef]
- Lau, M.-T.; Leung, P.C.K. The PI3K/Akt/mTOR signaling pathway mediates insulin-like growth factor 1-induced E-cadherin down-regulation and cell proliferation in ovarian cancer cells. Cancer Lett. 2012, 326, 191–198. [Google Scholar] [CrossRef]
- Cáceres-Gorriti, K.Y.; Carmona, E.; Barrès, V.; Rahimi, K.; Létourneau, I.J.; Tonin, P.N.; Provencher, D.; Mes-Masson, A.-M. RAN Nucleo-Cytoplasmic Transport and Mitotic Spindle Assembly Partners XPO7 and TPX2 Are New Prognostic Biomarkers in Serous Epithelial Ovarian Cancer. PLoS ONE 2014, 9, e91000. [Google Scholar] [CrossRef]
- Barrès, V.; Ouellet, V.; Lafontaine, J.; Tonin, P.; Provencher, D.; Mes-Masson, A.-M. An essential role for Ran GTPase in epithelial ovarian cancer cell survival. Mol. Cancer 2010, 9, 272. [Google Scholar] [CrossRef]
- Lorenzato, A.; Biolatti, M.; Delogu, G.; Capobianco, G.; Farace, C.; Dessole, S.; Cossu, A.G.M.; Tanda, F.; Madeddu, R.; Olivero, M.; et al. AKT activation drives the nuclear localization of CSE1L and a pro-oncogenic transcriptional activation in ovarian cancer cells. Exp. Cell Res. 2013, 319, 2627–2636. [Google Scholar] [CrossRef]
- Zaoui, K.; Boudhraa, Z.; Khalifé, P.; Carmona, E.; Provencher, D.; Mes-Masson, A.-M. Ran promotes membrane targeting and stabilization of RhoA to orchestrate ovarian cancer cell invasion. Nat. Commun. 2019, 10, 2666. [Google Scholar] [CrossRef] [PubMed]
- Moldovan, G.-L.; Pfander, B.; Jentsch, S. PCNA, the Maestro of the Replication Fork. Cell 2007, 129, 665–679. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.-H.; Lu, D.-L.; Wang, N.; Liu, L.-Y.; Wang, Y.; Li, Y.-Q.; Yan, T.-B.; Sun, X.-G.; Hu, P.; Zhang, T.-C. Estrogen receptor α mediates proliferation of breast cancer MCF-7 cells via a p21/PCNA/E2F1-dependent pathway. FEBS J. 2014, 281, 927–942. [Google Scholar] [CrossRef] [PubMed]
- Thomas, H.; Nasim, M.; Sarraf, C.; Alison, M.; Love, S.; Lambert, H.; Price, P. Proliferating cell nuclear antigen (PCNA) immunostaining—A prognostic factor in ovarian cancer? Br. J. Cancer 1995, 71, 357–362. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Skanderup, A.J.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401-4. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Skanderup, A.J.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- cBioPortal for Cancer Genomics. Available online: https://www.cbioportal.org/ (accessed on 10 May 2020).
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A. MYC-Driven Pathways in Breast Cancer Subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef]
- Cheng, K.W.; Agarwal, R.; Mills, G.B. Ras-Superfamily GTP-ases in Ovarian Cancer. Cancer Treat. Res. 2009, 149, 229–240. [Google Scholar] [CrossRef]
- Brennan, D.; Brändstedt, J.; Rexhepaj, E.; Foley, M.E.; Pontén, F.; Uhlén, M.; Gallagher, W.; O’Connor, D.; O’Herlihy, C.; Jirström, K. Tumour-specific HMG-CoAR is an independent predictor of recurrence free survival in epithelial ovarian cancer. BMC Cancer 2010, 10, 125. [Google Scholar] [CrossRef] [PubMed]
- Friedman, G.D.; Flick, E.D.; Udaltsova, N.; Chan Pharm, D.J.; Quesenberry, C.P.; Habel, L.A. Screening statins for possible carcinogenic risk: Up to 9 years of follow-up of 361 859 recipients. Pharmacoepidemiol. Drug Saf. 2007, 17, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.; Boudreau, D.M.; Buist, D.S.M.; Miglioretti, D.L. Statin use and female reproductive organ cancer risk in a large population-based setting. Cancer Causes Control 2008, 20, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Qin, A.; Li, T.; Qin, X.; Li, S. Effect of statin on risk of gynecologic cancers: A meta-analysis of observational studies and randomized controlled trials. Gynecol. Oncol. 2014, 133, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Stine, J.E.; Guo, H.; Sheng, X.; Han, X.; Schointuch, M.N.; Gilliam, T.P.; Gehrig, P.A.; Zhou, C.; Bae-Jump, V. The HMG-CoA reductase inhibitor, simvastatin, exhibits anti-metastatic and anti-tumorigenic effects in ovarian cancer. Oncotarget 2015, 7, 946–960. [Google Scholar] [CrossRef]
- Martirosyan, A.; Clendening, J.W.A.; Goard, C.; Penn, L.Z. Lovastatin induces apoptosis of ovarian cancer cells and synergizes with doxorubicin: Potential therapeutic relevance. BMC Cancer 2010, 10, 103. [Google Scholar] [CrossRef]
- Robinson, E.; Nandi, M.; Wilkinson, L.L.; Arrowsmith, D.M.; Curtis, A.; Richardson, A. Preclinical evaluation of statins as a treatment for ovarian cancer. Gynecol. Oncol. 2013, 129, 417–424. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kashima, H.; Wu, R.-C.; Jung, J.-G.; Kuan, J.-C.; Gu, J.; Xuan, J.; Sokoll, L.; Visvanathan, K.; Shih, I.-M.; et al. Mevalonate Pathway Antagonist Suppresses Formation of Serous Tubal Intraepithelial Carcinoma and Ovarian Carcinoma in Mouse Models. Clin. Cancer Res. 2015, 21, 4652–4662. [Google Scholar] [CrossRef]
- Greenaway, J.B.; Virtanen, C.; Osz, K.; Revay, T.; Hardy, D.; Shepherd, T.; DiMattia, G.; Petrik, J. Ovarian tumour growth is characterized by mevalonate pathway gene signature in an orthotopic, syngeneic model of epithelial ovarian cancer. Oncotarget 2016, 7, 47343–47365. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Kashima, H.; Rahmanto, Y.S.; Banno, K.; Yu, Y.; Matoba, Y.; Watanabe, K.; Iijima, M.; Takeda, T.; Kunitomi, H.; et al. Drug repositioning of mevalonate pathway inhibitors as antitumor agents for ovarian cancer. Oncotarget 2017, 8, 72147–72156. [Google Scholar] [CrossRef]
- Abdullah, M.I.; Abed, M.N.; Richardson, A. Inhibition of the mevalonate pathway augments the activity of pitavastatin against ovarian cancer cells. Sci. Rep. 2017, 7, 8090. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.; Epel, E.; Lin, J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science 2015, 350, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Xin, H.; Liu, D.; Songyang, Z. The telosome/shelterin complex and its functions. Genome Biol. 2008, 9, 232. [Google Scholar] [CrossRef] [PubMed]
- Counter, C.M.; Hirte, H.W.; Bacchetti, S.; Harley, C.B. Telomerase activity in human ovarian carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 2900–2904. [Google Scholar] [CrossRef] [PubMed]
- Huda, N.; Xu, Y.; Bates, A.M.; Rankin, D.A.; Kannan, N.; Gilley, D. Onset of Telomere Dysfunction and Fusions in Human Ovarian Carcinoma. Cells 2019, 8, 414. [Google Scholar] [CrossRef]
- Kotsopoulos, J.; Prescott, J.; De Vivo, I.; Fan, I.; McLaughlin, J.; Rosen, B.; Risch, H.; Sun, P.A.; Narod, S. Telomere length and mortality following a diagnosis of ovarian cancer. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2603–2606. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Tao, W.; Huang, M.; Wu, X.; Gu, J. Genetic variants in telomere-maintenance genes are associated with ovarian cancer risk and outcome. J. Cell. Mol. Med. 2016, 21, 510–518. [Google Scholar] [CrossRef]
- Martínez-Delgado, B.; Yanowsky, K.; Inglada, L.; De La Hoya, M.; Caldes, T.; Vega, A.; Blanco, A.; Martin, T.; González-Sarmiento, R.; Blasco, M.A.; et al. Shorter telomere length is associated with increased ovarian cancer risk in both familial and sporadic cases. J. Med. Genet. 2012, 49, 341–344. [Google Scholar] [CrossRef]
- Kunifuji, Y.; Gotoh, S.; Abe, T.; Miura, M.; Karasaki, Y. Down-regulation of telomerase activity by anticancer drugs in human ovarian cancer cells. Anti-Cancer Drugs 2002, 13, 595–598. [Google Scholar] [CrossRef]
- Shay, J.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Patel, K.P.; Vonderheide, R.H. Telomerase as a tumor-associated antigen for cancer immunotherapy. Cytotechnology 2004, 45, 91–99. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saretzki, G. Telomerase inhibition as cancer therapy. Cancer Lett. 2003, 194, 209–219. [Google Scholar] [CrossRef]
- Chen, M.; Xing, L.-N. siRNA-mediated inhibition of hTERC enhances radiosensitivity of cervical cancer. Asian Pac. J. Cancer Prev. 2012, 13, 5975–5979. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bidzinska, J.; Baginski, M.; Skladanowski, A. Novel anticancer strategy aimed at targeting shelterin complexes by the induction of structural changes in telomeric DNA: Hitting two birds with one stone. Curr. Cancer Drug Targets 2014, 14, 201–216. [Google Scholar] [CrossRef]
- Bejarano, L.; Schuhmacher, A.J.; Méndez, M.; Megias, D.; Blanco-Aparicio, C.; Martínez, S.; Pastor, J.; Squatrito, M.; Blasco, M.A. Inhibition of TRF1 Telomere Protein Impairs Tumor Initiation and Progression in Glioblastoma Mouse Models and Patient-Derived Xenografts. Cancer Cell 2017, 32, 590–607.e4. [Google Scholar] [CrossRef]
- Lanzetti, L.; Di Fiore, P.P. Endocytosis and Cancer: An ‘Insider’ Network with Dangerous Liaisons. Traffic 2008, 9, 2011–2021. [Google Scholar] [CrossRef]
- Arakel, E.C.; Schwappach, B. Formation of COPI-coated vesicles at a glance. J. Cell Sci. 2018, 131, jcs209890. [Google Scholar] [CrossRef] [PubMed]
- Claerhout, S.; Dutta, B.; Bossuyt, W.; Zhang, F.; Nguyen-Charles, C.; Dennison, J.B.; Yu, Q.; Yu, S.; Balazsi, G.; Lu, Y.; et al. Abortive Autophagy Induces Endoplasmic Reticulum Stress and Cell Death in Cancer Cells. PLoS ONE 2012, 7, e39400. [Google Scholar] [CrossRef]
- Shtutman, M.; Roninson, I.B. A subunit of coatomer protein complex offers a novel tumor-specific target through a surprising mechanism. Autophagy 2011, 7, 1551–1552. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ovarian Cancer Subtype | Cell Lines |
|---|---|
| High Grade Serous | Caov-3, COV318, COV362, HEY A8, JHOS-2, JHOS-4, Kuramochi, OAW28, ONCO-DG-1, OV-90, OVCAR-8, Caov-4, HEY, OVCAR-5*, TYK-nu, OVCAR-3, OVMIU, PEO1, PEO4 |
| Clear Cell | JHOC-5, OVISE, OVMANA, OVTOKO, ES-2, RMG-I, TOV-21G |
| Endometrioid | A2780, TOV-112D, A2780ADR, IGROV-1, OVK18, A2780cis |
| Mucinous | COV644, JHOM-1, RMUG-S, EFO-27, MCAS |
| Serous | SNU-8, UWB1.289, OAW42, OC 314, OVCA420 |
| Mixed | 59M, OV7 |
| Brenner Tumor | SNU-840 |
| Granulosa Cells Tumor | COV434 |
| Unspecified | DOV13, EFO-21 |
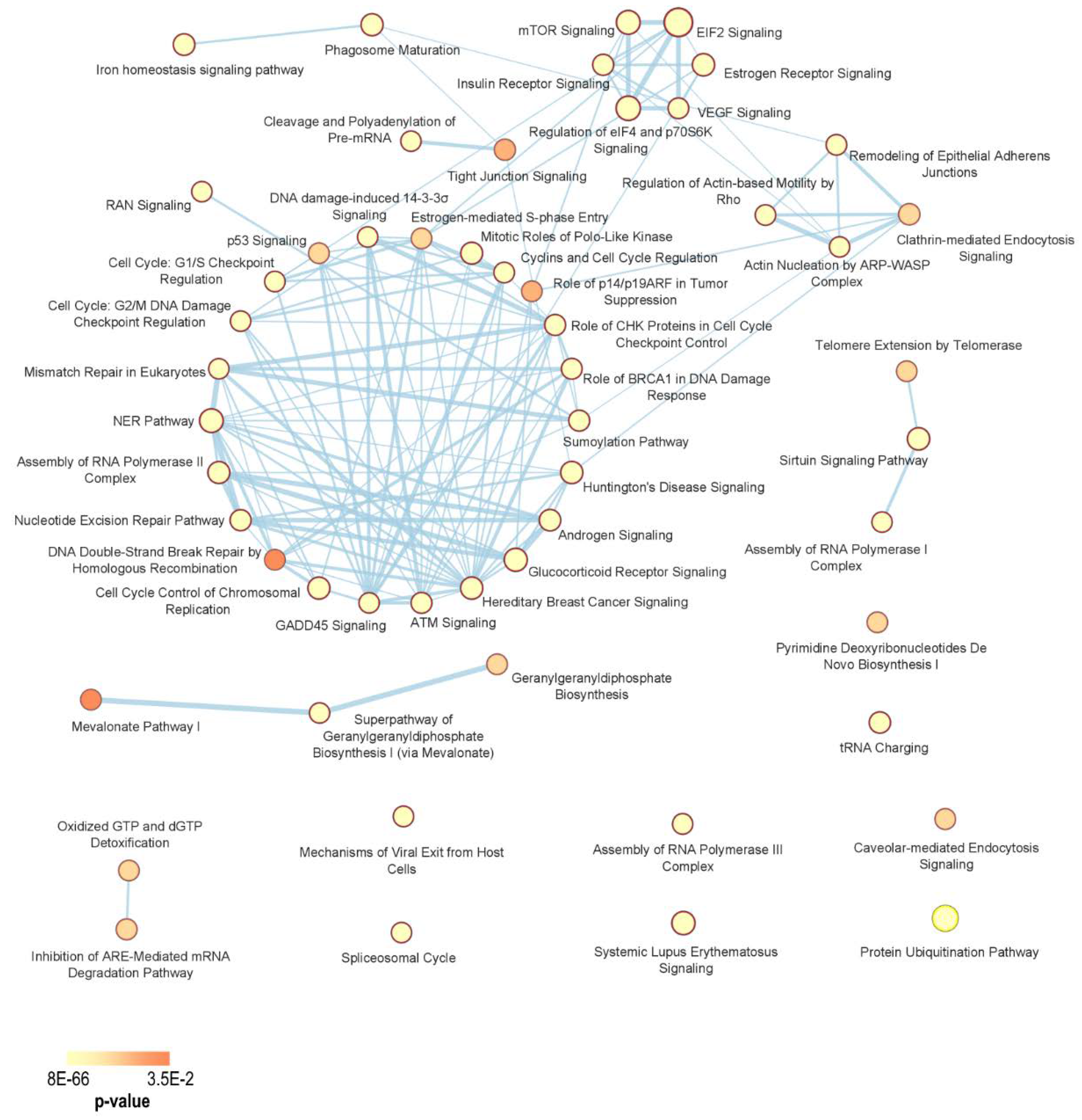
| Pathway | p-Value | Genes |
|---|---|---|
| Cell cycle regulation and DNA damage response (DDR) | ||
| NER Pathway | 1.58E−32 | CCNH, CDK7, CHAF1A, CHAF1B, COPS2, COPS4, COPS5, COPS6, COPS8, DDB1, ERCC2, ERCC3, GPS1, GTF2H1, NEDD8, PCNA, POLA1, POLA2, POLD1, POLD2, POLD3, POLE, POLE2, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, PRIM1, RBX1, RFC2, RFC3, RFC4, RFC5, RPA1, RPA2, RPA3, TOP2A, UBE2I, UBE2N, USP7, XAB2 |
| Cell Cycle Control of Chromosomal Replication | 1.26E−22 | CDC45, CDC6, CDC7, CDK1, CDK11A, CDK2, CDK7, CDK9, CDT1, DBF4, MCM2, MCM3, MCM4, MCM5, MCM6, MCM7, ORC1, ORC5, ORC6, PCNA, POLA1, POLA2, POLD1, POLE, PRIM1, RPA1, RPA2, RPA3, TOP2A |
| Mitotic Roles of Polo-Like Kinase | 1.00E−17 | ANAPC1, ANAPC10, ANAPC11, ANAPC2, ANAPC4, ANAPC5, CCNB1, CDC16, CDC20, CDC23, CDC26, CDC27, CDC7, CDK1, ESPL1, FBXO5, KIF11, KIF23, PKMYT1, PLK1, PLK4, PPP2R1A, PRC1, RAD21, SMC1A, SMC3, WEE1 |
| Nucleotide Excision Repair Pathway | 2.00E−14 | CCNH, CDK7, ERCC2, ERCC3, GTF2H1, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, RPA1, RPA2, RPA3 |
| Role of CHK Proteins in Cell Cycle Checkpoint Control | 3.47E−09 | ATR, CDK1, CDK2, CHEK1, CLSPN, HUS1, PCNA, PLK1, PPP2R1A, RAD1, RAD17, RAD9A, RFC2, RFC3, RFC4, RFC5, RPA1 |
| DNA damage-induced 14-3-3σ Signaling | 2.75E−06 | ATR, CCNB1, CDK1, CDK2, HUS1, RAD1, RAD17, RAD9A |
| Mismatch Repair in Eukaryotes | 8.71E−06 | PCNA, POLD1, RFC2, RFC3, RFC4, RFC5, RPA1 |
| Role of BRCA1 in DNA Damage Response | 1.74E−05 | ACTB, ATR, ATRIP, CHEK1, PLK1, RAD51, RBBP8, RFC2, RFC3, RFC4, RFC5, RPA1, SMARCB1, SMARCE1, TOPBP1 |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 4.07E−05 | ATR, AURKA, CCNB1, CDK1, CDK7, CHEK1, PKMYT1, PLK1, SKP1, TOP2A, WEE1 |
| ATM Signaling | 4.90E−05 | ATR, CCNB1, CDK1, CDK2, CHEK1, PPP2R1A, RAD17, RAD51, RAD9A, RBBP8, SMC1A, SMC2, SMC3, TOPBP1, TRRAP, USP7 |
| Cell Cycle: G1/S Checkpoint Regulation | 1.86E−04 | ATR, CCND1, CDK2, GNL3, HDAC3, MAX, MYC, PAK1IP1, RPL11, RPL5, SIN3A, SKP1 |
| Cyclins and Cell Cycle Regulation | 3.24E−04 | ATR, CCNA2, CCNB1, CCND1, CCNH, CDK1, CDK2, CDK7, HDAC3, PPP2R1A, SIN3A, SKP1, WEE1 |
| Estrogen-mediated S-phase Entry | 1.07E−02 | CCNA2, CCND1, CDK1, CDK2, MYC |
| p53 Signaling | 1.70E−02 | ACTB, CDC42, CPSF1, CPSF2, CPSF3, CPSF6, CSTF3, GOSR2, NAPA, NAPG, NSF, NUDT21, PPP2R1A, RAC1, SYMPK, YKT6 |
| Role of p14/p19ARF in Tumor Suppression | 1.70E−02 | NPM1, PIK3C3, RAC1, SF3A1, UBTF |
| DNA Double-Strand Break Repair by Homologous Recombination | 3.47E−02 | POLA1, RAD51, RPA1 |
| Hypoxia and Angiogenesis | ||
| EIF2 Signaling | 7.94E−66 | ACTB, CCND1, CDK11A, EIF1, EIF1AX, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S1, EIF2S2, EIF2S3, EIF3A, EIF3B, EIF3D, EIF3E, EIF3F, EIF3G, EIF3I, EIF3M, EIF4A1, EIF4A3, EIF4E, EIF4G1, EIF5, FAU, GRB2, HSPA5, MYC, PABPC1, PDPK1, PIK3C3, PPP1CB, RPL10A, RPL11, RPL12, RPL13, RPL13A, RPL14, RPL15, RPL17, RPL18, RPL18A, RPL19, RPL23, RPL23A, RPL24, RPL26, RPL27, RPL27A, RPL28, RPL3, RPL30, RPL31, RPL32, RPL34, RPL35, RPL36, RPL37, RPL37A, RPL38, RPL4, RPL5, RPL6, RPL7, RPL7A, RPL7L1, RPL8, RPLP0, RPLP1, RPLP2, RPS11, RPS12, RPS13, RPS14, RPS15, RPS15A, RPS16, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS25, RPS27A, RPS28, RPS29, RPS3, RPS4X, RPS5, RPS6, RPS7, RPS8, RPS9, RPSA, UBA52, WARS1 |
| Sirtuin Signaling Pathway | 8.13E−05 | GABPA, GTF3C2, MTOR, MYC, NDUFA11, NDUFAB1, NDUFB3, PAM16, POLR1A, POLR1B, POLR1C, POLR1E, POLR2F, RBBP8, RPTOR, RRP9, SDHC, SF3A1, SOD1, SOD2, TIMM10, TIMM13, TIMM23, TIMM44, TIMM9, TOMM22, TOMM40, TUBA1B, TUBA1C, UQCRFS1, XRCC5, XRCC6 |
| VEGF Signaling | 7.24E−04 | ACTB, BCL2L1, EIF1, EIF1AX, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S1, EIF2S2, EIF2S3, GRB2, PIK3C3, PTPN11 |
| Proliferative Signaling | ||
| Regulation of eIF4 and p70S6K Signaling | 1.26E−29 | EIF1, EIF1AX, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF2S1, EIF2S2, EIF2S3, EIF3A, EIF3B, EIF3D, EIF3E, EIF3F, EIF3G, EIF3I, EIF3M, EIF4A1, EIF4A3, EIF4E, EIF4G1, FAU, GRB2, MTOR, PABPC1, PDPK1, PIK3C3, PPP2R1A, RPS11, RPS12, RPS13, RPS14, RPS15, RPS15A, RPS16, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS25, RPS27A, RPS28, RPS29, RPS3, RPS4X, RPS5, RPS6, RPS7, RPS8, RPS9, RPSA |
| mTOR Signaling | 3.98E−18 | CDC42, EIF3A, EIF3B, EIF3D, EIF3E, EIF3F, EIF3G, EIF3I, EIF3M, EIF4A1, EIF4A3, EIF4E, EIF4G1, FAU, GNB1L, MTOR, PDPK1, PIK3C3, PPP2R1A, RAC1, RHOQ, RPS11, RPS12, RPS13, RPS14, RPS15, RPS15A, RPS16, RPS18, RPS19, RPS2, RPS20, RPS21, RPS23, RPS24, RPS25, RPS27A, RPS28, RPS29, RPS3, RPS4X, RPS5, RPS6, RPS7, RPS8, RPS9, RPSA, RPTOR |
| Hereditary Breast Cancer Signaling | 7.94E−12 | ACTB, ATR, CCNB1, CCND1, CDK1, CHEK1, HDAC3, NPM1, PIK3C3, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, RAD51, RFC2, RFC3, RFC4, RFC5, RPA1, RPS27A, SMARCB1, SMARCE1, TUBG1, UBA52, WEE1 |
| Iron homeostasis signaling pathway | 2.24E−07 | ACO2, ATP6AP1, ATP6V0B, ATP6V0C, ATP6V0D1, ATP6V1A, ATP6V1B2, ATP6V1C1, ATP6V1D, ATP6V1E1, ATP6V1F, ATP6V1G1, ATP6V1H, CIAO1, HSCB, HSPA9, ISCU, LYRM4, MMS19, NFS1, NUBP1, NUBP2, PCBP1, SKP1 |
| Androgen Signaling | 7.59E−07 | CCND1, CCNH, CDK7, ERCC2, ERCC3, GNB1L, GTF2A1, GTF2B, GTF2E1, GTF2E2, GTF2F1, GTF2H1, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, TAF2 |
| Glucocorticoid Receptor Signaling | 1.58E−06 | ACTB, BCL2L1, CCNH, CDK7, ERCC2, ERCC3, GRB2, GTF2A1, GTF2A2, GTF2B, GTF2E1, GTF2E2, GTF2F1, GTF2F2, GTF2H1, HSPA5, HSPA9, MED14, PIK3C3, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, RAC1, SMARCB1, SMARCE1, TAF1, TAF10, TAF12, TAF2, TAF6, TAF7, TSG101, UBE2I |
| RAN Signaling | 1.62E−04 | CSE1L, KPNB1, RAN, RANGAP1, RCC1, XPO1 |
| Insulin Receptor Signaling | 4.47E−04 | CRKL, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF4E, GRB2, MTOR, PDPK1, PIK3C3, PPP1CB, PPP1R10, PPP1R11, PPP1R12A, PPP1R7, PTPN11, RHOQ, RPTOR |
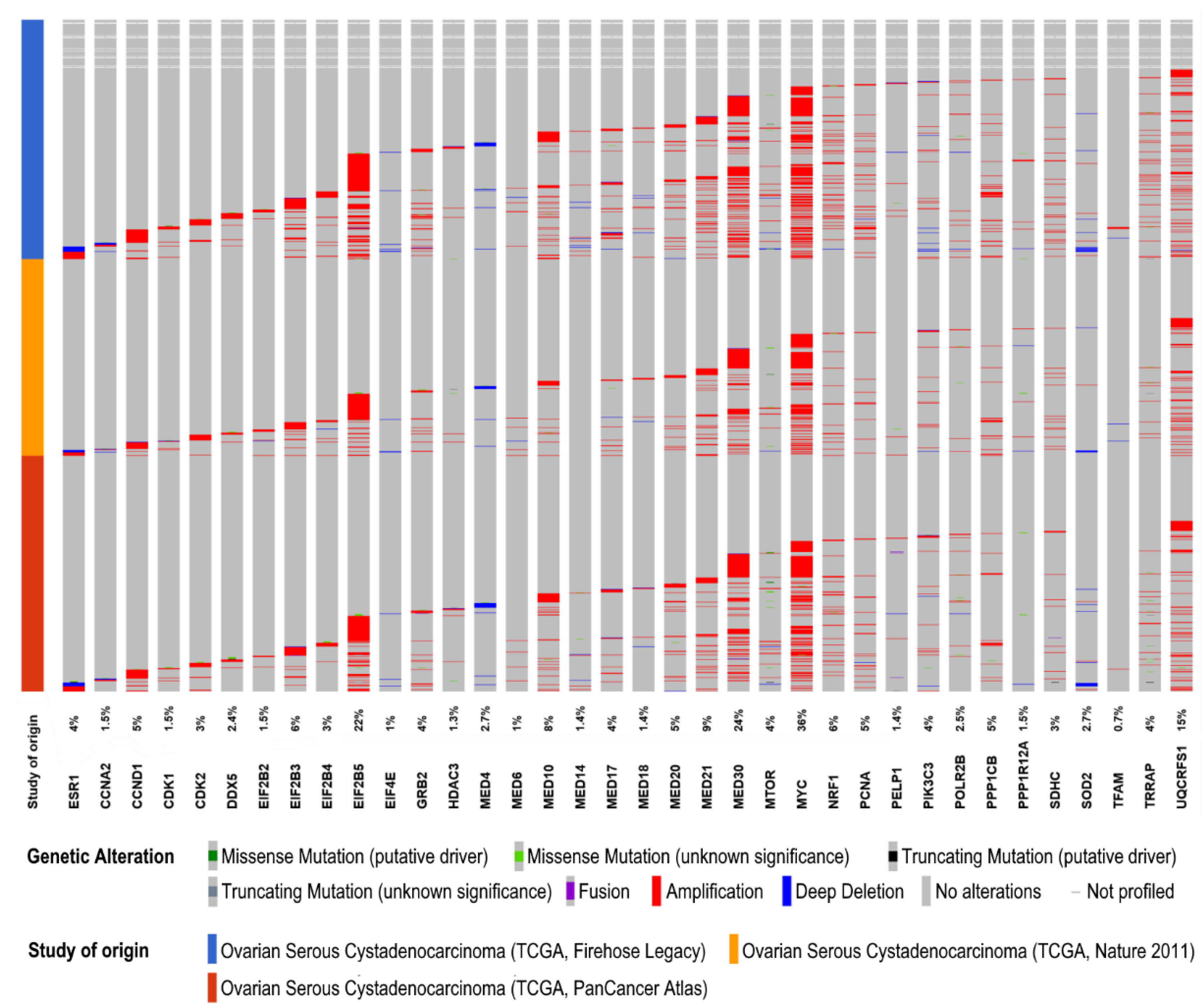
| Estrogen Receptor Signaling | 6.92E−04 | CCND1, DDX5, EIF2B2, EIF2B3, EIF2B4, EIF2B5, EIF4E, GRB2, HDAC3, MED10, MED14, MED17, MED18, MED20, MED21, MED30, MED4, MED6, MTOR, MYC, NRF1, PCNA, PELP1, PIK3C3, POLR2B, PPP1CB, PPP1R12A, SDHC, SOD2, TFAM, TRRAP, UQCRFS1 |
| Translation and post-translational modifications | ||
| Protein Ubiquitination Pathway | 2.51E−24 | ANAPC1, ANAPC10, ANAPC11, ANAPC2, ANAPC4, ANAPC5, BAP1, CDC20, CDC23, DNAJC17, DNAJC8, DNAJC9, HSCB, HSPA5, HSPA9, HSPD1, HSPE1, MED20, PSMA1, PSMA2, PSMA3, PSMA4, PSMA5, PSMA6, PSMA7, PSMB1, PSMB2, PSMB3, PSMB4, PSMB5, PSMB6, PSMB7, PSMC1, PSMC2, PSMC3, PSMC4, PSMC6, PSMD1, PSMD11, PSMD12, PSMD13, PSMD14, PSMD2, PSMD3, PSMD4, PSMD6, PSMD7, RBX1, RPS27A, SKP1, UBA1, UBA52, UBE2D3, UBE2I, UBE2L3, UBE2M, UBE2N, USP10, USP36, USP37, USP39, USP5, USP7, USP8 |
| Assembly of RNA Polymerase II Complex | 3.98E−23 | CCNH, CDK7, DR1, ERCC2, ERCC3, GTF2A1, GTF2A2, GTF2B, GTF2E1, GTF2E2, GTF2F1, GTF2H1, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, TAF1, TAF10, TAF12, TAF2, TAF6, TAF7 |
| tRNA Charging | 3.16E−17 | AARS1, CARS1, DARS1, EPRS1, FARSA, FARSB, GARS1, HARS1, IARS1, IARS2, KARS1, LARS1, MARS1, MARS2, NARS1, RARS1, SARS1, TARS1, VARS, WARS1, YARS1 |
| Cleavage and Polyadenylation of Pre-mRNA | 2.51E−08 | CPSF1, CPSF2, CPSF3, CPSF6, CSTF3, NUDT21, PABPN1, WDR33 |
| Assembly of RNA Polymerase I Complex | 2.51E−08 | POLR1A, POLR1B, POLR1C, POLR1E, POLR2F, TAF1B, TAF1C, UBTF |
| Assembly of RNA Polymerase III Complex | 6.17E−08 | BRF1, BRF2, GTF3A, GTF3C1, GTF3C2, GTF3C4, GTF3C5, SF3A1 |
| Sumoylation Pathway | 2.82E−05 | CDC42, PCNA, RAC1, RAN, RANGAP1, RCC1, RFC2, RFC3, RFC4, RFC5, RHOQ, RNF4, RPA1, SAE1, SENP6, UBA2, UBE2I |
| Spliceosomal Cycle | 2.82E−03 | U2AF1/U2AF1L5, U2AF2 |
| Others | ||
| Systemic Lupus Erythematosus Signaling | 1.58E−12 | EFTUD2, GRB2, HNRNPC, LSM11, LSM2, LSM3, LSM4, LSM5, LSM6, LSM7, MTOR, PIK3C3, PRPF18, PRPF19, PRPF3, PRPF31, PRPF38A, PRPF38B, PRPF4, PRPF40A, PRPF4B, PRPF6, PRPF8, RNPC3, SART1, SF3B4, SNRNP200, SNRNP25, SNRNP27, SNRNP35, SNRNP40, SNRNP70, SNRPA1, SNRPB, SNRPD1, SNRPD2, SNRPD3, SNRPE, SNRPF, SNRPG, TXNL4A, ZMAT5 |
| Phagosome Maturation | 1.41E−09 | ATP6AP1, ATP6V0B, ATP6V0C, ATP6V0D1, ATP6V1A, ATP6V1B2, ATP6V1C1, ATP6V1D, ATP6V1E1, ATP6V1F, ATP6V1G1, ATP6V1H, DYNC1H1, DYNC1I2, DYNLRB1, GOSR2, NAPA, NAPG, NSF, PIK3C3, TSG101, TUBA1B, TUBA1C, TUBB, TUBG1, VPS18, VPS28, VPS37A, YKT6 |
| Huntington’s Disease Signaling | 3.39E−06 | BCL2L1, CLTC, DNM1L, DNM2, DYNC1I2, GNB1L, GOSR2, GRB2, HDAC3, HSPA5, HSPA9, MTOR, NAPA, NAPG, NSF, PDPK1, PIK3C3, POLR2B, POLR2C, POLR2D, POLR2E, POLR2F, POLR2G, POLR2H, POLR2I, POLR2K, POLR2L, RPS27A, SIN3A, UBA52, YKT6 |
| Mechanisms of Viral Exit from Host Cells | 4.27E−05 | ACTB, CHMP2A, CHMP3, CHMP4B, CHMP6, SNF8, TSG101, VPS25, VPS28, XPO1 |
| Remodeling of Epithelial Adherens Junctions | 5.13E−05 | ACTB, ACTR2, ACTR3, ARPC2, ARPC3, ARPC4, DNM1L, DNM2, HGS, TUBA1B, TUBA1C, TUBB, TUBG1 |
| Superpathway of Geranylgeranyldiphosphate Biosynthesis I (via Mevalonate) | 2.00E−03 | FNTB, GGPS1, HMGCR, HMGCS1, MVK |
| Regulation of Actin-based Motility by Rho | 4.07E−03 | ACTB, ACTR2, ACTR3, ARPC2, ARPC3, ARPC4, CDC42, PFN1, PPP1CB, PPP1R12A, RAC1, RHOQ |
| Actin Nucleation by ARP-WASP Complex | 4.57E−03 | ACTR2, ACTR3, ARPC2, ARPC3, ARPC4, CDC42, GRB2, PPP1R12A, RAC1, RHOQ |
| Caveolar-mediated Endocytosis Signaling | 5.01E−03 | ACTB, ARCN1, COPA, COPB1, COPB2, COPE, COPG1, COPZ1, DNM2, ITGAV |
| Pyrimidine Deoxyribonucleotides De Novo Biosynthesis I | 5.13E−03 | CMPK1, DTYMK, DUT, RRM1, RRM2 |
| Inhibition of ARE-Mediated mRNA Degradation Pathway | 5.25E−03 | CNOT1, CNOT3, DDX6, EXOSC2, EXOSC3, EXOSC4, EXOSC5, EXOSC6, EXOSC7, EXOSC8, EXOSC9, PABPN1, PPP2R1A, XRN1 |
| Telomere Extension by Telomerase | 6.76E−03 | TERF1, TINF2, XRCC5, XRCC6 |
| Clathrin-mediated Endocytosis Signaling | 7.08E−03 | ACTB, ACTR2, ACTR3, ARPC2, ARPC3, ARPC4, CDC42, CLTC, CSNK2B, DNM1L, DNM2, GAK, GRB2, HGS, PIK3C3, RAC1, RPS27A, TSG101, UBA52 |
| Oxidized GTP and dGTP Detoxification | 8.13E−03 | DDX6, RUVBL2 |
| Geranylgeranyldiphosphate Biosynthesis | 8.13E−03 | FNTB, GGPS1 |
| Tight Junction Signaling | 1.70E−02 | ACTB, CDC42, CPSF1, CPSF2, CPSF3, CPSF6, CSTF3, GOSR2, NAPA, NAPG, NSF, NUDT21, PPP2R1A, RAC1, SYMPK, YKT6 |
| Mevalonate Pathway I | 3.47E−02 | HMGCR, HMGCS1, MVK |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alexandrova, E.; Pecoraro, G.; Sellitto, A.; Melone, V.; Ferravante, C.; Rocco, T.; Guacci, A.; Giurato, G.; Nassa, G.; Rizzo, F.; et al. An Overview of Candidate Therapeutic Target Genes in Ovarian Cancer. Cancers 2020, 12, 1470. https://doi.org/10.3390/cancers12061470
Alexandrova E, Pecoraro G, Sellitto A, Melone V, Ferravante C, Rocco T, Guacci A, Giurato G, Nassa G, Rizzo F, et al. An Overview of Candidate Therapeutic Target Genes in Ovarian Cancer. Cancers. 2020; 12(6):1470. https://doi.org/10.3390/cancers12061470
Chicago/Turabian StyleAlexandrova, Elena, Giovanni Pecoraro, Assunta Sellitto, Viola Melone, Carlo Ferravante, Teresa Rocco, Anna Guacci, Giorgio Giurato, Giovanni Nassa, Francesca Rizzo, and et al. 2020. "An Overview of Candidate Therapeutic Target Genes in Ovarian Cancer" Cancers 12, no. 6: 1470. https://doi.org/10.3390/cancers12061470
APA StyleAlexandrova, E., Pecoraro, G., Sellitto, A., Melone, V., Ferravante, C., Rocco, T., Guacci, A., Giurato, G., Nassa, G., Rizzo, F., Weisz, A., & Tarallo, R. (2020). An Overview of Candidate Therapeutic Target Genes in Ovarian Cancer. Cancers, 12(6), 1470. https://doi.org/10.3390/cancers12061470