Pharmacogenomic Analysis Reveals CCNA2 as a Predictive Biomarker of Sensitivity to Polo-Like Kinase I Inhibitor in Gastric Cancer

 , , and
, , and
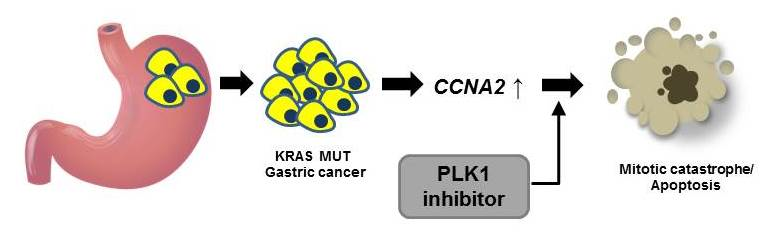
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Pharmacogenomic Analysis Highlights Novel Drug–Biomarker Relationships Among Gastric Cancer Cells
2.2. CCNA2 Upregulation is Causally Linked to BI-2536 Induced Cytotoxicity in Gastric Cancer Cells
2.3. CCNA2 is Required for BI-2536-Induced Mitotic Catastrophe and Apoptosis
2.4. KRAS Driven Upregulation of CCNA2 Confers Sensitivity to PLK1 Inhibitors Among KRAS Mutant Cancers
3. Discussion
4. Materials and Methods
4.1. Pharmacological Characterization
4.2. RNA Sequencing
4.3. Pharmacogenomic Analysis
4.4. siRNA Transfection
4.5. cDNA Transfection
4.6. Immunoblot Analysis
4.7. The Cancer Genome Atlas Analysis
4.8. Flow CytometricAnalysis
4.9. Immunofluorescence Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Apicella, M.; Corso, S.; Giordano, S. Targeted therapies for gastric cancer: Failures and hopes from clinical trials. Oncotarget 2017, 8, 57654–57669. [Google Scholar] [CrossRef] [PubMed]
- Glover, D.M.; Hagan, I.M.; Tavares, A.A. Polo-like kinases: A team that plays throughout mitosis. Genes Dev. 1998, 12, 3777–3787. [Google Scholar] [CrossRef] [PubMed]
- Eckerdt, F.; Yuan, J.; Strebhardt, K. Polo-like kinases and oncogenesis. Oncogene 2005, 24, 267–276. [Google Scholar] [CrossRef]
- Dietzmann, K.; Kirches, E.; von, B.; Jachau, K.; Mawrin, C. Increased human polo-like kinase-1 expression in gliomas. J. Neurooncol. 2001, 53, 1–11. [Google Scholar] [CrossRef]
- Gutteridge, R.E.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef]
- Lenart, P.; Petronczki, M.; Steegmaier, M.; Di Fiore, B.; Lipp, J.J.; Hoffmann, M.; Rettig, W.J.; Kraut, N.; Peters, J.M. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of polo-like kinase 1. Curr. Biol. 2007, 17, 304–315. [Google Scholar] [CrossRef]
- Steegmaier, M.; Hoffmann, M.; Baum, A.; Lenart, P.; Petronczki, M.; Krssak, M.; Gurtler, U.; Garin-Chesa, P.; Lieb, S.; Quant, J.; et al. BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr. Biol. 2007, 17, 316–322. [Google Scholar] [CrossRef]
- Frost, A.; Mross, K.; Steinbild, S.; Hedbom, S.; Unger, C.; Kaiser, R.; Trommeshauser, D.; Munzert, G. Phase i study of the Plk1 inhibitor BI 2536 administered intravenously on three consecutive days in advanced solid tumours. Curr. Oncol. 2012, 19, e28–e35. [Google Scholar] [CrossRef]
- Sebastian, M.; Reck, M.; Waller, C.F.; Kortsik, C.; Frickhofen, N.; Schuler, M.; Fritsch, H.; Gaschler-Markefski, B.; Hanft, G.; Munzert, G.; et al. The efficacy and safety of BI 2536, a novel Plk-1 inhibitor, in patients with stage IIIB/IV non-small cell lung cancer who had relapsed after, or failed, chemotherapy: Results from an open-label, randomized phase II clinical trial. J. Thorac. Oncol. 2010, 5, 1060–1067. [Google Scholar] [CrossRef]
- Mross, K.; Dittrich, C.; Aulitzky, W.E.; Strumberg, D.; Schutte, J.; Schmid, R.M.; Hollerbach, S.; Merger, M.; Munzert, G.; Fleischer, F.; et al. A randomised phase II trial of the Polo-like kinase inhibitor BI 2536 in chemo-naive patients with unresectable exocrine adenocarcinoma of the pancreas—A study within the Central European Society Anticancer Drug Research (CESAR) collaborative network. Br. J. Cancer 2012, 107, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, H.; Lee, J.E.; Shin, S.J.; Oh, S.; Kwon, G.; Kim, H.; Choi, Y.Y.; White, M.A.; Paik, S.; et al. Selective Cytotoxicity of the NAMPT Inhibitor FK866 Toward Gastric Cancer Cells With Markers of the Epithelial-Mesenchymal Transition, Due to Loss of NAPRT. Gastroenterology 2018, 155, 799–814 e713. [Google Scholar] [CrossRef]
- Pagano, M.; Pepperkok, R.; Verde, F.; Ansorge, W.; Draetta, G. Cyclin A is required at two points in the human cell cycle. EMBO J. 1992, 11, 961–971. [Google Scholar] [CrossRef]
- Seong, Y.S.; Kamijo, K.; Lee, J.S.; Fernandez, E.; Kuriyama, R.; Miki, T.; Lee, K.S. A spindle checkpoint arrest and a cytokinesis failure by the dominant-negative polo-box domain of Plk1 in U-2 OS cells. J. Biol. Chem. 2002, 277, 32282–32293. [Google Scholar] [CrossRef]
- Arnaud, L.; Pines, J.; Nigg, E.A. GFP tagging reveals human Polo-like kinase 1 at the kinetochore/centromere region of mitotic chromosomes. Chromosoma 1998, 107, 424–429. [Google Scholar] [CrossRef]
- Lee, K.S.; Yuan, Y.L.; Kuriyama, R.; Erikson, R.L. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol. Cell Biol. 1995, 15, 7143–7151. [Google Scholar] [CrossRef]
- Golsteyn, R.M.; Mundt, K.E.; Fry, A.M.; Nigg, E.A. Cell cycle regulation of the activity and subcellular localization of Plk1, a human protein kinase implicated in mitotic spindle function. J. Cell Biol. 1995, 129, 1617–1628. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Liu, C.J.; Huang, Y.C.; Wu, S.H.; Fang, H.W.; Chen, Y.J. BI2536 induces mitotic catastrophe and radiosensitization in human oral cancer cells. Oncotarget 2018, 9, 21231–21243. [Google Scholar] [CrossRef]
- Choi, M.; Kim, W.; Cheon, M.G.; Lee, C.W.; Kim, J.E. Polo-like kinase 1 inhibitor BI2536 causes mitotic catastrophe following activation of the spindle assembly checkpoint in non-small cell lung cancer cells. Cancer Lett. 2015, 357, 591–601. [Google Scholar] [CrossRef] [PubMed]
- Gheghiani, L.; Loew, D.; Lombard, B.; Mansfeld, J.; Gavet, O. PLK1 Activation in Late G2 Sets Up Commitment to Mitosis. Cell Rep. 2017, 19, 2060–2073. [Google Scholar] [CrossRef] [PubMed]
- Suryadinata, R.; Sadowski, M.; Sarcevic, B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010, 30, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Sonke, G.S.; Hart, L.L.; Campone, M.; Erdkamp, F.; Janni, W.; Verma, S.; Villanueva, C.; Jakobsen, E.; Alba, E.; Wist, E.; et al. Ribociclib with letrozole vs letrozole alone in elderly patients with hormone receptor-positive, HER2-negative breast cancer in the randomized MONALEESA-2 trial. Breast Cancer Res. Treat. 2018, 167, 659–669. [Google Scholar] [CrossRef]
- Kwapisz, D. Cyclin-dependent kinase 4/6 inhibitors in breast cancer: Palbociclib, ribociclib, and abemaciclib. Breast Cancer Res. Treat. 2017, 166, 41–54. [Google Scholar] [CrossRef]
- Kim, E.S.; Scott, L.J. Palbociclib: A Review in HR-Positive, HER2-Negative, Advanced or Metastatic Breast Cancer. Target Oncol. 2017, 12, 373–383. [Google Scholar] [CrossRef]
- Tripathy, D.; Bardia, A.; Sellers, W.R. Ribociclib (LEE011): Mechanism of Action and Clinical Impact of This Selective Cyclin-Dependent Kinase 4/6 Inhibitor in Various Solid Tumors. Clin. Cancer Res. 2017, 23, 3251–3262. [Google Scholar] [CrossRef]
- de Gramont, A.; Herrera, A.; de Gramont, A. Ribociclib for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2017, 376, 288–289. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget 2017, 8, 624–643. [Google Scholar] [CrossRef]
- Wang, X.; Simon, R. Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med.Genom. 2013, 6, 30. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hu, K.; Guo, J.; Cheng, F.; Lv, J.; Jiang, W.; Lu, W.; Liu, J.; Pang, X.; Liu, M. Suppression of KRas-mutant cancer through the combined inhibition of KRAS with PLK1 and ROCK. Nat. Commun. 2016, 7, 11363. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Ferrell, J.E., Jr. The roles of cyclin A2, B1, and B2 in early and late mitotic events. Mol. Biol. Cell 2010, 21, 3149–3161. [Google Scholar] [CrossRef]
- Jia, L.; Li, B.; Yu, H. The Bub1-Plk1 kinase complex promotes spindle checkpoint signalling through Cdc20 phosphorylation. Nat. Commun. 2016, 7, 10818. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic lethality in KRas-driven cancer cells created by glutamine deprivation. Oncoscience 2015, 2, 807–808. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Moten, A.; Lin, H.K. Akt: A new activation mechanism. Cell Res. 2014, 24, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Begley, M.; Michowski, W.; Inuzuka, H.; Ginzberg, M.; Gao, D.; Tsou, P.; Gan, W.; Papa, A.; Kim, B.M.; et al. Cell-cycle-regulated activation of Akt kinase by phosphorylation at its carboxyl terminus. Nature 2014, 508, 541–545. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Mendiratta, S.; Kim, J.; Pecot, C.V.; Larsen, J.E.; Zubovych, I.; Seo, B.Y.; Kim, J.; Eskiocak, B.; Chung, H.; et al. Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell 2013, 155, 552–566. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.; Lee, C.E.; Oh, S.; Kim, H.; Lee, J.; Kim, S.B.; Kim, H.S. Pharmacogenomic Analysis Reveals CCNA2 as a Predictive Biomarker of Sensitivity to Polo-Like Kinase I Inhibitor in Gastric Cancer. Cancers 2020, 12, 1418. https://doi.org/10.3390/cancers12061418
Lee Y, Lee CE, Oh S, Kim H, Lee J, Kim SB, Kim HS. Pharmacogenomic Analysis Reveals CCNA2 as a Predictive Biomarker of Sensitivity to Polo-Like Kinase I Inhibitor in Gastric Cancer. Cancers. 2020; 12(6):1418. https://doi.org/10.3390/cancers12061418
Chicago/Turabian StyleLee, Yunji, Chae Eun Lee, Sejin Oh, Hakhyun Kim, Jooyoung Lee, Sang Bum Kim, and Hyun Seok Kim. 2020. "Pharmacogenomic Analysis Reveals CCNA2 as a Predictive Biomarker of Sensitivity to Polo-Like Kinase I Inhibitor in Gastric Cancer" Cancers 12, no. 6: 1418. https://doi.org/10.3390/cancers12061418
APA StyleLee, Y., Lee, C. E., Oh, S., Kim, H., Lee, J., Kim, S. B., & Kim, H. S. (2020). Pharmacogenomic Analysis Reveals CCNA2 as a Predictive Biomarker of Sensitivity to Polo-Like Kinase I Inhibitor in Gastric Cancer. Cancers, 12(6), 1418. https://doi.org/10.3390/cancers12061418