The Pivotal Role of the Dysregulation of Cholesterol Homeostasis in Cancer: Implications for Therapeutic Targets


Abstract
1. Introduction
2. Physiological and Functional Roles of Cholesterol Homeostasis
3. The Relationship between Cholesterol and Cancer Incidence
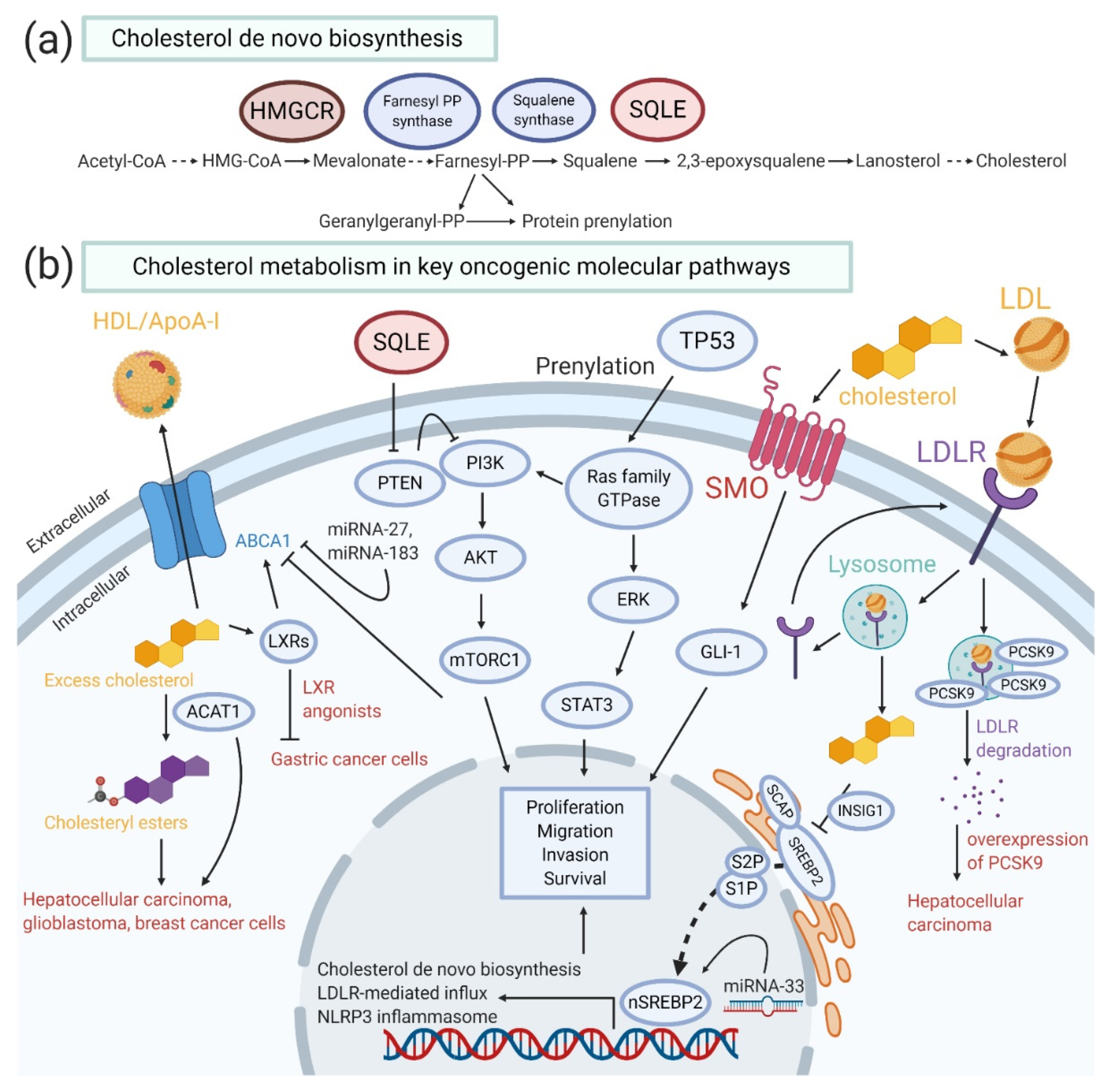
4. Critical Oncogenic Pathways in Cholesterol Homeostasis
5. The Role of Cholesterol Metabolism in the Regulation of Cancer Stem Cells
6. The Role of Cholesterol Metabolism in Immune Cells
7. Effect of Anti-Cancer Drugs and Natural Compounds on Cholesterol Homeostasis Pathway in Cancer Cells
8. Molecular Targeted Drugs Targeting Cholesterol Homeostasis Pathway for Cancer Prevention and Treatment
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- DuBroff, R.; de Lorgeril, M. Cholesterol confusion and statin controversy. World J. Cardiol. 2015, 7, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Zhang, W.; Li, S.; Yang, H. The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 2019, 9, 219–227. [Google Scholar] [PubMed]
- Pelton, K.; Freeman, M.R.; Solomon, K.R. Cholesterol and prostate cancer. Curr Opin. Pharmacol. 2012, 12, 751–759. [Google Scholar] [CrossRef] [PubMed]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef]
- Heir, T.; Falk, R.S.; Robsahm, T.E.; Sandvik, L.; Erikssen, J.; Tretli, S. Cholesterol and prostate cancer risk: A long-term prospective cohort study. BMC Cancer 2016, 16, 643. [Google Scholar] [CrossRef]
- Asano, K.; Kubo, M.; Yonemoto, K.; Doi, Y.; Ninomiya, T.; Tanizaki, Y.; Arima, H.; Shirota, T.; Matsumoto, T.; Iida, M.; et al. Impact of serum total cholesterol on the incidence of gastric cancer in a population-based prospective study: The Hisayama study. Int. J. Cancer 2008, 122, 909–914. [Google Scholar] [CrossRef]
- Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat. Rev. Mol. Cell Biol. 2008, 9, 125–138. [Google Scholar] [CrossRef]
- Berndt, N.; Hamilton, A.D.; Sebti, S.M. Targeting protein prenylation for cancer therapy. Nat. Rev. Cancer 2011, 11, 775–791. [Google Scholar] [CrossRef]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef]
- Sato, R. Sterol metabolism and SREBP activation. Arch. Biochem. Biophys. 2010, 501, 177–181. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. The SREBP pathway: Regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 1997, 89, 331–340. [Google Scholar] [CrossRef]
- Shimano, H. Sterol regulatory element-binding proteins (SREBPs): Transcriptional regulators of lipid synthetic genes. Prog. Lipid Res. 2001, 40, 439–452. [Google Scholar] [CrossRef]
- Brown, A.J.; Sun, L.; Feramisco, J.D.; Brown, M.S.; Goldstein, J.L. Cholesterol addition to ER membranes alters conformation of SCAP, the SREBP escort protein that regulates cholesterol metabolism. Mol. Cell 2002, 10, 237–245. [Google Scholar] [CrossRef]
- Sun, L.P.; Li, L.; Goldstein, J.L.; Brown, M.S. Insig required for sterol-mediated inhibition of Scap/SREBP binding to COPII proteins in vitro. J. Biol. Chem. 2005, 280, 26483–26490. [Google Scholar] [CrossRef]
- Espenshade, P.J.; Li, W.P.; Yabe, D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proc. Natl. Acad. Sci. USA 2002, 99, 11694–11699. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Loh, K.; Tam, S.; Murray-Segal, L.; Huynh, K.; Meikle, P.J.; Scott, J.W.; van Denderen, B.; Chen, Z.; Steel, R.; LeBlond, N.D.; et al. Inhibition of Adenosine Monophosphate-Activated Protein Kinase-3-Hydroxy-3-Methylglutaryl Coenzyme A Reductase Signaling Leads to Hypercholesterolemia and Promotes Hepatic Steatosis and Insulin Resistance. Hepatol. Commun. 2019, 3, 84–98. [Google Scholar] [CrossRef]
- Yoshioka, H.; Coates, H.W.; Chua, N.K.; Hashimoto, Y.; Brown, A.J.; Ohgane, K. A key mammalian cholesterol synthesis enzyme, squalene monooxygenase, is allosterically stabilized by its substrate. Proc. Natl. Acad. Sci. USA 2020, 117, 7150–7158. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Tveten, K.; Strom, T.B.; Berge, K.E.; Leren, T.P. PCSK9-mediated degradation of the LDL receptor generates a 17 kDa C-terminal LDL receptor fragment. J. Lipid Res. 2013, 54, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Betters, J.L.; Yu, L. NPC1L1 and cholesterol transport. FEBS Lett. 2010, 584, 2740–2747. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Betters, J.L.; Yu, L. Niemann-pick C1-like 1 (NPC1L1) protein in intestinal and hepatic cholesterol transport. Annu. Rev. Physiol. 2011, 73, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Gelissen, I.C.; Harris, M.; Rye, K.A.; Quinn, C.; Brown, A.J.; Kockx, M.; Cartland, S.; Packianathan, M.; Kritharides, L.; Jessup, W. ABCA1 and ABCG1 synergize to mediate cholesterol export to apoA-I. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Daniil, G.; Zannis, V.I.; Chroni, A. Effect of apoA-I Mutations in the Capacity of Reconstituted HDL to Promote ABCG1-Mediated Cholesterol Efflux. PLoS ONE 2013, 8, e67993. [Google Scholar] [CrossRef]
- Lorenzi, I.; von Eckardstein, A.; Radosavljevic, S.; Rohrer, L. Lipidation of apolipoprotein A-I by ATP-binding cassette transporter (ABC) A1 generates an interaction partner for ABCG1 but not for scavenger receptor BI. Biochim. Biophys. Acta 2008, 1781, 306–313. [Google Scholar] [CrossRef]
- Ouvrier, A.; Cadet, R.; Vernet, P.; Laillet, B.; Chardigny, J.M.; Lobaccaro, J.M.; Drevet, J.R.; Saez, F. LXR and ABCA1 control cholesterol homeostasis in the proximal mouse epididymis in a cell-specific manner. J. Lipid Res. 2009, 50, 1766–1775. [Google Scholar] [CrossRef]
- Chang, T.Y.; Li, B.L.; Chang, C.C.; Urano, Y. Acyl-coenzyme A:cholesterol acyltransferases. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1–E9. [Google Scholar] [CrossRef]
- Fernandez-Hernando, C.; Moore, K.J. MicroRNA modulation of cholesterol homeostasis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2378–2382. [Google Scholar] [CrossRef]
- Najafi-Shoushtari, S.H.; Kristo, F.; Li, Y.; Shioda, T.; Cohen, D.E.; Gerszten, R.E.; Naar, A.M. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 2010, 328, 1566–1569. [Google Scholar] [CrossRef]
- Vickers, K.C.; Landstreet, S.R.; Levin, M.G.; Shoucri, B.M.; Toth, C.L.; Taylor, R.C.; Palmisano, B.T.; Tabet, F.; Cui, H.L.; Rye, K.A.; et al. MicroRNA-223 coordinates cholesterol homeostasis. Proc. Natl. Acad. Sci. USA 2014, 111, 14518–14523. [Google Scholar] [CrossRef]
- Rotllan, N.; Fernandez-Hernando, C. MicroRNA Regulation of Cholesterol Metabolism. Cholesterol 2012, 2012. [Google Scholar] [CrossRef]
- Khan, A.A.; Agarwal, H.; Reddy, S.S.; Arige, V.; Natarajan, B.; Gupta, V.; Kalyani, A.; Barthwal, M.K.; Mahapatra, N.R. MicroRNA 27a Is a Key Modulator of Cholesterol Biosynthesis. Mol. Cell Biol. 2020, 40. [Google Scholar] [CrossRef]
- Wagschal, A.; Najafi-Shoushtari, S.H.; Wang, L.; Goedeke, L.; Sinha, S.; de Lemos, A.S.; Black, J.C.; Ramirez, C.M.; Li, Y.; Tewhey, R.; et al. Genome-wide identification of microRNAs regulating cholesterol and triglyceride homeostasis. Nat. Med. 2015, 21, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, R.; Knekt, P.; Hakulinen, T.; Rissanen, H.; Heliovaara, M. Dietary fat, cholesterol and colorectal cancer in a prospective study. Br. J. Cancer 2001, 85, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Shafique, K.; McLoone, P.; Qureshi, K.; Leung, H.; Hart, C.; Morrison, D.S. Cholesterol and the risk of grade-specific prostate cancer incidence: Evidence from two large prospective cohort studies with up to 37 years’ follow up. BMC Cancer 2012, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Strohmaier, S.; Edlinger, M.; Manjer, J.; Stocks, T.; Bjorge, T.; Borena, W.; Haggstrom, C.; Engeland, A.; Nagel, G.; Almquist, M.; et al. Total serum cholesterol and cancer incidence in the Metabolic syndrome and Cancer Project (Me-Can). PLoS ONE 2013, 8, e54242. [Google Scholar] [CrossRef]
- Jafri, H.; Alsheikh-Ali, A.A.; Karas, R.H. Baseline and on-treatment high-density lipoprotein cholesterol and the risk of cancer in randomized controlled trials of lipid-altering therapy. J. Am. Coll. Cardiol. 2010, 55, 2846–2854. [Google Scholar] [CrossRef]
- Lim, U.; Gayles, T.; Katki, H.A.; Stolzenberg-Solomon, R.; Weinstein, S.J.; Pietinen, P.; Taylor, P.R.; Virtamo, J.; Albanes, D. Serum high-density lipoprotein cholesterol and risk of non-hodgkin lymphoma. Cancer Res. 2007, 67, 5569–5574. [Google Scholar] [CrossRef]
- Chen, H.; Qin, S.; Wang, M.; Zhang, T.; Zhang, S. Association between cholesterol intake and pancreatic cancer risk: Evidence from a meta-analysis. Sci. Rep. 2015, 5, 8243. [Google Scholar] [CrossRef]
- Kitahara, C.M.; Berrington de Gonzalez, A.; Freedman, N.D.; Huxley, R.; Mok, Y.; Jee, S.H.; Samet, J.M. Total cholesterol and cancer risk in a large prospective study in Korea. J. Clin. Oncol 2011, 29, 1592–1598. [Google Scholar] [CrossRef] [PubMed]
- Mamtani, R.; Lewis, J.D.; Scott, F.I.; Ahmad, T.; Goldberg, D.S.; Datta, J.; Yang, Y.X.; Boursi, B. Disentangling the Association between Statins, Cholesterol, and Colorectal Cancer: A Nested Case-Control Study. PLoS Med. 2016, 13, e1002007. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Chen, J.; Gao, Y.T.; Rashid, A.; Chang, S.C.; Shen, M.C.; Wang, B.S.; Han, T.Q.; Zhang, B.H.; Danforth, K.N.; et al. Serum lipid levels and the risk of biliary tract cancers and biliary stones: A population-based study in China. Int. J. Cancer 2008, 122, 2322–2329. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, X.Q.; Lin, H.C.; Wang, D.S.; Wang, Z.Q.; Shao, Q.; Wang, F.H.; Yan, S.M.; Liang, J.Y.; Zeng, Z.L.; et al. Correlation between immune signature and high-density lipoprotein cholesterol level in stage II/III colorectal cancer. Cancer Med. 2019, 8, 1209–1217. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zheng, H.; Nie, B.; Gong, W.; Cui, X. Statin use and risk of liver cancer: An update meta-analysis. BMJ Open 2014, 4, e005399. [Google Scholar] [CrossRef] [PubMed]
- Bansal, D.; Undela, K.; D’Cruz, S.; Schifano, F. Statin use and risk of prostate cancer: A meta-analysis of observational studies. PLoS ONE 2012, 7, e46691. [Google Scholar] [CrossRef]
- Singh, P.P.; Singh, S. Statins are associated with reduced risk of gastric cancer: A systematic review and meta-analysis. Ann. Oncol. 2013, 24, 1721–1730. [Google Scholar] [CrossRef]
- Khurana, V.; Sheth, A.; Caldito, G.; Barkin, J.S. Statins reduce the risk of pancreatic cancer in humans: A case-control study of half a million veterans. Pancreas 2007, 34, 260–265. [Google Scholar] [CrossRef]
- Rodrigues Dos Santos, C.; Fonseca, I.; Dias, S.; Mendes de Almeida, J.C. Plasma level of LDL-cholesterol at diagnosis is a predictor factor of breast tumor progression. BMC Cancer 2014, 14, 132. [Google Scholar] [CrossRef]
- Ni, H.; Liu, H.; Gao, R. Serum Lipids and Breast Cancer Risk: A Meta-Analysis of Prospective Cohort Studies. PLoS ONE 2015, 10, e0142669. [Google Scholar] [CrossRef]
- Touvier, M.; Fassier, P.; His, M.; Norat, T.; Chan, D.S.; Blacher, J.; Hercberg, S.; Galan, P.; Druesne-Pecollo, N.; Latino-Martel, P. Cholesterol and breast cancer risk: A systematic review and meta-analysis of prospective studies. Br. J. Nutr. 2015, 114, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, P.; Xuan, J.; Zhu, C.; Liu, J.; Shan, L.; Du, Q.; Ren, Y.; Ye, J. Cholesterol Enhances Colorectal Cancer Progression via ROS Elevation and MAPK Signaling Pathway Activation. Cell Physiol. Biochem. 2017, 42, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Kucharska-Newton, A.M.; Rosamond, W.D.; Mink, P.J.; Alberg, A.J.; Shahar, E.; Folsom, A.R. HDL-cholesterol and incidence of breast cancer in the ARIC cohort study. Ann. Epidemiol. 2008, 18, 671–677. [Google Scholar] [CrossRef]
- Al-Haidari, A.A.; Syk, I.; Thorlacius, H. HMG-CoA reductase regulates CCL17-induced colon cancer cell migration via geranylgeranylation and RhoA activation. Biochem. Biophys. Res. Commun. 2014, 446, 68–72. [Google Scholar] [CrossRef]
- Athavale, D.; Chouhan, S.; Pandey, V.; Mayengbam, S.S.; Singh, S.; Bhat, M.K. Hepatocellular carcinoma-associated hypercholesterolemia: Involvement of proprotein-convertase-subtilisin-kexin type-9 (PCSK9). Cancer Metab. 2018, 6, 16. [Google Scholar] [CrossRef]
- Wang, Q.; Feng, F.; Wang, J.; Ren, M.; Shi, Z.; Mao, X.; Zhang, H.; Ju, X. Liver X receptor activation reduces gastric cancer cell proliferation by suppressing Wnt signalling via LXRbeta relocalization. J. Cell Mol. Med. 2019, 23, 789–797. [Google Scholar] [CrossRef]
- Jiang, Y.; Sun, A.; Zhao, Y.; Ying, W.; Sun, H.; Yang, X.; Xing, B.; Sun, W.; Ren, L.; Hu, B.; et al. Proteomics identifies new therapeutic targets of early-stage hepatocellular carcinoma. Nature 2019, 567, 257–261. [Google Scholar] [CrossRef]
- Geng, F.; Cheng, X.; Wu, X.; Yoo, J.Y.; Cheng, C.; Guo, J.Y.; Mo, X.; Ru, P.; Hurwitz, B.; Kim, S.H.; et al. Inhibition of SOAT1 Suppresses Glioblastoma Growth via Blocking SREBP-1-Mediated Lipogenesis. Clin. Cancer Res. 2016, 22, 5337–5348. [Google Scholar] [CrossRef]
- Antalis, C.J.; Arnold, T.; Rasool, T.; Lee, B.; Buhman, K.K.; Siddiqui, R.A. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res. Treat. 2010, 122, 661–670. [Google Scholar] [CrossRef]
- Saraon, P.; Trudel, D.; Kron, K.; Dmitromanolakis, A.; Trachtenberg, J.; Bapat, B.; van der Kwast, T.; Jarvi, K.A.; Diamandis, E.P. Evaluation and prognostic significance of ACAT1 as a marker of prostate cancer progression. Prostate 2014, 74, 372–380. [Google Scholar] [CrossRef]
- Muller, P.A.; Vousden, K.H.; Norman, J.C. p53 and its mutants in tumor cell migration and invasion. J. Cell Biol. 2011, 192, 209–218. [Google Scholar] [CrossRef]
- Moon, S.H.; Huang, C.H.; Houlihan, S.L.; Regunath, K.; Freed-Pastor, W.A.; Morris, J.P.t.; Tschaharganeh, D.F.; Kastenhuber, E.R.; Barsotti, A.M.; Culp-Hill, R.; et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 2019, 176, 564–580.e59. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260. [Google Scholar] [CrossRef]
- Yamauchi, Y.; Furukawa, K.; Hamamura, K.; Furukawa, K. Positive feedback loop between PI3K-Akt-mTORC1 signaling and the lipogenic pathway boosts Akt signaling: Induction of the lipogenic pathway by a melanoma antigen. Cancer Res. 2011, 71, 4989–4997. [Google Scholar] [CrossRef]
- Dong, F.; Mo, Z.; Eid, W.; Courtney, K.C.; Zha, X. Akt inhibition promotes ABCA1-mediated cholesterol efflux to ApoA-I through suppressing mTORC1. PLoS ONE 2014, 9, e113789. [Google Scholar] [CrossRef]
- Castellano, B.M.; Thelen, A.M.; Moldavski, O.; Feltes, M.; van der Welle, R.E.; Mydock-McGrane, L.; Jiang, X.; van Eijkeren, R.J.; Davis, O.B.; Louie, S.M.; et al. Lysosomal cholesterol activates mTORC1 via an SLC38A9-Niemann-Pick C1 signaling complex. Science 2017, 355, 1306–1311. [Google Scholar] [CrossRef]
- Yue, S.; Li, J.; Lee, S.Y.; Lee, H.J.; Shao, T.; Song, B.; Cheng, L.; Masterson, T.A.; Liu, X.; Ratliff, T.L.; et al. Cholesteryl ester accumulation induced by PTEN loss and PI3K/AKT activation underlies human prostate cancer aggressiveness. Cell Metab. 2014, 19, 393–406. [Google Scholar] [CrossRef]
- Liu, D.; Wong, C.C.; Fu, L.; Chen, H.; Zhao, L.; Li, C.; Zhou, Y.; Zhang, Y.; Xu, W.; Yang, Y.; et al. Squalene epoxidase drives NAFLD-induced hepatocellular carcinoma and is a pharmaceutical target. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Huang, P.; Nedelcu, D.; Watanabe, M.; Jao, C.; Kim, Y.; Liu, J.; Salic, A. Cellular Cholesterol Directly Activates Smoothened in Hedgehog Signaling. Cell 2016, 166, 1176–1187.e1114. [Google Scholar] [CrossRef]
- Carpenter, R.L.; Lo, H.W. Hedgehog pathway and GLI1 isoforms in human cancer. Discov. Med. 2012, 13, 105–113. [Google Scholar]
- Gordon, R.E.; Zhang, L.; Peri, S.; Kuo, Y.M.; Du, F.; Egleston, B.L.; Ng, J.M.Y.; Andrews, A.J.; Astsaturov, I.; Curran, T.; et al. Statins Synergize with Hedgehog Pathway Inhibitors for Treatment of Medulloblastoma. Clin. Cancer Res. 2018, 24, 1375–1388. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nat. Rev. Immunol. 2015, 15, 104–116. [Google Scholar] [CrossRef]
- Moossavi, M.; Parsamanesh, N.; Bahrami, A.; Atkin, S.L.; Sahebkar, A. Role of the NLRP3 inflammasome in cancer. Mol. Cancer 2018, 17, 158. [Google Scholar] [CrossRef]
- Bae, J.Y.; Lee, S.W.; Shin, Y.H.; Lee, J.H.; Jahng, J.W.; Park, K. P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8, 48972–48982. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Q.; Zhang, H.; Wang, H.; Chen, W.; Meng, G.; Chen, F. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J. Exp. Clin. Cancer Res. 2017, 36, 81. [Google Scholar] [CrossRef]
- Weichand, B.; Popp, R.; Dziumbla, S.; Mora, J.; Strack, E.; Elwakeel, E.; Frank, A.C.; Scholich, K.; Pierre, S.; Syed, S.N.; et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J. Exp. Med. 2017, 214, 2695–2713. [Google Scholar] [CrossRef]
- Du, Q.; Wang, Q.; Fan, H.; Wang, J.; Liu, X.; Wang, H.; Wang, Y.; Hu, R. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem. Pharmacol. 2016, 105, 42–54. [Google Scholar] [CrossRef]
- He, M.; Zhang, W.; Dong, Y.; Wang, L.; Fang, T.; Tang, W.; Lv, B.; Chen, G.; Yang, B.; Huang, P.; et al. Pro-inflammation NF-kappaB signaling triggers a positive feedback via enhancing cholesterol accumulation in liver cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 15. [Google Scholar] [CrossRef]
- Lin, T.Y.; Wei, T.W.; Li, S.; Wang, S.C.; He, M.; Martin, M.; Zhang, J.; Shentu, T.P.; Xiao, H.; Kang, J.; et al. TIFA as a crucial mediator for NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2016, 113, 15078–15083. [Google Scholar] [CrossRef]
- Xiao, H.; Lu, M.; Lin, T.Y.; Chen, Z.; Chen, G.; Wang, W.C.; Marin, T.; Shentu, T.P.; Wen, L.; Gongol, B.; et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation 2013, 128, 632–642. [Google Scholar] [CrossRef]
- Lee, Y.S.; Dutta, A. MicroRNAs in cancer. Annu. Rev. Pathol. 2009, 4, 199–227. [Google Scholar] [CrossRef]
- Stahlhut Espinosa, C.E.; Slack, F.J. The role of microRNAs in cancer. Yale J. Biol. Med. 2006, 79, 131–140. [Google Scholar]
- Luna, J.M.; Scheel, T.K.; Danino, T.; Shaw, K.S.; Mele, A.; Fak, J.J.; Nishiuchi, E.; Takacs, C.N.; Catanese, M.T.; de Jong, Y.P.; et al. Hepatitis C virus RNA functionally sequesters miR-122. Cell 2015, 160, 1099–1110. [Google Scholar] [CrossRef]
- Schult, P.; Roth, H.; Adams, R.L.; Mas, C.; Imbert, L.; Orlik, C.; Ruggieri, A.; Pyle, A.M.; Lohmann, V. microRNA-122 amplifies hepatitis C virus translation by shaping the structure of the internal ribosomal entry site. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Bi, D.P.; Yin, C.H.; Zhang, X.Y.; Yang, N.N.; Xu, J.Y. MiR-183 functions as an oncogene by targeting ABCA1 in colon cancer. Oncol. Rep. 2016, 35, 2873–2879. [Google Scholar] [CrossRef]
- Su, C.; Huang, D.P.; Liu, J.W.; Liu, W.Y.; Cao, Y.O. miR-27a-3p regulates proliferation and apoptosis of colon cancer cells by potentially targeting BTG1. Oncol. Lett. 2019, 18, 2825–2834. [Google Scholar] [CrossRef]
- Wang, X.; Huang, Z.; Wu, Q.; Prager, B.C.; Mack, S.C.; Yang, K.; Kim, L.J.Y.; Gimple, R.C.; Shi, Y.; Lai, S.; et al. MYC-Regulated Mevalonate Metabolism Maintains Brain Tumor-Initiating Cells. Cancer Res. 2017, 77, 4947–4960. [Google Scholar] [CrossRef]
- Liu, Y.; Lu, L.L.; Wen, D.; Liu, D.L.; Dong, L.L.; Gao, D.M.; Bian, X.Y.; Zhou, J.; Fan, J.; Wu, W.Z. MiR-612 regulates invadopodia of hepatocellular carcinoma by HADHA-mediated lipid reprogramming. J. Hematol. Oncol. 2020, 13, 12. [Google Scholar] [CrossRef]
- Kang, M.; Li, Y.; Zhao, Y.; He, S.; Shi, J. miR-33a inhibits cell proliferation and invasion by targeting CAND1 in lung cancer. Clin. Transl. Oncol. 2018, 20, 457–466. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Y.; Ding, W.; Lin, Y.; Huang, Z.; Luo, Q. MiR-33a suppresses breast cancer cell proliferation and metastasis by targeting ADAM9 and ROS1. Protein Cell 2015, 6, 881–889. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, C.; Hu, L. Cholesterol regulates cell proliferation and apoptosis of colorectal cancer by modulating miR-33a-PIM3 pathway. Biochem. Biophys. Res. Commun. 2019, 511, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer Stem Cells and Their Microenvironment: Biology and Therapeutic Implications. Stem Cells Int. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [CrossRef]
- Bono, B.; Ostano, P.; Peritore, M.; Gregnanin, I.; Belgiovine, C.; Liguori, M.; Allavena, P.; Chiorino, G.; Chiodi, I.; Mondello, C. Cells with stemness features are generated from in vitro transformed human fibroblasts. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Kim, H.Y.; Kim, D.K.; Bae, S.H.; Gwak, H.; Jeon, J.H.; Kim, J.K.; Lee, B.I.; You, H.J.; Shin, D.H.; Kim, Y.H.; et al. Farnesyl diphosphate synthase is important for the maintenance of glioblastoma stemness. Exp. Mol. Med. 2018, 50, 1–12. [Google Scholar] [CrossRef]
- Ginestier, C.; Monville, F.; Wicinski, J.; Cabaud, O.; Cervera, N.; Josselin, E.; Finetti, P.; Guille, A.; Larderet, G.; Viens, P.; et al. Mevalonate metabolism regulates Basal breast cancer stem cells and is a potential therapeutic target. Stem Cells 2012, 30, 1327–1337. [Google Scholar] [CrossRef]
- Sharma, A.; Bandyopadhayaya, S.; Chowdhury, K.; Sharma, T.; Maheshwari, R.; Das, A.; Chakrabarti, G.; Kumar, V.; Mandal, C.C. Metformin exhibited anticancer activity by lowering cellular cholesterol content in breast cancer cells. PLoS ONE 2019, 14, e0209435. [Google Scholar] [CrossRef]
- Saito, T.; Chiba, T.; Yuki, K.; Zen, Y.; Oshima, M.; Koide, S.; Motoyama, T.; Ogasawara, S.; Suzuki, E.; Ooka, Y.; et al. Metformin, a diabetes drug, eliminates tumor-initiating hepatocellular carcinoma cells. PLoS ONE 2013, 8, e70010. [Google Scholar] [CrossRef]
- Wang, B.; Rong, X.; Palladino, E.N.D.; Wang, J.; Fogelman, A.M.; Martin, M.G.; Alrefai, W.A.; Ford, D.A.; Tontonoz, P. Phospholipid Remodeling and Cholesterol Availability Regulate Intestinal Stemness and Tumorigenesis. Cell Stem Cell 2018, 22, 206–220.e204. [Google Scholar] [CrossRef]
- Simons, K.; Ikonen, E. How cells handle cholesterol. Science 2000, 290, 1721–1726. [Google Scholar] [CrossRef]
- Fessler, M.B.; Parks, J.S. Intracellular lipid flux and membrane microdomains as organizing principles in inflammatory cell signaling. J. Immunol. 2011, 187, 1529–1535. [Google Scholar] [CrossRef]
- Fox, C.J.; Hammerman, P.S.; Thompson, C.B. Fuel feeds function: Energy metabolism and the T-cell response. Nat. Rev. Immunol. 2005, 5, 844–852. [Google Scholar] [CrossRef]
- Bensinger, S.J.; Bradley, M.N.; Joseph, S.B.; Zelcer, N.; Janssen, E.M.; Hausner, M.A.; Shih, R.; Parks, J.S.; Edwards, P.A.; Jamieson, B.D.; et al. LXR signaling couples sterol metabolism to proliferation in the acquired immune response. Cell 2008, 134, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, E.D.; Daige, C.L.; Petrowski, M.; Dedman, H.; Pattison, J.; Juliano, J.; Li, A.C.; Schulman, I.G. Non-redundant roles for LXRalpha and LXRbeta in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J. Lipid Res. 2010, 51, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Araki, K.; Turner, A.P.; Shaffer, V.O.; Gangappa, S.; Keller, S.A.; Bachmann, M.F.; Larsen, C.P.; Ahmed, R. mTOR regulates memory CD8 T-cell differentiation. Nature 2009, 460, 108–112. [Google Scholar] [CrossRef]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol regulatory element-binding proteins are essential for the metabolic programming of effector T cells and adaptive immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef]
- Verhaar, A.P.; Wildenberg, M.E.; te Velde, A.A.; Meijer, S.L.; Vos, A.C.; Duijvestein, M.; Peppelenbosch, M.P.; Hommes, D.W.; van den Brink, G.R. Miltefosine suppresses inflammation in a mouse model of inflammatory bowel disease. Inflamm. Bowel Dis. 2013, 19, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Surls, J.; Nazarov-Stoica, C.; Kehl, M.; Olsen, C.; Casares, S.; Brumeanu, T.D. Increased membrane cholesterol in lymphocytes diverts T-cells toward an inflammatory response. PLoS ONE 2012, 7, e38733. [Google Scholar] [CrossRef]
- Chyu, K.Y.; Lio, W.M.; Dimayuga, P.C.; Zhou, J.; Zhao, X.; Yano, J.; Trinidad, P.; Honjo, T.; Cercek, B.; Shah, P.K. Cholesterol lowering modulates T cell function in vivo and in vitro. PLoS ONE 2014, 9, e92095. [Google Scholar] [CrossRef]
- Wang, L.; Chi, P.D.; Chen, H.; Xiang, J.; Xia, Z.J.; Zhang, Y.J. Low level of high-density lipoprotein cholesterol correlates with poor prognosis in extranodal natural killer/T cell lymphoma. Tumour. Biol. 2014, 35, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [PubMed]
- Baek, A.E.; Yu, Y.A.; He, S.; Wardell, S.E.; Chang, C.Y.; Kwon, S.; Pillai, R.V.; McDowell, H.B.; Thompson, J.W.; Dubois, L.G.; et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef]
- Kidani, Y.; Bensinger, S.J. Modulating Cholesterol Homeostasis to Build a Better T Cell. Cell Metab. 2016, 23, 963–964. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Lu, Y.; Su, P.; Huang, C.; Liu, L.; Wang, Q.; Yang, M.; Kalady, M.F.; Qian, J.; et al. Cholesterol Induces CD8(+) T Cell Exhaustion in the Tumor Microenvironment. Cell Metab. 2019, 30, 143–156.e145. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Bi, E.; Huang, C.; Lu, Y.; Xue, G.; Guo, X.; Wang, A.; Yang, M.; Qian, J.; Dong, C.; et al. Cholesterol negatively regulates IL-9-producing CD8(+) T cell differentiation and antitumor activity. J. Exp. Med. 2018, 215, 1555–1569. [Google Scholar] [CrossRef]
- Soncini, M.; Corna, G.; Moresco, M.; Coltella, N.; Restuccia, U.; Maggioni, D.; Raccosta, L.; Lin, C.Y.; Invernizzi, F.; Crocchiolo, R.; et al. 24-Hydroxycholesterol participates in pancreatic neuroendocrine tumor development. Proc. Natl. Acad. Sci. USA 2016, 113, E6219–E6227. [Google Scholar] [CrossRef]
- Wang, S.; Yao, Y.; Rao, C.; Zheng, G.; Chen, W. 25-HC decreases the sensitivity of human gastric cancer cells to 5-fluorouracil and promotes cells invasion via the TLR2/NF-kappaB signaling pathway. Int. J. Oncol. 2019, 54, 966–980. [Google Scholar] [CrossRef]
- Qin, W.H.; Yang, Z.S.; Li, M.; Chen, Y.; Zhao, X.F.; Qin, Y.Y.; Song, J.Q.; Wang, B.B.; Yuan, B.; Cui, X.L.; et al. High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology 2020. [Google Scholar] [CrossRef]
- Villablanca, E.J.; Raccosta, L.; Zhou, D.; Fontana, R.; Maggioni, D.; Negro, A.; Sanvito, F.; Ponzoni, M.; Valentinis, B.; Bregni, M.; et al. Tumor-mediated liver X receptor-alpha activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med. 2010, 16, 98–105. [Google Scholar] [CrossRef]
- Zhang, R.; Tang, L.; Tian, Y.; Ji, X.; Hu, Q.; Zhou, B.; Zhenyu, D.; Heng, X.; Yang, L. Cholesterol-modified DP7 enhances the effect of individualized cancer immunotherapy based on neoantigens. Biomaterials 2020, 241, 119852. [Google Scholar] [CrossRef] [PubMed]
- Yun, U.J.; Lee, J.H.; Shim, J.; Yoon, K.; Goh, S.H.; Yi, E.H.; Ye, S.K.; Lee, J.S.; Lee, H.; Park, J.; et al. Anti-cancer effect of doxorubicin is mediated by downregulation of HMG-Co A reductase via inhibition of EGFR/Src pathway. Lab Investig. 2019, 99, 1157–1172. [Google Scholar] [CrossRef] [PubMed]
- Segala, G.; de Medina, P.; Iuliano, L.; Zerbinati, C.; Paillasse, M.R.; Noguer, E.; Dalenc, F.; Payre, B.; Jordan, V.C.; Record, M.; et al. 5,6-Epoxy-cholesterols contribute to the anticancer pharmacology of tamoxifen in breast cancer cells. Biochem. Pharmacol. 2013, 86, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Tonini, C.; Colardo, M.; Colella, B.; Di Bartolomeo, S.; Berardinelli, F.; Caretti, G.; Pallottini, V.; Segatto, M. Inhibition of Bromodomain and Extraterminal Domain (BET) Proteins by JQ1 Unravels a Novel Epigenetic Modulation to Control Lipid Homeostasis. Int. J. Mol. Sci. 2020, 21, 1297. [Google Scholar] [CrossRef]
- Parker, R.A.; Pearce, B.C.; Clark, R.W.; Gordon, D.A.; Wright, J.J. Tocotrienols regulate cholesterol production in mammalian cells by post-transcriptional suppression of 3-hydroxy-3-methylglutaryl-coenzyme A reductase. J. Biol. Chem. 1993, 268, 11230–11238. [Google Scholar]
- Jones, S.; Fernandes, N.V.; Yeganehjoo, H.; Katuru, R.; Qu, H.; Yu, Z.; Mo, H. beta-ionone induces cell cycle arrest and apoptosis in human prostate tumor cells. Nutr. Cancer 2013, 65, 600–610. [Google Scholar] [CrossRef]
- Fernandes, N.V.; Yeganehjoo, H.; Katuru, R.; DeBose-Boyd, R.A.; Morris, L.L.; Michon, R.; Yu, Z.L.; Mo, H. Geranylgeraniol suppresses the viability of human DU145 prostate carcinoma cells and the level of HMG CoA reductase. Exp. Biol. Med. (Maywood) 2013, 238, 1265–1274. [Google Scholar] [CrossRef]
- Houten, S.M.; Schneiders, M.S.; Wanders, R.J.; Waterham, H.R. Regulation of isoprenoid/cholesterol biosynthesis in cells from mevalonate kinase-deficient patients. J. Biol. Chem. 2003, 278, 5736–5743. [Google Scholar] [CrossRef]
- Kim, G.H.; Kan, S.Y.; Kang, H.; Lee, S.; Ko, H.M.; Kim, J.H.; Lim, J.H. Ursolic Acid Suppresses Cholesterol Biosynthesis and Exerts Anti-Cancer Effects in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2019, 20, 4767. [Google Scholar] [CrossRef]
- Bursill, C.A.; Roach, P.D. Modulation of cholesterol metabolism by the green tea polyphenol (-)-epigallocatechin gallate in cultured human liver (HepG2) cells. J. Agric. Food Chem. 2006, 54, 1621–1626. [Google Scholar] [CrossRef]
- Singh, D.K.; Banerjee, S.; Porter, T.D. Green and black tea extracts inhibit HMG-CoA reductase and activate AMP kinase to decrease cholesterol synthesis in hepatoma cells. J. Nutr. Biochem. 2009, 20, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Momose, Y.; Maeda-Yamamoto, M.; Nabetani, H. Systematic review of green tea epigallocatechin gallate in reducing low-density lipoprotein cholesterol levels of humans. Int. J. Food Sci. Nutr. 2016, 67, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Porter, T.D. Inhibition of sterol 4alpha-methyl oxidase is the principal mechanism by which garlic decreases cholesterol synthesis. J. Nutr. 2006, 136, 759S–764S. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.P.; Kim, H.G.; Choi, J.H.; Do, M.T.; Chung, Y.C.; Jeong, T.C.; Jeong, H.G. S-allyl cysteine attenuates free fatty acid-induced lipogenesis in human HepG2 cells through activation of the AMP-activated protein kinase-dependent pathway. J. Nutr. Biochem. 2013, 24, 1469–1478. [Google Scholar] [CrossRef]
- Einbond, L.S.; Manservisi, F.; Wu, H.A.; Balick, M.; Antonetti, V.; Vornoli, A.; Menghetti, I.; Belpoggi, F.; Redenti, S.; Roter, A. A transcriptomic analysis of turmeric: Curcumin represses the expression of cholesterol biosynthetic genes and synergizes with simvastatin. Pharmacol. Res. 2018, 132, 176–187. [Google Scholar] [CrossRef]
- Feng, D.; Ohlsson, L.; Duan, R.D. Curcumin inhibits cholesterol uptake in Caco-2 cells by down-regulation of NPC1L1 expression. Lipids Health Dis. 2010, 9, 40. [Google Scholar] [CrossRef]
- Endo, A. A historical perspective on the discovery of statins. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 484–493. [Google Scholar] [CrossRef]
- Bytyci, I.; Bajraktari, G.; Bhatt, D.L.; Morgan, C.J.; Ahmed, A.; Aronow, W.S.; Banach, M. Hydrophilic vs lipophilic statins in coronary artery disease: A meta-analysis of randomized controlled trials. J. Clin. Lipidol. 2017, 11, 624–637. [Google Scholar] [CrossRef]
- Kato, S.; Smalley, S.; Sadarangani, A.; Chen-Lin, K.; Oliva, B.; Branes, J.; Carvajal, J.; Gejman, R.; Owen, G.I.; Cuello, M. Lipophilic but not hydrophilic statins selectively induce cell death in gynaecological cancers expressing high levels of HMGCoA reductase. J. Cell Mol. Med. 2010, 14, 1180–1193. [Google Scholar] [CrossRef]
- Kim, M.C.; Ahn, Y.; Jang, S.Y.; Cho, K.H.; Hwang, S.H.; Lee, M.G.; Ko, J.S.; Park, K.H.; Sim, D.S.; Yoon, N.S.; et al. Comparison of clinical outcomes of hydrophilic and lipophilic statins in patients with acute myocardial infarction. Korean J. Int. Med. 2011, 26, 294–303. [Google Scholar] [CrossRef]
- Simon, T.G.; Butt, A.A. Lipid dysregulation in hepatitis C virus, and impact of statin therapy upon clinical outcomes. World J. Gastroenterol. 2015, 21, 8293–8303. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, G.A.; Fisher, R.K.; Georgiadis, G.S.; Antoniou, S.A.; Torella, F. Statin therapy in lower limb peripheral arterial disease: Systematic review and meta-analysis. Vasc. Pharmacol. 2014, 63, 79–87. [Google Scholar] [CrossRef]
- Farwell, W.R.; Scranton, R.E.; Lawler, E.V.; Lew, R.A.; Brophy, M.T.; Fiore, L.D.; Gaziano, J.M. The association between statins and cancer incidence in a veterans population. J. Natl. Cancer Inst. 2008, 100, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Park, S.; Park, J.; Shim, M.; Kim, A.; Jeong, I.G.; Hong, J.H.; Kim, C.S.; Ahn, H. Statin use after radical prostatectomy reduces biochemical recurrence in men with prostate cancer. Prostate 2015, 75, 211–217. [Google Scholar] [CrossRef] [PubMed]
- McKay, R.R.; Lin, X.; Albiges, L.; Fay, A.P.; Kaymakcalan, M.D.; Mickey, S.S.; Ghoroghchian, P.P.; Bhatt, R.S.; Kaffenberger, S.D.; Simantov, R.; et al. Statins and survival outcomes in patients with metastatic renal cell carcinoma. Eur. J. Cancer 2016, 52, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Gbelcova, H.; Lenicek, M.; Zelenka, J.; Knejzlik, Z.; Dvorakova, G.; Zadinova, M.; Pouckova, P.; Kudla, M.; Balaz, P.; Ruml, T.; et al. Differences in antitumor effects of various statins on human pancreatic cancer. Int. J. Cancer 2008, 122, 1214–1221. [Google Scholar] [CrossRef] [PubMed]
- Benakanakere, I.; Johnson, T.; Sleightholm, R.; Villeda, V.; Arya, M.; Bobba, R.; Freter, C.; Huang, C. Targeting cholesterol synthesis increases chemoimmuno-sensitivity in chronic lymphocytic leukemia cells. Exp. Hematol. Oncol. 2014, 3, 24. [Google Scholar] [CrossRef]
- Bjarnadottir, O.; Romero, Q.; Bendahl, P.O.; Jirstrom, K.; Ryden, L.; Loman, N.; Uhlen, M.; Johannesson, H.; Rose, C.; Grabau, D.; et al. Targeting HMG-CoA reductase with statins in a window-of-opportunity breast cancer trial. Breast Cancer Res. Treat. 2013, 138, 499–508. [Google Scholar] [CrossRef]
- Szajnman, S.H.; Ravaschino, E.L.; Docampo, R.; Rodriguez, J.B. Synthesis and biological evaluation of 1-amino-1,1-bisphosphonates derived from fatty acids against Trypanosoma cruzi targeting farnesyl pyrophosphate synthase. Bioorg. Med. Chem. Lett. 2005, 15, 4685–4690. [Google Scholar] [CrossRef]
- Nishimoto, T.; Ishikawa, E.; Anayama, H.; Hamajyo, H.; Nagai, H.; Hirakata, M.; Tozawa, R. Protective effects of a squalene synthase inhibitor, lapaquistat acetate (TAK-475), on statin-induced myotoxicity in guinea pigs. Toxicol. Appl. Pharmacol. 2007, 223, 39–45. [Google Scholar] [CrossRef]
- Ryder, N.S. Terbinafine: Mode of action and properties of the squalene epoxidase inhibition. Br. J. Dermatol. 1992, 126 (Suppl. 39), 2–7. [Google Scholar] [CrossRef] [PubMed]
- Lanterna, C.; Musumeci, A.; Raccosta, L.; Corna, G.; Moresco, M.; Maggioni, D.; Fontana, R.; Doglioni, C.; Bordignon, C.; Traversari, C.; et al. The administration of drugs inhibiting cholesterol/oxysterol synthesis is safe and increases the efficacy of immunotherapeutic regimens in tumor-bearing mice. Cancer Immunol. Immunother. 2016, 65, 1303–1315. [Google Scholar] [CrossRef]
- Maione, F.; Oliaro-Bosso, S.; Meda, C.; Di Nicolantonio, F.; Bussolino, F.; Balliano, G.; Viola, F.; Giraudo, E. The cholesterol biosynthesis enzyme oxidosqualene cyclase is a new target to impair tumour angiogenesis and metastasis dissemination. Sci. Rep. 2015, 5, 9054. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, D.M.; Yu, O.; Johnson, J. Statin use and cancer risk: A comprehensive review. Exp. Opin. Drug Saf. 2010, 9, 603–621. [Google Scholar] [CrossRef]
- Roelofs, A.J.; Thompson, K.; Ebetino, F.H.; Rogers, M.J.; Coxon, F.P. Bisphosphonates: Molecular mechanisms of action and effects on bone cells, monocytes and macrophages. Curr. Pharm. Des. 2010, 16, 2950–2960. [Google Scholar] [CrossRef] [PubMed]
- Shiomi, M.; Yamada, S.; Amano, Y.; Nishimoto, T.; Ito, T. Lapaquistat acetate, a squalene synthase inhibitor, changes macrophage/lipid-rich coronary plaques of hypercholesterolaemic rabbits into fibrous lesions. Br. J. Pharmacol. 2008, 154, 949–957. [Google Scholar] [CrossRef]
- Bergstrom, J.D.; Kurtz, M.M.; Rew, D.J.; Amend, A.M.; Karkas, J.D.; Bostedor, R.G.; Bansal, V.S.; Dufresne, C.; VanMiddlesworth, F.L.; Hensens, O.D.; et al. Zaragozic acids: A family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc. Natl. Acad. Sci. USA 1993, 90, 80–84. [Google Scholar] [CrossRef]
- Liang, Y.; Goyette, S.; Hyder, S.M. Cholesterol biosynthesis inhibitor RO 48-8071 reduces progesterone receptor expression and inhibits progestin-dependent stem cell-like cell growth in hormone-dependent human breast cancer cells. Breast Cancer (Dove Med. Press) 2017, 9, 487–494. [Google Scholar] [CrossRef]
- Liang, Y.; Mafuvadze, B.; Aebi, J.D.; Hyder, S.M. Cholesterol biosynthesis inhibitor RO 48-8071 suppresses growth of hormone-dependent and castration-resistant prostate cancer cells. Onco Targets Ther. 2016, 9, 3223–3232. [Google Scholar] [CrossRef]
- Hammersley, D.; Signy, M. Ezetimibe: An update on its clinical usefulness in specific patient groups. Ther. Adv. Chronic Dis. 2017, 8, 4–11. [Google Scholar] [CrossRef]
- Raal, F.J.; Marais, A.D.; Klepack, E.; Lovalvo, J.; McLain, R.; Heinonen, T. Avasimibe, an ACAT inhibitor, enhances the lipid lowering effect of atorvastatin in subjects with homozygous familial hypercholesterolemia. Atherosclerosis 2003, 171, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.A.; de Vries, A.H.; Marrink, S.J. Molecular mechanism of cyclodextrin mediated cholesterol extraction. PLoS Comput. Biol. 2011, 7, e1002020. [Google Scholar] [CrossRef] [PubMed]
- Solomon, K.R.; Pelton, K.; Boucher, K.; Joo, J.; Tully, C.; Zurakowski, D.; Schaffner, C.P.; Kim, J.; Freeman, M.R. Ezetimibe is an inhibitor of tumor angiogenesis. Am. J. Pathol. 2009, 174, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Nutescu, E.A.; Shapiro, N.L. Ezetimibe: A selective cholesterol absorption inhibitor. Pharmacotherapy 2003, 23, 1463–1474. [Google Scholar] [CrossRef]
- Mulas, M.F.; Abete, C.; Pulisci, D.; Pani, A.; Massidda, B.; Dessi, S.; Mandas, A. Cholesterol esters as growth regulators of lymphocytic leukaemia cells. Cell Prolif. 2011, 44, 360–371. [Google Scholar] [CrossRef]
- Shim, S.H.; Sur, S.; Steele, R.; Albert, C.J.; Huang, C.; Ford, D.A.; Ray, R.B. Disrupting cholesterol esterification by bitter melon suppresses triple-negative breast cancer cell growth. Mol. Carcinog. 2018, 57, 1599–1607. [Google Scholar] [CrossRef]
- Li, J.; Gu, D.; Lee, S.S.; Song, B.; Bandyopadhyay, S.; Chen, S.; Konieczny, S.F.; Ratliff, T.L.; Liu, X.; Xie, J.; et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene 2016, 35, 6378–6388. [Google Scholar] [CrossRef]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118. [Google Scholar] [CrossRef]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef]
- Mollinedo, F.; Gajate, C. Lipid rafts as major platforms for signaling regulation in cancer. Adv. Biol. Regul. 2015, 57, 130–146. [Google Scholar] [CrossRef] [PubMed]
- Kong, Y.; Cheng, L.; Mao, F.; Zhang, Z.; Zhang, Y.; Farah, E.; Bosler, J.; Bai, Y.; Ahmad, N.; Kuang, S.; et al. Inhibition of cholesterol biosynthesis overcomes enzalutamide resistance in castration-resistant prostate cancer (CRPC). J. Biol. Chem. 2018, 293, 14328–14341. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Q.; Wu, Y.; Wang, D.; Xu, L.; Zhang, Y.; Wang, S.; Wang, T.; Liu, F.; Zaky, M.Y.; et al. Cholesterol content in cell membrane maintains surface levels of ErbB2 and confers a therapeutic vulnerability in ErbB2-positive breast cancer. Cell Commun. Signal. 2019, 17, 15. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Wang, H.; Zhu, D.; Wan, Y.; Yin, L. Combined effects of avasimibe immunotherapy, doxorubicin chemotherapy, and metal-organic frameworks nanoparticles on breast cancer. J. Cell Physiol. 2020, 235, 4814–4823. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Song, Q.; Xia, L.; Xu, X. Synergy of Dendritic Cell Vaccines and Avasimibe in Treatment of Head and Neck Cancer in Mice. Med. Sci. Monit. 2017, 23, 4471–4476. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Year | Study Design | Population Group | Number | Main Findings | Reference |
|---|---|---|---|---|---|
| 1967–1999 | Prospective cohort study | Finland | >9000 | High dietary cholesterol intake was associated with increased risk of colorectal cancer | [35] |
| 1970–2007 | Prospective cohort study | Scotland, United Kingdom | >12,000 | Plasma cholesterol was positively related to risk of high-grade prostate cancer incidence | [36] |
| 1972–2005 | Cohort study | Norway, Austria, and Sweden | >500,000 | Total serum cholesterol level was inversely associated with risk of overall cancer in females and with risk of liver, pancreas, and melanoma cancers in males | [37] |
| 1972–2012 | Prospective cohort study | Norway | >2000 | An inverse association was found between cholesterol level and risk of prostate cancer | [5] |
| 1984–2009 | Randomized controlled trials | 28 pharmacologic intervention arms and 23 control arms | >600,000 | An inverse association was found between HDL-C level and risk of cancer incidence | [38] |
| 1985–2002 | Prospective cohort study | Finland | >20,000 | No association between total cholesterol and risk of non-Hodgkin lymphoma (NHL), but an inverse association between HDL-C and NHL | [39] |
| 1988–2002 | Prospective study | Japan | >2000 | An inverse correlation between serum cholesterol level and the incidence of gastric cancer | [6] |
| 1990–2012 | Meta-analysis | 12 case-control studies and 4 cohort studies | >4000 | Dietary cholesterol intake contributed to higher risk of pancreatic cancer | [40] |
| 1992–2006 | Prospective study | Korea | >1,000,000 | High cholesterol was related to prostate, colon, and breast cancers and inversely related to lung, liver, and stomach cancers. | [41] |
| 1995–2013 | Case-control study | United Kingdom | >100,000 | A decreased risk of colorectal cancer with statin use | [42] |
| 1997–2001 | Population-based case-control study | Shanghai, China | >800 | Low HDL-C was related to higher risk of gallbladder and bile duct cancers. A U-shape relationship was found between total cholesterol level and LDL-C with biliary tract cancers | [43] |
| 2002–2012 | Retrospective study | Guangzhou, China | >600 | Low HDL-C was correlated with poorer disease-free survival and overall survival in stage II/III colorectal cancer | [44] |
| 2014 | Meta-analysis | 22 randomized controlled trials, 5 cohorts, and 6 case-control studies | >5,000,000 | A significant risk reduction of liver cancer in all statin users, regardless of the type of statin used | [45] |
| 1993–2011 | Meta-analysis | 15 cohort and 12 case-control studies | >1,000,000 | A decreased risk of prostate cancer in statin users, though long-term statin use did not affect the total risk of prostate cancer | [46] |
| 2012 | Meta-analysis | 8 observational and 3 post-hoc analyses of 26 clinical trials | >5000 | A significant drop of over 30% in gastric cancer with statin use; significance remains in both Asian and Western populations | [47] |
| 1998–2014 | Retrospective case-control study | United States | >400,000 | A drop of over 60% risk in pancreatic cancer with statin use of more than 6 months | [48] |
| Chemical | Inhibitory Target | Effects | Reference | |
|---|---|---|---|---|
| Cholesterol biosynthesis | Statins | HMGCR | Block the formation of mevalonate from HMG-CoA | [137,154] |
| δ-, γ-, and α-tocotrienol | [125] | |||
| β-ionone | [126] | |||
| geranylgeraniol | [127] | |||
| geraniol | [128] | |||
| S-allylcysteine | [134] | |||
| Bisphosphonate | FPP synthase | Prevent the prenylation of small GTPases | [155] | |
| Lapaquistat | Squalene synthase | Block the conversion from FPP to squalene | [156] | |
| Zaragozic acid | [157] | |||
| RO 48-8071 | OSC | Block the synthesis of lanosterol from 2,3-monoepoxysqualene | [158,159] | |
| Curcumin | SQLE | Block the synthesis of squalene epoxide | [135] | |
| Garlic extract | Sterol 4α-methyl oxidase | Prevent the formation of zymosterol | [133] | |
| Cholesterol intake | Ezetimibe | NPC1L1 | Reduce LDL-C levels | [160] |
| Curcumin | [136] | |||
| EGCG | LDLR | [130,131,132] | ||
| Cholesterol esterification | Avasimibe | ACAT1 | Reduce the formation of cholesteryl esters | [161] |
| Cholesterol depletion agent | Methyl-β-cyclodextrin | Lipid rafts | Facilitate the depletion of cholesterol from membranes | [162] |
| Immunotherapy | Dendritic cell vaccine | Increase antigen presentation efficiency | [121] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mok, E.H.K.; Lee, T.K.W. The Pivotal Role of the Dysregulation of Cholesterol Homeostasis in Cancer: Implications for Therapeutic Targets. Cancers 2020, 12, 1410. https://doi.org/10.3390/cancers12061410
Mok EHK, Lee TKW. The Pivotal Role of the Dysregulation of Cholesterol Homeostasis in Cancer: Implications for Therapeutic Targets. Cancers. 2020; 12(6):1410. https://doi.org/10.3390/cancers12061410
Chicago/Turabian StyleMok, Etienne Ho Kit, and Terence Kin Wah Lee. 2020. "The Pivotal Role of the Dysregulation of Cholesterol Homeostasis in Cancer: Implications for Therapeutic Targets" Cancers 12, no. 6: 1410. https://doi.org/10.3390/cancers12061410
APA StyleMok, E. H. K., & Lee, T. K. W. (2020). The Pivotal Role of the Dysregulation of Cholesterol Homeostasis in Cancer: Implications for Therapeutic Targets. Cancers, 12(6), 1410. https://doi.org/10.3390/cancers12061410