Clinical Implications of DNA Repair Defects in High-Grade Serous Ovarian Carcinomas



Abstract
1. DNA Repair Defects: the “Achille’s heel” of High-Grade Serous Ovarian Cancers
2. Homologous Recombination Deficiency in EOC
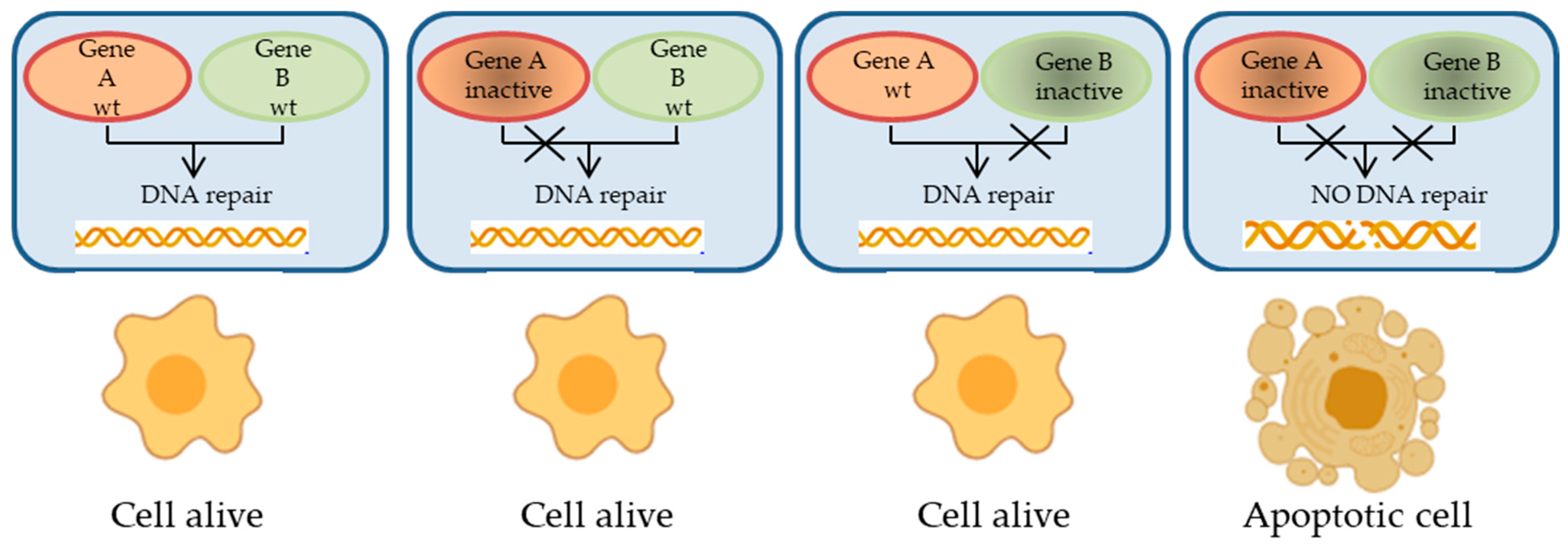
2.1. Relevant Proteins and Pathways
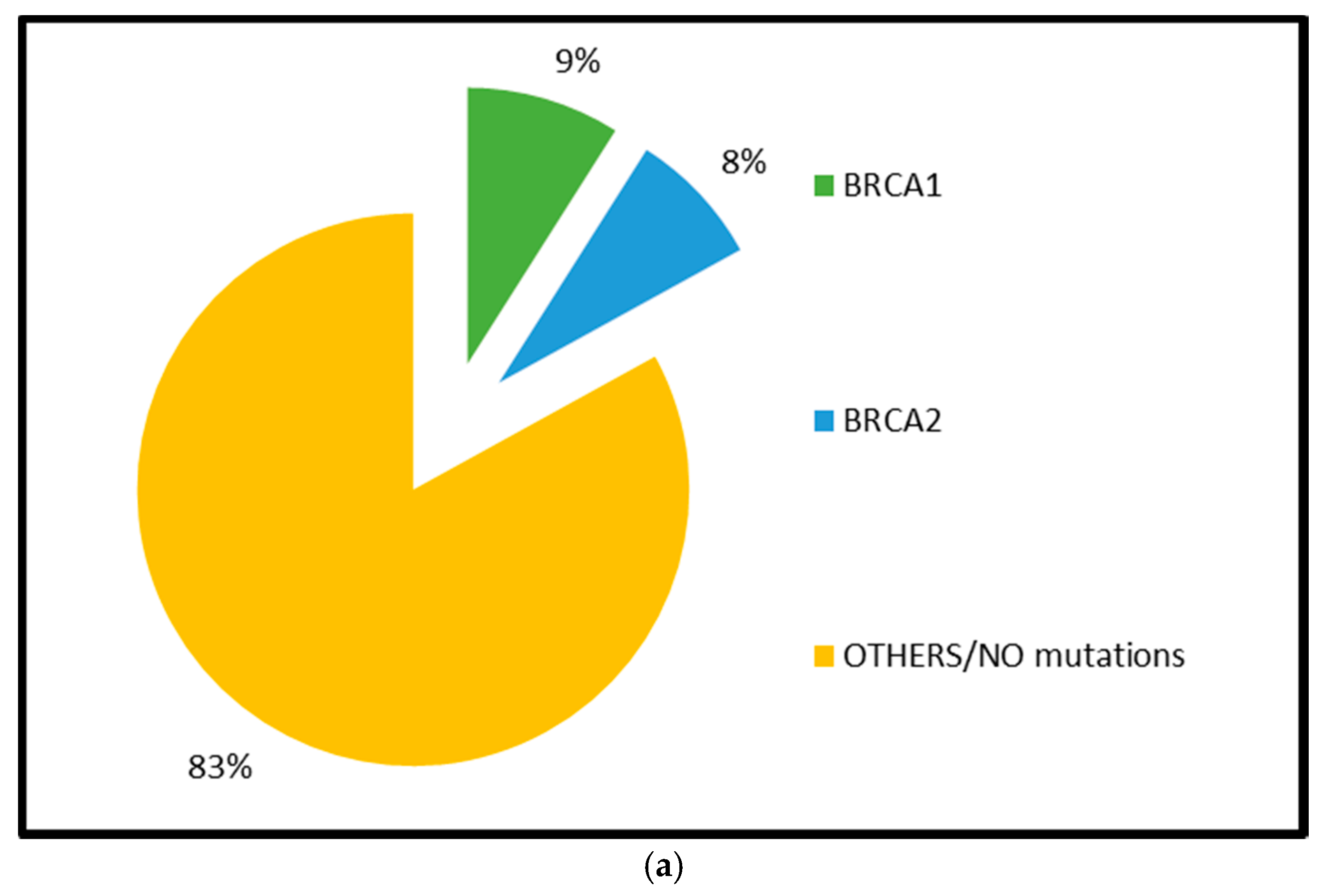
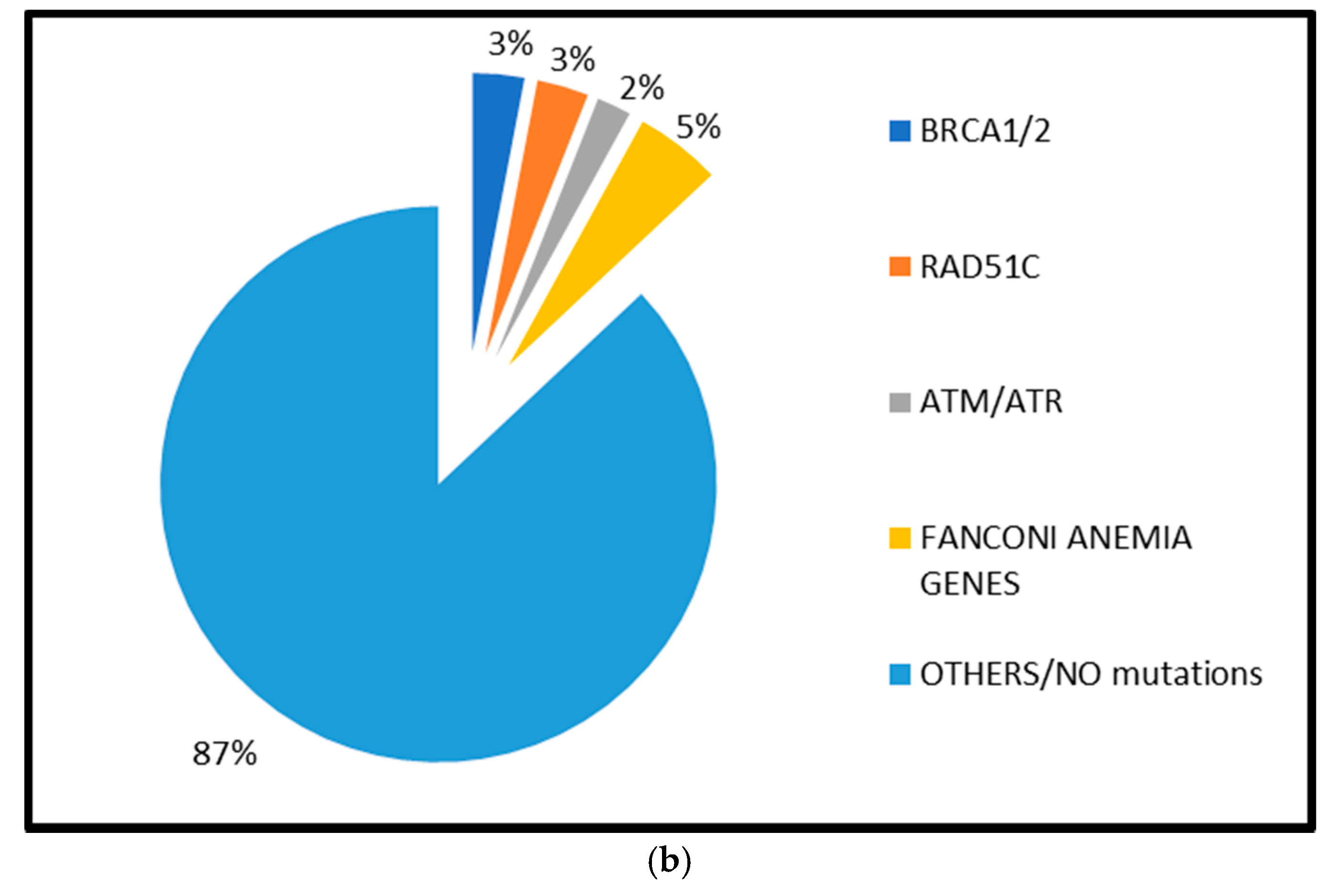
2.1.1. BRCA1 and BRCA2 Mutations
2.1.2. Role of Poly (ADP-ribose) Polymerase (PARP) in HR deficiency
2.1.3. Focus on p53 Protein
2.2. Therapeutic Implications
2.3. Immunologic Features and Related Clinical Implications in HR-Deficient EOC
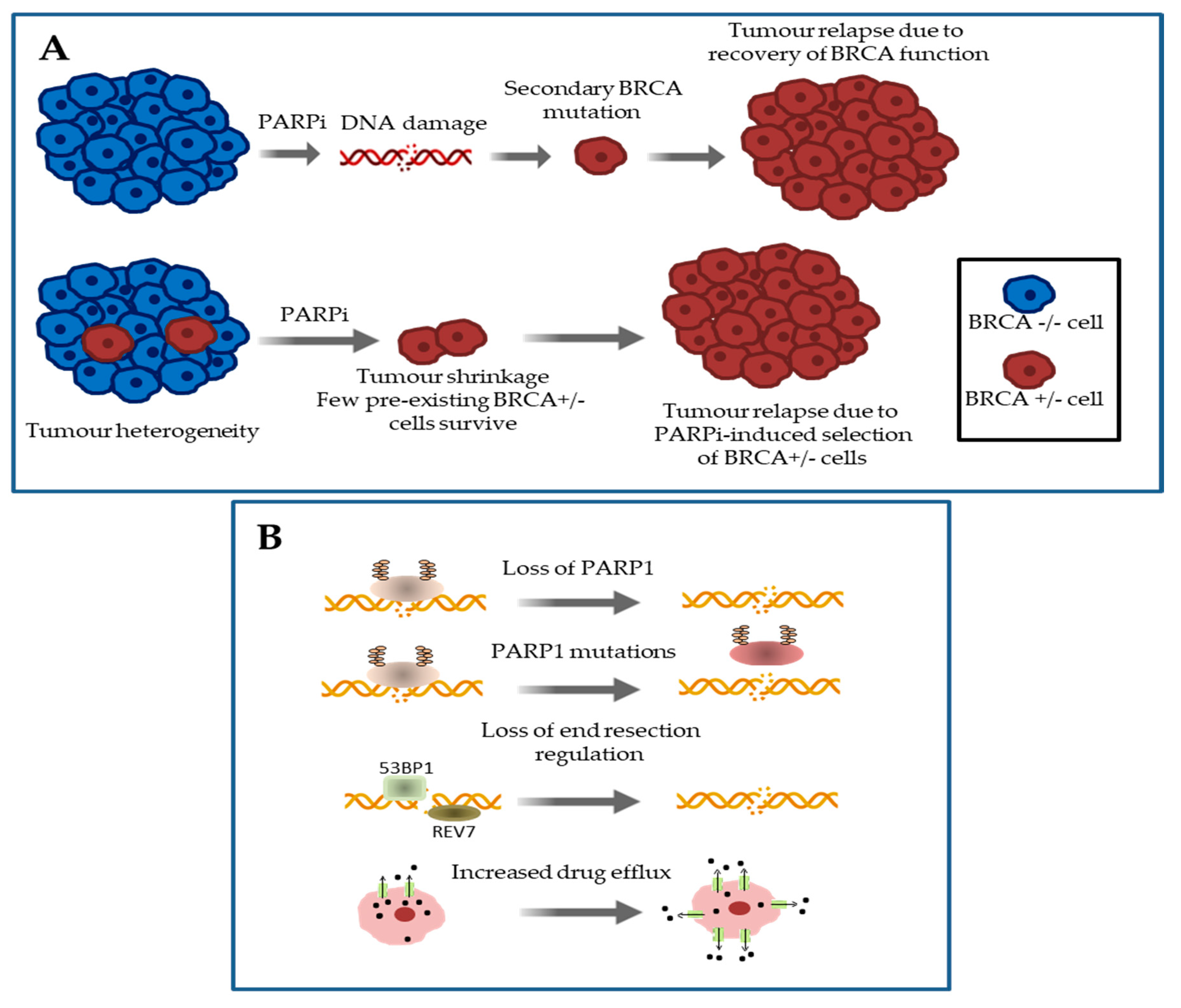
3. Mechanisms of Primary and Acquired Resistance to Platinum Salts and PARP Inhibitors in HGSOC
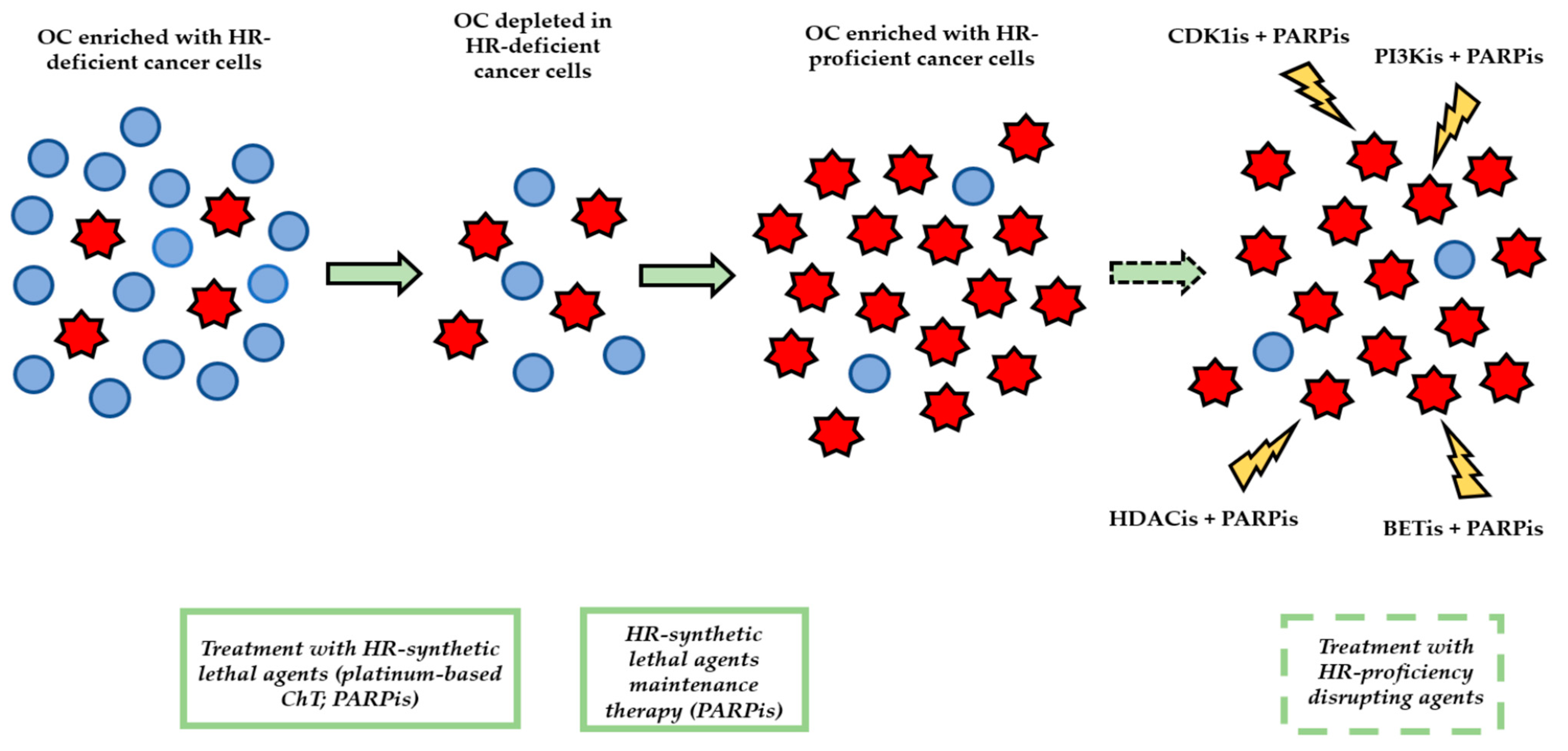
4. Issues with the Management of HR Proficiency in HGSOC
5. DNA Mismatch Repair Deficiency in EOC and Deriving Clinical Implications
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Network, C.G.A.R. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Kim, A.; Ueda, Y.; Naka, T.; Enomoto, T. Therapeutic strategies in epithelial ovarian cancer. J. Exp. Clin. Cancer Res. 2012, 31, 14. [Google Scholar] [CrossRef] [PubMed]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-genome characterization of chemoresistant ovarian cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Han, Y.; Kim, S.I.; Kim, H.S.; Kim, S.J.; Song, Y.S. Tumor evolution and chemoresistance in ovarian cancer. NPJ Precis Oncol. 2018, 2, 20. [Google Scholar] [CrossRef]
- Minchom, A.; Aversa, C.; Lopez, J. Dancing with the DNA damage response: Next-generation anti-cancer therapeutic strategies. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786658. [Google Scholar] [CrossRef]
- Rojas, V.; Hirshfield, K.M.; Ganesan, S.; Rodriguez-Rodriguez, L. Molecular Characterization of Epithelial Ovarian Cancer: Implications for Diagnosis and Treatment. Int. J. Mol. Sci. 2016, 17, 2113. [Google Scholar] [CrossRef]
- Murphy, M.A.; Wentzensen, N. Frequency of mismatch repair deficiency in ovarian cancer: A systematic review This article is a US Government work and, as such, is in the public domain of the United States of America. Int. J. Cancer 2011, 129, 1914–1922. [Google Scholar] [CrossRef]
- Helder-Woolderink, J.M.; Blok, E.A.; Vasen, H.F.; Hollema, H.; Mourits, M.J.; De Bock, G.H. Ovarian cancer in Lynch syndrome; a systematic review. Eur. J. Cancer 2016, 55, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Scambia, G.; Ferrandina, G. A turning point in the fight against ovarian cancer? Lancet Oncol. 2018, 19, 154–156. [Google Scholar] [CrossRef]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell. Biol. 2016, 19, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, B.T.; Timms, K.M.; Carey, M.S.; Gutin, A.; Meyer, L.A.; Flake, D.D.; Abkevich, V.; Potter, J.; Pruss, D.; Glenn, P.; et al. Somatic mutations in BRCA1 and BRCA2 could expand the number of patients that benefit from poly (ADP ribose) polymerase inhibitors in ovarian cancer. J. Clin. Oncol. 2010, 28, 3570–3576. [Google Scholar] [CrossRef]
- Pennington, K.P.; Walsh, T.; Harrell, M.I.; Lee, M.K.; Pennil, C.C.; Rendi, M.H.; Thornton, A.; Norquist, B.M.; Casadei, S.; Nord, A.S.; et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 2014, 20, 764–775. [Google Scholar] [CrossRef]
- Cunningham, J.M.; Cicek, M.S.; Larson, N.B.; Davila, J.; Wang, C.; Larson, M.C.; Song, H.; Dicks, E.M.; Harrington, P.; Wick, M.; et al. Clinical characteristics of ovarian cancer classified by BRCA1, BRCA2, and RAD51C status. Sci. Rep. 2014, 4, 4026. [Google Scholar] [CrossRef] [PubMed]
- Beltrame, L.; Di Marino, M.; Fruscio, R.; Calura, E.; Chapman, B.; Clivio, L.; Sina, F.; Mele, C.; Iatropoulos, P.; Grassi, T.; et al. Profiling cancer gene mutations in longitudinal epithelial ovarian cancer biopsies by targeted next-generation sequencing: A retrospective study. Ann. Oncol. 2015, 26, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wiese, C.; Kwon, Y.; Hromas, R.; Sung, P. The BRCA Tumor Suppressor Network in Chromosome Damage Repair by Homologous Recombination. Annu. Rev. Biochem. 2019, 88, 221–245. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.; Sonoda, Y.; Federici, M.G.; Bogomolniy, F.; Rhei, E.; Maresco, D.L.; Saigo, P.E.; Almadrones, L.A.; Barakat, R.R.; Brown, C.L.; et al. Clinicopathologic features of BRCA-linked and sporadic ovarian cancer. JAMA 2000, 283, 2260–2265. [Google Scholar] [CrossRef]
- Birkbak, N.J.; Kochupurakkal, B.; Izarzugaza, J.M.; Eklund, A.C.; Li, Y.; Liu, J.; Szallasi, Z.; Matulonis, U.A.; Richardson, A.L.; Iglehart, J.D.; et al. Tumor mutation burden forecasts outcome in ovarian cancer with BRCA1 or BRCA2 mutations. PLoS ONE 2013, 8, e80023. [Google Scholar] [CrossRef]
- Foulkes, W.D. BRCA1 and BRCA2: Chemosensitivity, treatment outcomes and prognosis. Fam. Cancer 2006, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Bolton, K.L.; Chenevix-Trench, G.; Goh, C.; Sadetzki, S.; Ramus, S.J.; Karlan, B.Y.; Lambrechts, D.; Despierre, E.; Barrowdale, D.; McGuffog, L.; et al. Association between BRCA1 and BRCA2 mutations and survival in women with invasive epithelial ovarian cancer. JAMA 2012, 307, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’Andrea, A.D.; et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget 2016, 7, 13587–13598. [Google Scholar] [CrossRef] [PubMed]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, T.; Christie, E.L.; Ahuja, A.; Buros, J.; Aksoy, B.A.; Bowtell, D.D.L.; Snyder, A.; Hammerbacher, J. Chemotherapy weakly contributes to predicted neoantigen expression in ovarian cancer. BMC Cancer 2018, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP inhibitors: Clinical utility and possibilities of overcoming resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef]
- Mittica, G.; Ghisoni, E.; Giannone, G.; Genta, S.; Aglietta, M.; Sapino, A.; Valabrega, G. PARP Inhibitors in Ovarian Cancer. Recent Pat. Anticancer Drug Discov. 2018, 13, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Pascal, J.M. The comings and goings of PARP-1 in response to DNA damage. DNA Repair (Amst) 2018, 71, 177–182. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Wood, R.D. DNA polymerase POLQ and cellular defense against DNA damage. DNA Repair (Amst) 2013, 12, 1–9. [Google Scholar] [CrossRef]
- Lieber, M.R. NHEJ and its backup pathways in chromosomal translocations. Nat. Struct. Mol. Biol. 2010, 17, 393–395. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.; Renaud, A.; Zhang, Y.J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef] [PubMed]
- Soong, T.R.; Kolin, D.L.; Teschan, N.J.; Crum, C.P. Back to the Future? The Fallopian Tube, Precursor Escape and a Dualistic Model of High-Grade Serous Carcinogenesis. Cancers 2018, 10, 468. [Google Scholar] [CrossRef]
- Vazquez, A.; Bond, E.E.; Levine, A.J.; Bond, G.L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discov. 2008, 7, 979–987. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Del Campo, J.M.; Matulonis, U.A.; Malander, S.; Provencher, D.; Mahner, S.; Follana, P.; Waters, J.; Berek, J.S.; Woie, K.; Oza, A.M.; et al. Niraparib Maintenance Therapy in Patients with Recurrent Ovarian Cancer After a Partial Response to the Last Platinum-Based Chemotherapy in the ENGOT-OV16/NOVA Trial. J. Clin. Oncol. 2019, 37, 2968–2973. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Fagotti, A.; Scambia, G. Fighting against the challenge of treating patients with late-line ovarian cancer: Are we there yet? Lancet Oncol. 2019, 20, 603–605. [Google Scholar] [CrossRef]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Lacchetti, C.; Annunziata, C.M. Germline and Somatic Tumor Testing in Epithelial Ovarian Cancer: ASCO Guideline Summary. JCO Oncol. Pract. 2020, JOP1900773. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Zhang, L.; Conejo-Garcia, J.R.; Katsaros, D.; Gimotty, P.A.; Massobrio, M.; Regnani, G.; Makrigiannakis, A.; Gray, H.; Schlienger, K.; Liebman, M.N.; et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003, 348, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Odunsi, K. Immunotherapy in ovarian cancer. Ann. Oncol. 2017, 28 (Suppl. 8), viii1–viii7. [Google Scholar] [CrossRef]
- Bobisse, S.; Genolet, R.; Roberti, A.; Tanyi, J.L.; Racle, J.; Stevenson, B.J.; Iseli, C.; Michel, A.; Le Bitoux, M.A.; Guillaume, P.; et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8. Nat. Commun. 2018, 9, 1092. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Shapira-Frommer, R.; Santin, A.D.; Lisyanskaya, A.S.; Pignata, S.; Vergote, I.; Raspagliesi, F.; Sonke, G.S.; Birrer, M.; Provencher, D.M.; et al. Antitumor activity and safety of pembrolizumab in patients with advanced recurrent ovarian cancer: Results from the phase II KEYNOTE-100 study. Ann. Oncol. 2019, 30, 1080–1087. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Taylor, M.H.; Kelly, K.; Beck, J.T.; Gordon, M.; Moore, K.M.; Patel, M.R.; Chaves, J.; Park, H.; Mita, A.C.; et al. Efficacy and Safety of Avelumab for Patients with Recurrent or Refractory Ovarian Cancer: Phase 1b Results from the JAVELIN Solid Tumor Trial. JAMA Oncol. 2019, 5, 393–401. [Google Scholar] [CrossRef]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Pérez Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef]
- Boussios, S.; Karihtala, P.; Moschetta, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Combined Strategies with Poly (ADP-Ribose) Polymerase (PARP) Inhibitors for the Treatment of Ovarian Cancer: A Literature Review. Diagnostics 2019, 9, 87. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef]
- Norquist, B.; Wurz, K.A.; Pennil, C.C.; Garcia, R.; Gross, J.; Sakai, W.; Karlan, B.Y.; Taniguchi, T.; Swisher, E.M. Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J. Clin. Oncol. 2011, 29, 3008–3015. [Google Scholar] [CrossRef]
- Barber, L.J.; Sandhu, S.; Chen, L.; Campbell, J.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Rodrigues, D.N.; Reis Filho, J.S.; Moreno, V.; et al. Secondary mutations in BRCA2 associated with clinical resistance to a PARP inhibitor. J. Pathol. 2013, 229, 422–429. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Rehman, F.L.; Bajrami, I.; Brough, R.; Wallberg, F.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Chen, L.; Campbell, J.; et al. A genetic screen using the PiggyBac transposon in haploid cells identifies Parp1 as a mediator of olaparib toxicity. PLoS ONE 2013, 8, e61520. [Google Scholar] [CrossRef]
- Choi, Y.H.; Yu, A.M. ABC transporters in multidrug resistance and pharmacokinetics, and strategies for drug development. Curr. Pharm. Des. 2014, 20, 793–807. [Google Scholar] [CrossRef]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for stalled replication fork stabilization: New targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [PubMed]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; van der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.J.; Aly, A.; Doroshow, J.H.; et al. Loss of 53BP1 causes PARP inhibitor resistance in Brca1-mutated mouse mammary tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef]
- Liang, L.; Feng, J.; Zuo, P.; Yang, J.; Lu, Y.; Yin, Y. Molecular basis for assembly of the shieldin complex and its implications for NHEJ. Nat. Commun. 2020, 11, 1972. [Google Scholar] [CrossRef]
- Xu, G.; Chapman, J.R.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef]
- Johnson, N.; Li, Y.C.; Walton, Z.E.; Cheng, K.A.; Li, D.; Rodig, S.J.; Moreau, L.A.; Unitt, C.; Bronson, R.T.; Thomas, H.D.; et al. Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat. Med. 2011, 17, 875–882. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Wilson, A.J.; Saskowski, J.; Wass, E.; Khabele, D. Suberoylanilide hydroxamic acid (SAHA) enhances olaparib activity by targeting homologous recombination DNA repair in ovarian cancer. Gynecol. Oncol. 2014, 133, 599–606. [Google Scholar] [CrossRef]
- Wilson, A.J.; Sarfo-Kantanka, K.; Barrack, T.; Steck, A.; Saskowski, J.; Crispens, M.A.; Khabele, D. Panobinostat sensitizes cyclin E high, homologous recombination-proficient ovarian cancer to olaparib. Gynecol. Oncol. 2016, 143, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Baratta, M.G.; Schinzel, A.C.; Zwang, Y.; Bandopadhayay, P.; Bowman-Colin, C.; Kutt, J.; Curtis, J.; Piao, H.; Wong, L.C.; Kung, A.L.; et al. An in-tumor genetic screen reveals that the BET bromodomain protein, BRD4, is a potential therapeutic target in ovarian carcinoma. Proc. Natl. Acad. Sci. USA 2015, 112, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Stubbs, M.; Liu, P.; Ruggeri, B.; Khabele, D. The BET inhibitor INCB054329 reduces homologous recombination efficiency and augments PARP inhibitor activity in ovarian cancer. Gynecol. Oncol. 2018, 149, 575–584. [Google Scholar] [CrossRef]
- Gabbasov, R.; Benrubi, I.D.; O’Brien, S.W.; Krais, J.J.; Johnson, N.; Litwin, S.; Connolly, D.C. Targeted blockade of HSP90 impairs DNA-damage response proteins and increases the sensitivity of ovarian carcinoma cells to PARP inhibition. Cancer Biol. Ther. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Zhong, Q.; Hu, Z.; Li, Q.; Yi, T.; Li, J.; Yang, H. Cyclin D1 silencing impairs DNA double strand break repair, sensitizes BRCA1 wildtype ovarian cancer cells to olaparib. Gynecol. Oncol. 2019, 152, 157–165. [Google Scholar] [CrossRef]
- Lim, J.J.; Yang, K.; Taylor-Harding, B.; Wiedemeyer, W.R.; Buckanovich, R.J. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia 2014, 16, e1–e2. [Google Scholar] [CrossRef]
- Xiao, X.; Melton, D.W.; Gourley, C. Mismatch repair deficiency in ovarian cancer -- molecular characteristics and clinical implications. Gynecol. Oncol. 2014, 132, 506–512. [Google Scholar] [CrossRef]
- Guillotin, D.; Martin, S.A. Exploiting DNA mismatch repair deficiency as a therapeutic strategy. Exp. Cell. Res. 2014, 329, 110–115. [Google Scholar] [CrossRef]
- Li, G.M. Mechanisms and functions of DNA mismatch repair. Cell. Res. 2008, 18, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic review of microsatellite instability and colorectal cancer prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Radman, M.; Wagner, R. Carcinogenesis. Missing mismatch repair. Nature 1993, 366, 722. [Google Scholar] [CrossRef]
- Strathdee, G.; MacKean, M.J.; Illand, M.; Brown, R. A role for methylation of the hMLH1 promoter in loss of hMLH1 expression and drug resistance in ovarian cancer. Oncogene 1999, 18, 2335–2341. [Google Scholar] [CrossRef] [PubMed]
- Zeller, C.; Dai, W.; Steele, N.L.; Siddiq, A.; Walley, A.J.; Wilhelm-Benartzi, C.S.; Rizzo, S.; van der Zee, A.; Plumb, J.A.; Brown, R. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene 2012, 31, 4567–4576. [Google Scholar] [CrossRef]
- Plumb, J.A.; Strathdee, G.; Sludden, J.; Kaye, S.B.; Brown, R. Reversal of drug resistance in human tumor xenografts by 2’-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000, 60, 6039–6044. [Google Scholar]
- Ercoli, A.; Ferrandina, G.; Raspaglio, G.; Marone, M.; Maggiano, N.; Del Mastro, P.; Benedetti Panici, P.; Mancuso, S.; Scambia, G. hMSH2 and GTBP expression in advanced stage epithelial ovarian cancer. Br. J. Cancer 1999, 80, 1665–1671. [Google Scholar] [CrossRef][Green Version]
- Marcelis, C.L.; van der Putten, H.W.; Tops, C.; Lutgens, L.C.; Moog, U. Chemotherapy resistant ovarian cancer in carriers of an hMSH2 mutation? Fam. Cancer 2001, 1, 107–109. [Google Scholar] [CrossRef]
- Samimi, G.; Fink, D.; Varki, N.M.; Husain, A.; Hoskins, W.J.; Alberts, D.S.; Howell, S.B. Analysis of MLH1 and MSH2 expression in ovarian cancer before and after platinum drug-based chemotherapy. Clin. Cancer Res. 2000, 6, 1415–1421. [Google Scholar]
- Martin, S.A.; Hewish, M.; Sims, D.; Lord, C.J.; Ashworth, A. Parallel high-throughput RNA interference screens identify PINK1 as a potential therapeutic target for the treatment of DNA mismatch repair-deficient cancers. Cancer Res. 2011, 71, 1836–1848. [Google Scholar] [CrossRef]
- Martin, S.A.; McCabe, N.; Mullarkey, M.; Cummins, R.; Burgess, D.J.; Nakabeppu, Y.; Oka, S.; Kay, E.; Lord, C.J.; Ashworth, A. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010, 17, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.A.; McCarthy, A.; Barber, L.J.; Burgess, D.J.; Parry, S.; Lord, C.J.; Ashworth, A. Methotrexate induces oxidative DNA damage and is selectively lethal to tumour cells with defects in the DNA mismatch repair gene MSH2. EMBO Mol. Med. 2009, 1, 323–337. [Google Scholar] [CrossRef] [PubMed]
- Howitt, B.E.; Strickland, K.C.; Sholl, L.M.; Rodig, S.; Ritterhouse, L.L.; Chowdhury, D.; D’Andrea, A.D.; Matulonis, U.A.; Konstantinopoulos, P.A. Clear cell ovarian cancers with microsatellite instability: A unique subset of ovarian cancers with increased tumor-infiltrating lymphocytes and PD-1/PD-L1 expression. Oncoimmunology 2017, 6, e1277308. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Maintenance Therapy after Response To First-Line Platinum-Based ChT | Maintenance Therapy after Response To Platinum-Based ChT in Recurrence Setting | Monotherapy in Recurrence Setting |
|---|---|---|---|
OLAPARIB | Germline or somatic BRCA1/2-mutated HGSOC | Germline or somatic BRCA1/2-mutated PS recurrent HGSOC BRCA1/2-wild-type PS recurrent HGSOC | Germline BRCA1/2-mutated recurrent HGSOC, following 3 or more prior ChT lines |
RUCAPARIB | - | PS recurrent HGSOC | Germline or somatic BRCA1/2-mutated PS recurrent HGSOC, following 2 or more prior platinum lines (pts not candidate to further platinum) Germline or somatic BRCA1/2-mutated recurrent HGSOC, following 2 or more prior ChT lines |
NIRAPARIB | BRCA1/2-mutated and BRCA1/2 WT HGSOC | PS recurrent HGSOC | - |
| Trial Name/Identifier | Phase | Condition | Therapeutic Setting | Drug Regimen(s) | Primary Endpoint | Status |
| IMagyn050/NCT03038100 | III | Stage III-IV EOC, FTC and PPT | Neoadjuvant/post-operative | Atezolizumab + Paclitaxel, Carboplatin and Bevacizumab vs. Placebo + Paclitaxel, Carboplatin and Bevacizumab | PFS and OS ITT/PDL-1+ population | Active, not recruiting |
| ATHENA/NCT03522246 | III | Stage III-IV EOC, FTC and PPT | Maintenance after CR or PR to first line platinum-based ChT | Rucaparib + Nivolumab vs. Rucaparib + Placebo vs. Placebo + Nivolumab vs. Placebo + Placebo | PFS | Recruiting |
| MITO 25/NCT 03462212 | I/II | Stage III-IV EOC, FTC and PPT | First line | Carboplatin, Paclitaxel + Rucaparib (only in maintenance) vs. Carboplatin, Paclitaxel + Bevacizumab (in combination and maintenance) vs. Carboplatin, Paclitaxel + Bevacizumab (in combination and maintenance) + Rucaparib (only in maintenance) | PFS and Safety | Recruiting |
| KEYNOTE-162/NCT02657889 | I/II | Advanced and metastatic TNBC, EOC, FTC and PPT | First and subsequent lines | Niraparib + Pembrolizumab | ORR and Safety | Active, not recruiting |
| NCT02873962 | II | Progressive or recurrent EOC, FTC and PPT | Second, third or fourth line | Nivolumab + Bevacizumab vs. Nivolumab + Bevacizumab and Rucaparib | ORR | Recruiting |
| ATALANTE/NCT02891824 | III | Progressive or recurrent EOC, FTC and PPT | Second or third line | Atezolizumab + Bevacizumab and platinum-based ChT followed by Atezolizumab maintenance vs. Placebo + Bevacizumab and platinum-based ChT followed by Placebo maintenance | PFS | Active, not recruiting |
| JAVELIN/NCT01772004 | I | Metastatic or locally advanced solid tumors | Progressive disease following last “standard-of-care” line of treatment | Avelumab | BOR and Safety | Completed |
| ANITA/NCT03598270 | III | Progressive or recurrent EOC, FTC and PTT | Second or third line | Atezolizumab + platinum-based ChT followed by Atezolizumab and Niraparib maintenance vs. Placebo + platinum-based ChT followed by placebo and Niraparib maintenance | PFS | Recruiting |
| MEDIOLA/NCT02734004 | I/II | Advanced solid tumors | Relapsed disease following “standard-of-care” treatment | Olaparib + MEDI4736 (Anti-PDL-1 Antibody)/Olaparib + MEDI4736 (Anti-PDL-1 Antibody) + Bevacizumab | DCR, ORR and Safety | Recruiting |
| MITO27/NCT03539328 | II | Progressive or recurrent EOC, FTC and PPT | Second or third line | Pegylated liposomal Doxorubicin or weekly Paclitaxel or Gemcitabine (at Physician’s discretion) vs. Pegylated liposomal Doxorubicin + Pembrolizumab or weekly Paclitaxel + Pembrolizumab or Gemcitabine + Pembrolizumab (at Physician’s discretion) | OS | Not yet recruiting |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milanesio, M.C.; Giordano, S.; Valabrega, G. Clinical Implications of DNA Repair Defects in High-Grade Serous Ovarian Carcinomas. Cancers 2020, 12, 1315. https://doi.org/10.3390/cancers12051315
Milanesio MC, Giordano S, Valabrega G. Clinical Implications of DNA Repair Defects in High-Grade Serous Ovarian Carcinomas. Cancers. 2020; 12(5):1315. https://doi.org/10.3390/cancers12051315
Chicago/Turabian StyleMilanesio, Michela Camilla, Silvia Giordano, and Giorgio Valabrega. 2020. "Clinical Implications of DNA Repair Defects in High-Grade Serous Ovarian Carcinomas" Cancers 12, no. 5: 1315. https://doi.org/10.3390/cancers12051315
APA StyleMilanesio, M. C., Giordano, S., & Valabrega, G. (2020). Clinical Implications of DNA Repair Defects in High-Grade Serous Ovarian Carcinomas. Cancers, 12(5), 1315. https://doi.org/10.3390/cancers12051315