HuRdling Senescence: HuR Breaks BRAF-Induced Senescence in Melanocytes and Supports Melanoma Growth


Abstract
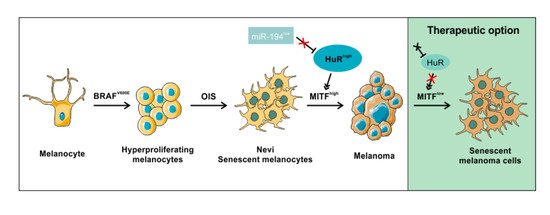
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
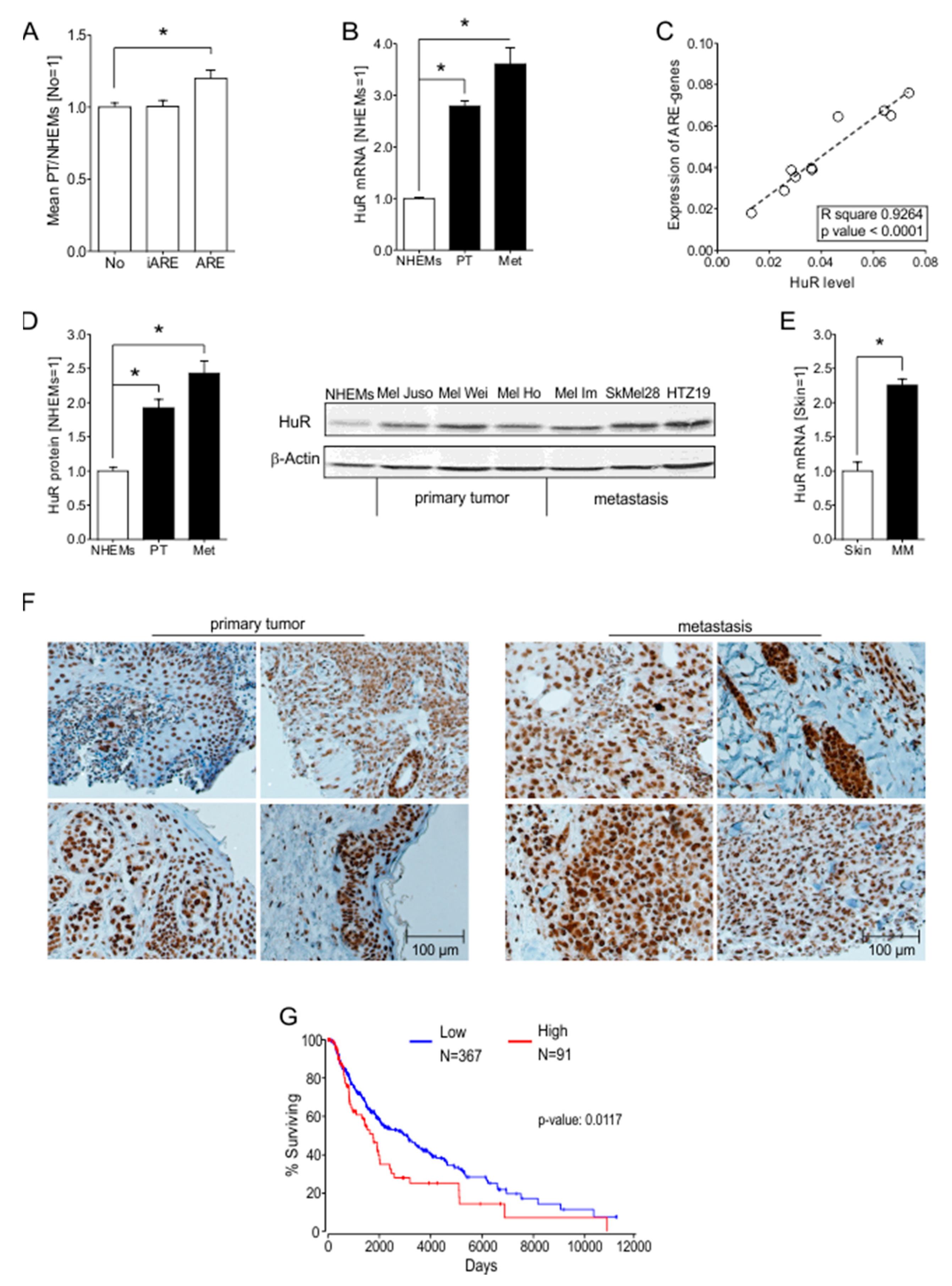
2.1. ARE Containing mRNAs Are More Abundant in Melanoma Cells Compared to NHEMs
2.2. HuR (ELAVL1) Is Upregulated in Malignant Melanoma In Vitro and In Vivo
2.3. The Loss of miR-194-5p Expression in Melanoma Promotes the Upregulation of HuR
2.4. HuR Expression and Localization Changes in the Progression of MM In Vitro and In Vivo
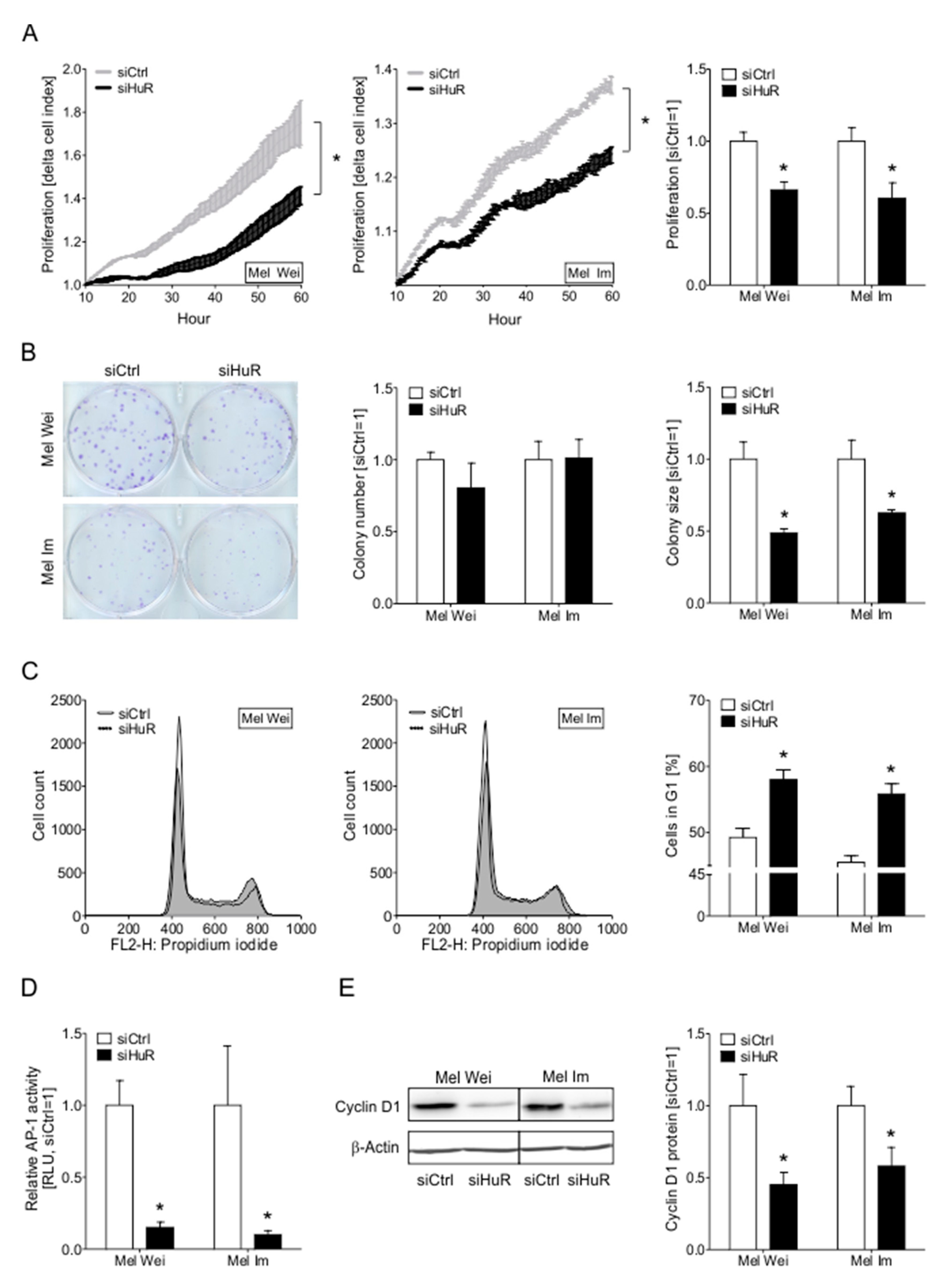
2.5. The Knockdown of HuR Leads to Decreased Proliferative Capacity of Melanoma Cells In Vitro
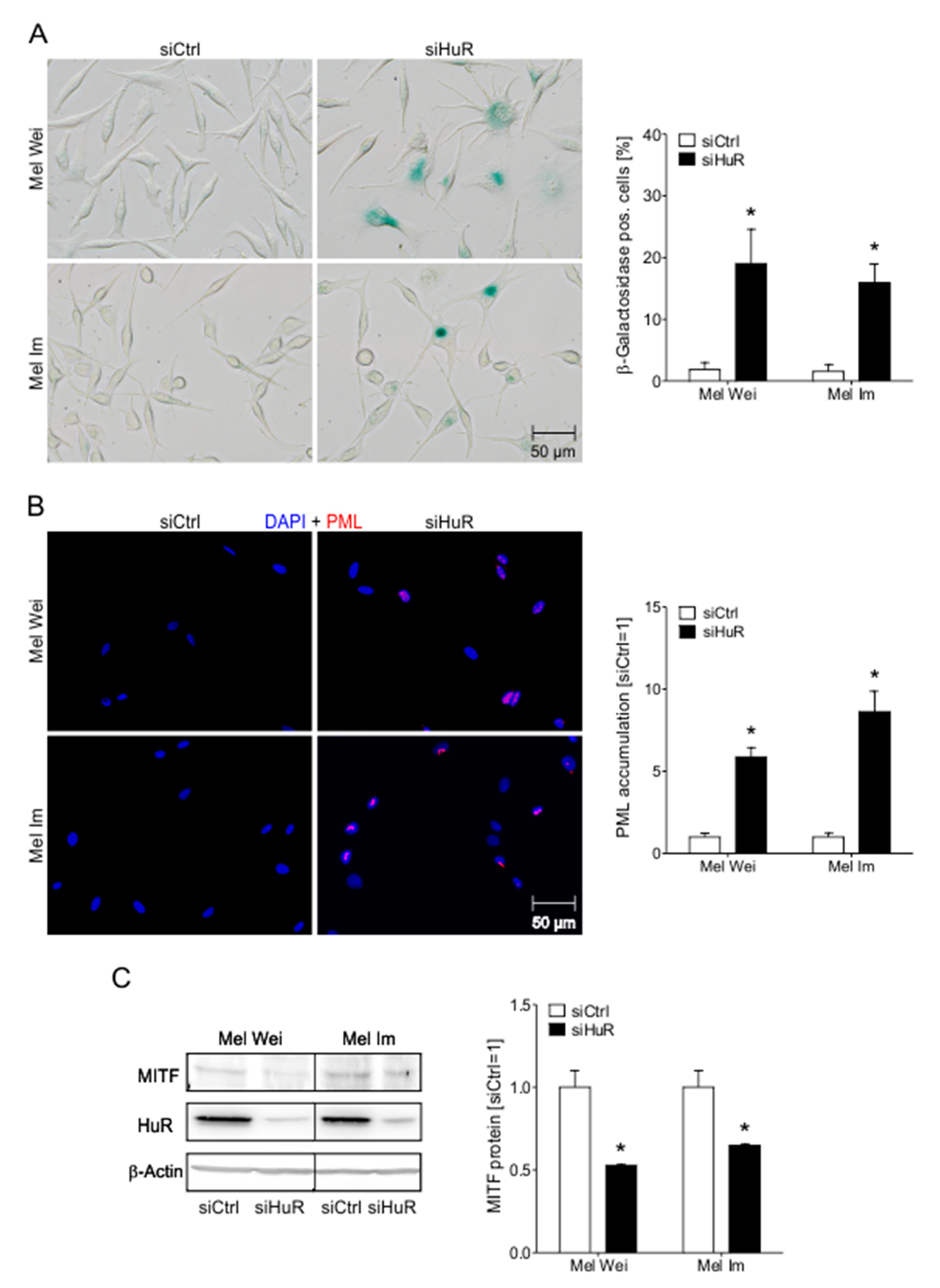
2.6. HuR Knockdown Induces Senescence Characteristics in Human Melanoma Cells
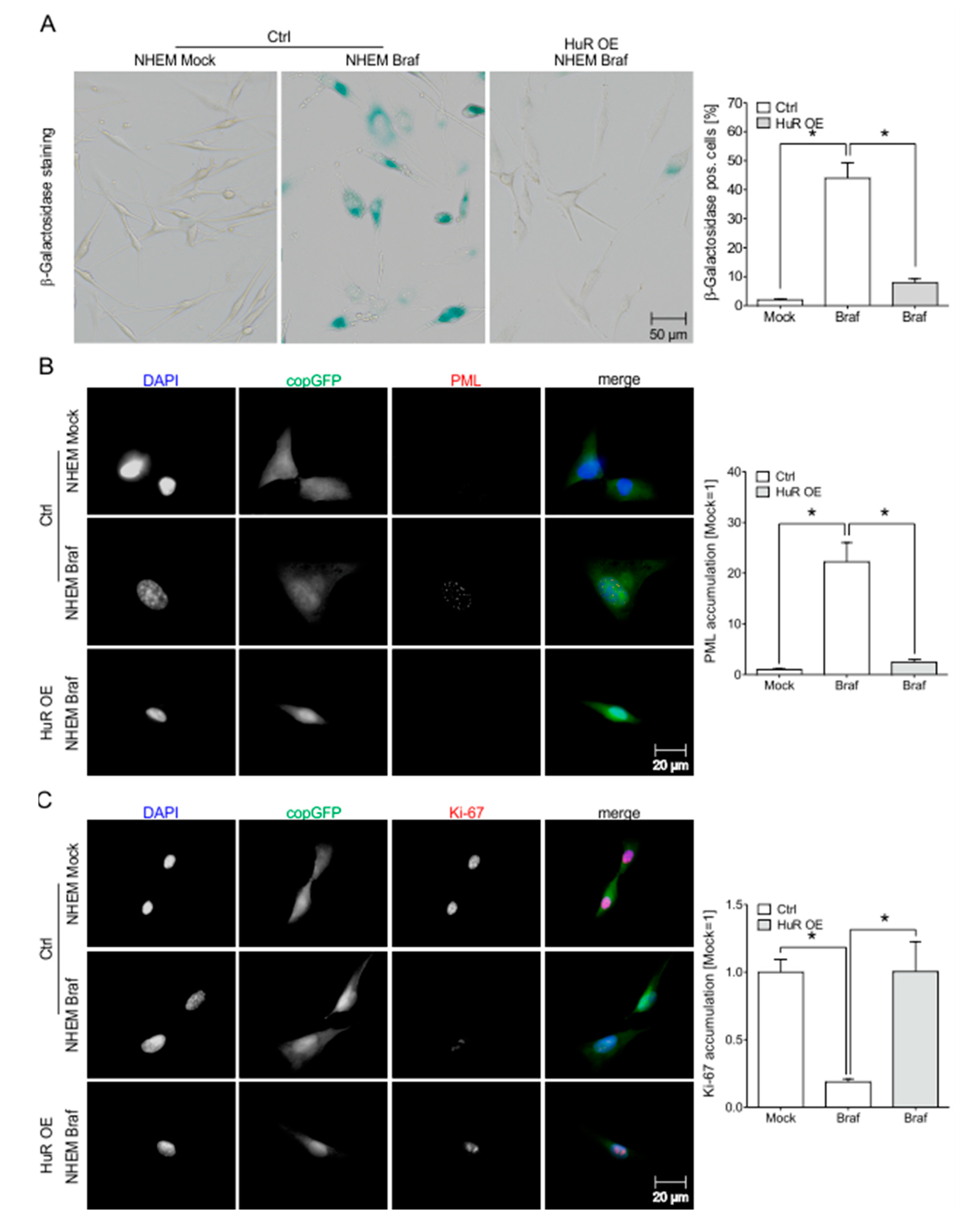
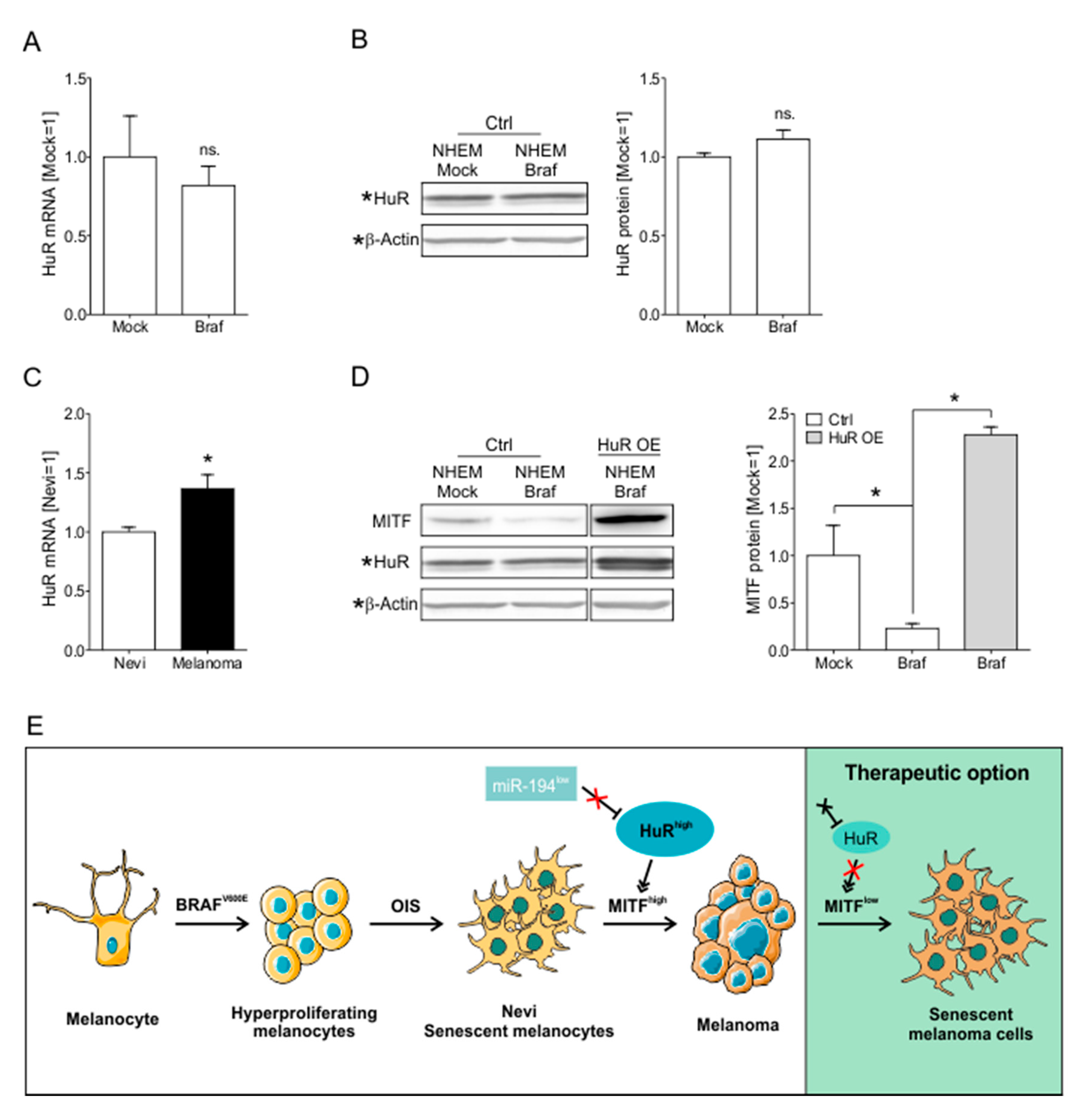
2.7. HuR Overexpression in NHEM BRAFV600E Cells Is Capable to Bypass Oncogene Induced Senescence by Induction of an MITFhigh Phenotype
3. Discussion
4. Material and Methods
4.1. Cell Lines and Cell Culture Conditions
4.2. Lentiviral Transduction
4.3. siRNA Transfection
4.4. miRNA Mimic Transfection
4.5. mRNA + miRNA Expression Analysis
4.6. Subcellular Fractionation
4.7. Western Blotting
4.8. Immunohistochemical Analysis
4.9. Immunofluorescence Staining
4.10. Analysis of Cell Proliferation
4.11. Clonogenic Assay
4.12. Analysis of Apoptosis by Flow Cytometry
4.13. Analysis of Intracellular ROS Content by Flow Cytometry
4.14. Luciferase Assay
4.15. Senescence-Associated ß-Galactosidase Staining
4.16. Schematic Illustrations
4.17. Statistical Analysis
4.18. Patients’ Consent
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Bernatchez, C.; Cooper, Z.A.; Wargo, J.A.; Hwu, P.; Lizee, G. Novel Treatments in Development for Melanoma. Cancer Treat. Res. 2016, 167, 371–416. [Google Scholar] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Pollock, P.M.; Harper, U.L.; Hansen, K.S.; Yudt, L.M.; Stark, M.; Robbins, C.M.; Moses, T.Y.; Hostetter, G.; Wagner, U.; Kakareka, J.; et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003, 33, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Abildgaard, C.; Guldberg, P. Molecular drivers of cellular metabolic reprogramming in melanoma. Trends Mol. Med. 2015, 21, 164–171. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The essence of senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, W.; Doller, A.; Akool, E.S.; Pfeilschifter, J. Modulation of mRNA stability as a novel therapeutic approach. Pharmacol. Ther. 2007, 114, 56–73. [Google Scholar] [CrossRef]
- Ivanov, P.; Anderson, P. Post-transcriptional regulatory networks in immunity. Immunol. Rev. 2013, 253, 253–272. [Google Scholar] [CrossRef] [PubMed]
- Deschenes-Furry, J.; Angus, L.M.; Belanger, G.; Mwanjewe, J.; Jasmin, B.J. Role of ELAV-like RNA-binding proteins HuD and HuR in the post-transcriptional regulation of acetylcholinesterase in neurons and skeletal muscle cells. Chem. Biol. Interact. 2005, 157–158, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, M.; Bono, P.; Narko, K.; Chang, S.H.; Lundin, J.; Joensuu, H.; Furneaux, H.; Hla, T.; Haglund, C.; Ristimaki, A. Cytoplasmic HuR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. 2005, 65, 2157–2161. [Google Scholar] [CrossRef] [PubMed]
- Lopez de Silanes, I.; Fan, J.; Yang, X.; Zonderman, A.B.; Potapova, O.; Pizer, E.S.; Gorospe, M. Role of the RNA-binding protein HuR in colon carcinogenesis. Oncogene 2003, 22, 7146–7154. [Google Scholar] [CrossRef] [PubMed]
- Blaxall, B.C.; Dwyer-Nield, L.D.; Bauer, A.K.; Bohlmeyer, T.J.; Malkinson, A.M.; Port, J.D. Differential expression and localization of the mRNA binding proteins, AU-rich element mRNA binding protein (AUF1) and Hu antigen R (HuR), in neoplastic lung tissue. Mol. Carcinog. 2000, 28, 76–83. [Google Scholar] [CrossRef]
- Liaudet, N.; Fernandez, M.; Fontao, L.; Kaya, G.; Merat, R. Hu antigen R (HuR) heterogeneous expression quantification as a prognostic marker of melanoma. J. Cutan. Pathol. 2018, 45, 333–336. [Google Scholar] [CrossRef]
- Dietrich, P.; Kuphal, S.; Spruss, T.; Hellerbrand, C.; Bosserhoff, A.K. MicroRNA-622 is a novel mediator of tumorigenicity in melanoma by targeting Kirsten rat sarcoma. Pigment. Cell Melanoma Res. 2018, 31, 614–629. [Google Scholar] [CrossRef]
- Xu, F.; Zhang, X.; Lei, Y.; Liu, X.; Liu, Z.; Tong, T.; Wang, W. Loss of repression of HuR translation by miR-16 may be responsible for the elevation of HuR in human breast carcinoma. J. Cell Biochem. 2010, 111, 727–734. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Mercken, E.M.; Brennan, S.E.; Wilson, G.M.; Cabo, R.; Gorospe, M. miR-519 suppresses tumor growth by reducing HuR levels. Cell Cycle 2010, 9, 1354–1359. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Wu, Y.; Hartley, R.S. MicroRNA-125a represses cell growth by targeting HuR in breast cancer. RNA Biol. 2009, 6, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Al-Ahmadi, W.; Al-Ghamdi, M.; Al-Souhibani, N.; Khabar, K.S. miR-29a inhibition normalizes HuR over-expression and aberrant AU-rich mRNA stability in invasive cancer. J. Pathol. 2013, 230, 28–38. [Google Scholar] [CrossRef]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef]
- Haferkamp, S.; Borst, A.; Adam, C.; Becker, T.M.; Motschenbacher, S.; Windhovel, S.; Hufnagel, A.L.; Houben, R.; Meierjohann, S. Vemurafenib induces senescence features in melanoma cells. J. Investig. Dermatol. 2013, 133, 1601–1609. [Google Scholar] [CrossRef]
- Bernardi, R.; Pandolfi, P.P. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol 2007, 8, 1006–1016. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Kunz, M.; Loffler-Wirth, H.; Dannemann, M.; Willscher, E.; Doose, G.; Kelso, J.; Kottek, T.; Nickel, B.; Hopp, L.; Landsberg, J.; et al. RNA-seq analysis identifies different transcriptomic types and developmental trajectories of primary melanomas. Oncogene 2018, 37, 6136–6151. [Google Scholar] [CrossRef]
- Wurth, L. Versatility of RNA-Binding Proteins in Cancer. Comp. Funct. Genom. 2012, 2012, 178525. [Google Scholar] [CrossRef]
- Moore, M.J. From birth to death: The complex lives of eukaryotic mRNAs. Science 2005, 309, 1514–1518. [Google Scholar] [CrossRef]
- Kim, M.Y.; Hur, J.; Jeong, S. Emerging roles of RNA and RNA-binding protein network in cancer cells. BMB Rep. 2009, 42, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Silvera, D.; Formenti, S.C.; Schneider, R.J. Translational control in cancer. Nat. Rev. Cancer 2010, 10, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Moradi, F.; Berglund, P.; Linnskog, R.; Leandersson, K.; Andersson, T.; Prasad, C.P. Dual mechanisms of action of the RNA-binding protein human antigen R explains its regulatory effect on melanoma cell migration. Transl. Res. 2016, 172, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Hatanaka, T.; Higashino, F.; Tei, K.; Yasuda, M. The neural ELAVL protein HuB enhances endogenous proto-oncogene activation. Biochem. Biophys. Res. Commun. 2019, 517, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Battaglia-Hsu, S.F.; Ghemrawi, R.; Coelho, D.; Dreumont, N.; Mosca, P.; Hergalant, S.; Gauchotte, G.; Sequeira, J.M.; Ndiongue, M.; Houlgatte, R.; et al. Inherited disorders of cobalamin metabolism disrupt nucleocytoplasmic transport of mRNA through impaired methylation/phosphorylation of ELAVL1/HuR. Nucleic Acids Res. 2018, 46, 7844–7857. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, S.; Zheng, M.; Kuver, A.; Wan, X.; Dai, K.; Li, X. Phosphorylation of ELAVL1 (Ser219/Ser316) mediated by PKC is required for erythropoiesis. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 214–224. [Google Scholar] [CrossRef]
- Denkert, C.; Weichert, W.; Winzer, K.J.; Muller, B.M.; Noske, A.; Niesporek, S.; Kristiansen, G.; Guski, H.; Dietel, M.; Hauptmann, S. Expression of the ELAV-like protein HuR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin. Cancer Res. 2004, 10, 5580–5586. [Google Scholar] [CrossRef]
- Sohn, B.H.; Park, I.Y.; Lee, J.J.; Yang, S.J.; Jang, Y.J.; Park, K.C.; Kim, D.J.; Lee, D.C.; Sohn, H.A.; Kim, T.W.; et al. Functional switching of TGF-beta1 signaling in liver cancer via epigenetic modulation of a single CpG site in TTP promoter. Gastroenterology 2010, 138, 1898–1908. [Google Scholar] [CrossRef]
- Abdelmohsen, K.; Srikantan, S.; Kuwano, Y.; Gorospe, M. miR-519 reduces cell proliferation by lowering RNA-binding protein HuR levels. Proc. Natl. Acad. Sci. USA 2008, 105, 20297–20302. [Google Scholar] [CrossRef]
- Stark, M.S.; Tyagi, S.; Nancarrow, D.J.; Boyle, G.M.; Cook, A.L.; Whiteman, D.C.; Parsons, P.G.; Schmidt, C.; Sturm, R.A.; Hayward, N.K. Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS ONE 2010, 5, e9685. [Google Scholar] [CrossRef]
- Poell, J.B.; van Haastert, R.J.; de Gunst, T.; Schultz, I.J.; Gommans, W.M.; Verheul, M.; Cerisoli, F.; van Puijenbroek, A.; van Noort, P.I.; Prevost, G.P.; et al. Correction: A Functional Screen Identifies Specific MicroRNAs Capable of Inhibiting Human Melanoma Cell Viability. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, S.; Ma, P.; Jing, Y.; Peng, H.; Gao, W.Q.; Zhuang, G. Lin28B promotes melanoma growth by mediating a microRNA regulatory circuit. Carcinogenesis 2015, 36, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Fasano, E.; Piccioni, E.; Monego, G.; Cittadini, A.R.; Celleno, L.; Ranelletti, F.O.; Calviello, G. DHA induces apoptosis and differentiation in human melanoma cells in vitro: Involvement of HuR-mediated COX-2 mRNA stabilization and beta-catenin nuclear translocation. Carcinogenesis 2012, 33, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Yang, X.; Cristofalo, V.J.; Holbrook, N.J.; Gorospe, M. Loss of HuR is linked to reduced expression of proliferative genes during replicative senescence. Mol. Cell Biol. 2001, 21, 5889–5898. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lal, A.; Mazan-Mamczarz, K.; Kawai, T.; Yang, X.; Martindale, J.L.; Gorospe, M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004, 23, 3092–3102. [Google Scholar] [CrossRef]
- Del Vecchio, G.; De Vito, F.; Saunders, S.J.; Risi, A.; Mannironi, C.; Bozzoni, I.; Presutti, C. RNA-binding protein HuR and the members of the miR-200 family play an unconventional role in the regulation of c-Jun mRNA. RNA 2016, 22, 1510–1521. [Google Scholar] [CrossRef]
- Merat, R.; Bugi-Marteyn, A.; Wrobel, L.J.; Py, C.; Daali, Y.; Schwarzler, C.; Liaudet, N. Drug-induced expression of the RNA-binding protein HuR attenuates the adaptive response to BRAF inhibition in melanoma. Biochem. Biophys. Res. Commun. 2019, 517, 181–187. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, M.; Hong, J.; Kim, M.K.; Chung, I.K. Loss of RNA-binding protein HuR facilitates cellular senescence through posttranscriptional regulation of TIN2 mRNA. Nucleic Acids Res. 2018, 46, 4271–4285. [Google Scholar] [CrossRef]
- Hoek, K.S.; Eichhoff, O.M.; Schlegel, N.C.; Dobbeling, U.; Kobert, N.; Schaerer, L.; Hemmi, S.; Dummer, R. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 2008, 68, 650–656. [Google Scholar] [CrossRef]
- Bertolotto, C.; Lesueur, F.; Giuliano, S.; Strub, T.; de Lichy, M.; Bille, K.; Dessen, P.; d’Hayer, B.; Mohamdi, H.; Remenieras, A.; et al. A SUMOylation-defective MITF germline mutation predisposes to melanoma and renal carcinoma. Nature 2011, 480, 94–98. [Google Scholar] [CrossRef]
- Hartman, M.L.; Czyz, M. MITF in melanoma: Mechanisms behind its expression and activity. Cell Mol. Life Sci. 2015, 72, 1249–1260. [Google Scholar] [CrossRef] [PubMed]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: A role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Meriin, A.B.; Gabai, V.L.; Christians, E.; Benjamin, I.; Wilson, A.; Wolozin, B.; Sherman, M.Y. The heat shock transcription factor Hsf1 is downregulated in DNA damage-associated senescence, contributing to the maintenance of senescence phenotype. Aging Cell 2012, 11, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Meisner, N.C.; Hintersteiner, M.; Mueller, K.; Bauer, R.; Seifert, J.M.; Naegeli, H.U.; Ottl, J.; Oberer, L.; Guenat, C.; Moss, S.; et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat. Chem. Biol. 2007, 3, 508–515. [Google Scholar] [CrossRef]
- Wu, X.; Lan, L.; Wilson, D.M.; Marquez, R.T.; Tsao, W.C.; Gao, P.; Roy, A.; Turner, B.A.; McDonald, P.; Tunge, J.A.; et al. Identification and validation of novel small molecule disruptors of HuR-mRNA interaction. ACS Chem. Biol. 2015, 10, 1476–1484. [Google Scholar] [CrossRef]
- Blanco, F.F.; Preet, R.; Aguado, A.; Vishwakarma, V.; Stevens, L.E.; Vyas, A.; Padhye, S.; Xu, L.; Weir, S.J.; Anant, S.; et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget 2016, 7, 74043–74058. [Google Scholar] [CrossRef]
- Lang, M.; Berry, D.; Passecker, K.; Mesteri, I.; Bhuju, S.; Ebner, F.; Sedlyarov, V.; Evstatiev, R.; Dammann, K.; Loy, A.; et al. HuR Small-Molecule Inhibitor Elicits Differential Effects in Adenomatosis Polyposis and Colorectal Carcinogenesis. Cancer Res. 2017, 77, 2424–2438. [Google Scholar] [CrossRef]
- Braig, S.; Mueller, D.W.; Bosserhoff, A.K. Micro RNA miR-196a is a central regulator of HOX-B7 and BMP4 expression in malignant melanoma. Cell. Mol. Life Sci. 2010, 67, 3535–3548. [Google Scholar] [CrossRef]
- Feuerer, L.; Lamm, S.; Henz, I.; Kappelmann-Fenzl, M.; Haferkamp, S.; Meierjohann, S.; Hellerbrand, C.; Kuphal, S.; Bosserhoff, A.K. Role of melanoma inhibitory activity in melanocyte senescence. Pigment. Cell Melanoma Res. 2019, 32, 777–791. [Google Scholar] [CrossRef]
- Ma, W.; Mayr, C. A Membraneless Organelle Associated with the Endoplasmic Reticulum Enables 3′UTR-Mediated Protein-Protein Interactions. Cell 2018, 175, 1492–1506.e19. [Google Scholar] [CrossRef]
- Hannus, M.; Beitzinger, M.; Engelmann, J.C.; Weickert, M.T.; Spang, R.; Hannus, S.; Meister, G. siPools: Highly complex but accurately defined siRNA pools eliminate off-target effects. Nucleic Acids Res. 2014, 42, 8049–8061. [Google Scholar] [CrossRef]
- Schiffner, S.; Braunger, B.M.; de Jel, M.M.; Coupland, S.E.; Tamm, E.R.; Bosserhoff, A.K. Tg(Grm1) transgenic mice: A murine model that mimics spontaneous uveal melanoma in humans? Exp. Eye Res. 2014, 127, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Kuphal, S.; Spruss, T.; Hellerbrand, C.; Bosserhoff, A.K. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene 2018, 37, 897–911. [Google Scholar] [CrossRef]
- Ott, C.A.; Linck, L.; Kremmer, E.; Meister, G.; Bosserhoff, A.K. Induction of exportin-5 expression during melanoma development supports the cellular behavior of human malignant melanoma cells. Oncotarget 2016, 7, 62292–62304. [Google Scholar] [CrossRef]
- Linck, L.; Liebig, J.; Voller, D.; Eichner, N.; Lehmann, G.; Meister, G.; Bosserhoff, A. MicroRNA-sequencing data analyzing melanoma development and progression. Exp. Mol. Pathol. 2018, 105, 371–379. [Google Scholar] [CrossRef]
- Bohme, I.; Bosserhoff, A. Extracellular acidosis triggers a senescence-like phenotype in human melanoma cells. Pigment. Cell Melanoma Res. 2020, 33, 41–51. [Google Scholar] [CrossRef]
- Ruedel, A.; Dietrich, P.; Schubert, T.; Hofmeister, S.; Hellerbrand, C.; Bosserhoff, A.K. Expression and function of microRNA-188-5p in activated rheumatoid arthritis synovial fibroblasts. Int. J. Clin. Exp. Pathol. 2015, 8, 6607–6616. [Google Scholar]
- Arndt, S.; Wacker, E.; Li, Y.F.; Shimizu, T.; Thomas, H.M.; Morfill, G.E.; Karrer, S.; Zimmermann, J.L.; Bosserhoff, A.K. Cold atmospheric plasma, a new strategy to induce senescence in melanoma cells. Exp. Dermatol. 2013, 22, 284–289. [Google Scholar] [CrossRef]
- Kappelmann, M.; Kuphal, S.; Meister, G.; Vardimon, L.; Bosserhoff, A.K. MicroRNA miR-125b controls melanoma progression by direct regulation of c-Jun protein expression. Oncogene 2013, 32, 2984–2991. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Roff, A.N.; Craig, T.J.; August, A.; Stellato, C.; Ishmael, F.T. MicroRNA-570-3p regulates HuR and cytokine expression in airway epithelial cells. Am. J. Clin. Exp. Immunol. 2014, 3, 68–83. [Google Scholar] [PubMed]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput. Sci. 2016, 2, e67. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liebig, J.K.; Kuphal, S.; Bosserhoff, A.K. HuRdling Senescence: HuR Breaks BRAF-Induced Senescence in Melanocytes and Supports Melanoma Growth. Cancers 2020, 12, 1299. https://doi.org/10.3390/cancers12051299
Liebig JK, Kuphal S, Bosserhoff AK. HuRdling Senescence: HuR Breaks BRAF-Induced Senescence in Melanocytes and Supports Melanoma Growth. Cancers. 2020; 12(5):1299. https://doi.org/10.3390/cancers12051299
Chicago/Turabian StyleLiebig, Janika K., Silke Kuphal, and Anja Katrin Bosserhoff. 2020. "HuRdling Senescence: HuR Breaks BRAF-Induced Senescence in Melanocytes and Supports Melanoma Growth" Cancers 12, no. 5: 1299. https://doi.org/10.3390/cancers12051299
APA StyleLiebig, J. K., Kuphal, S., & Bosserhoff, A. K. (2020). HuRdling Senescence: HuR Breaks BRAF-Induced Senescence in Melanocytes and Supports Melanoma Growth. Cancers, 12(5), 1299. https://doi.org/10.3390/cancers12051299