Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome



Abstract
1. Introduction
2. Results
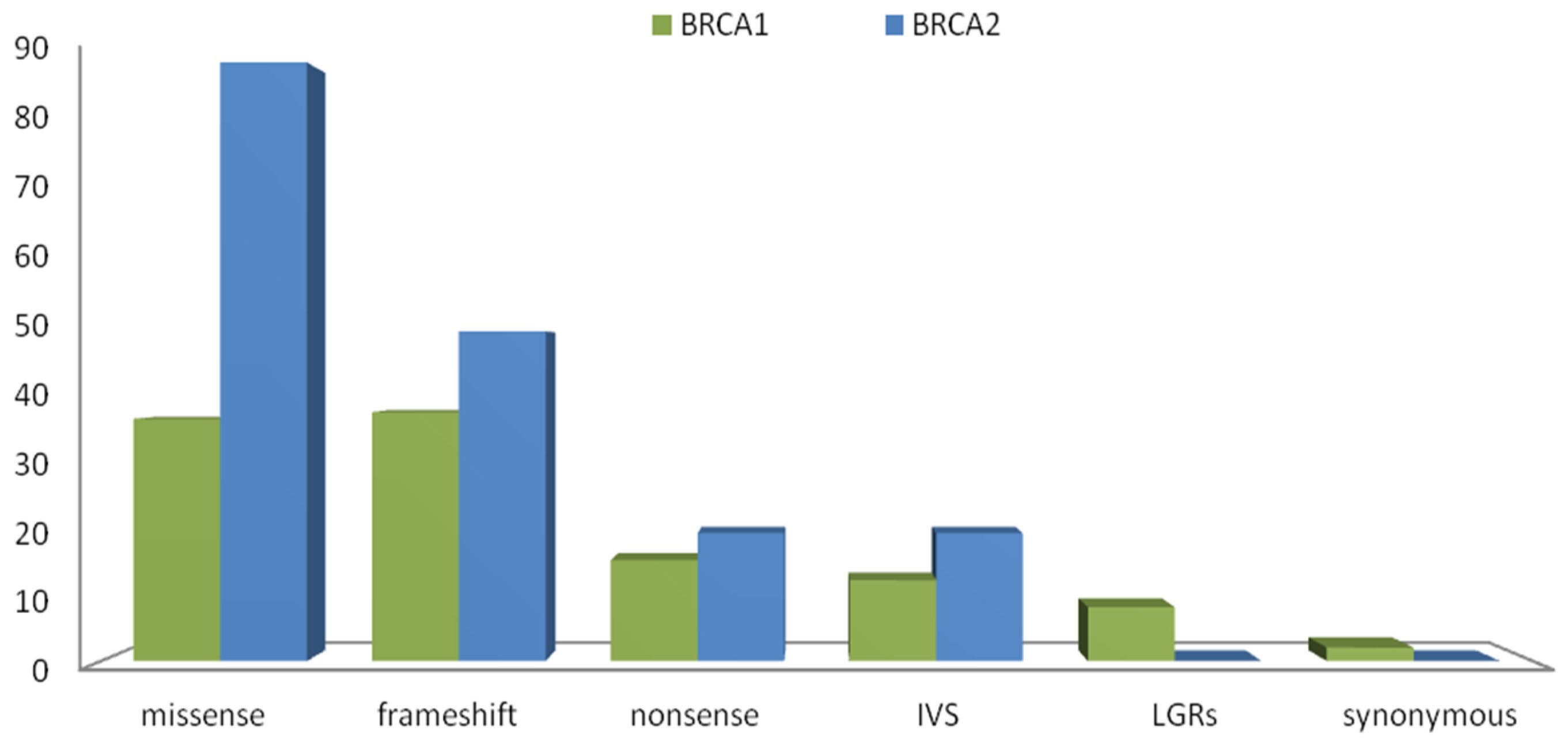
2.1. Results Next-Generation Sequencing
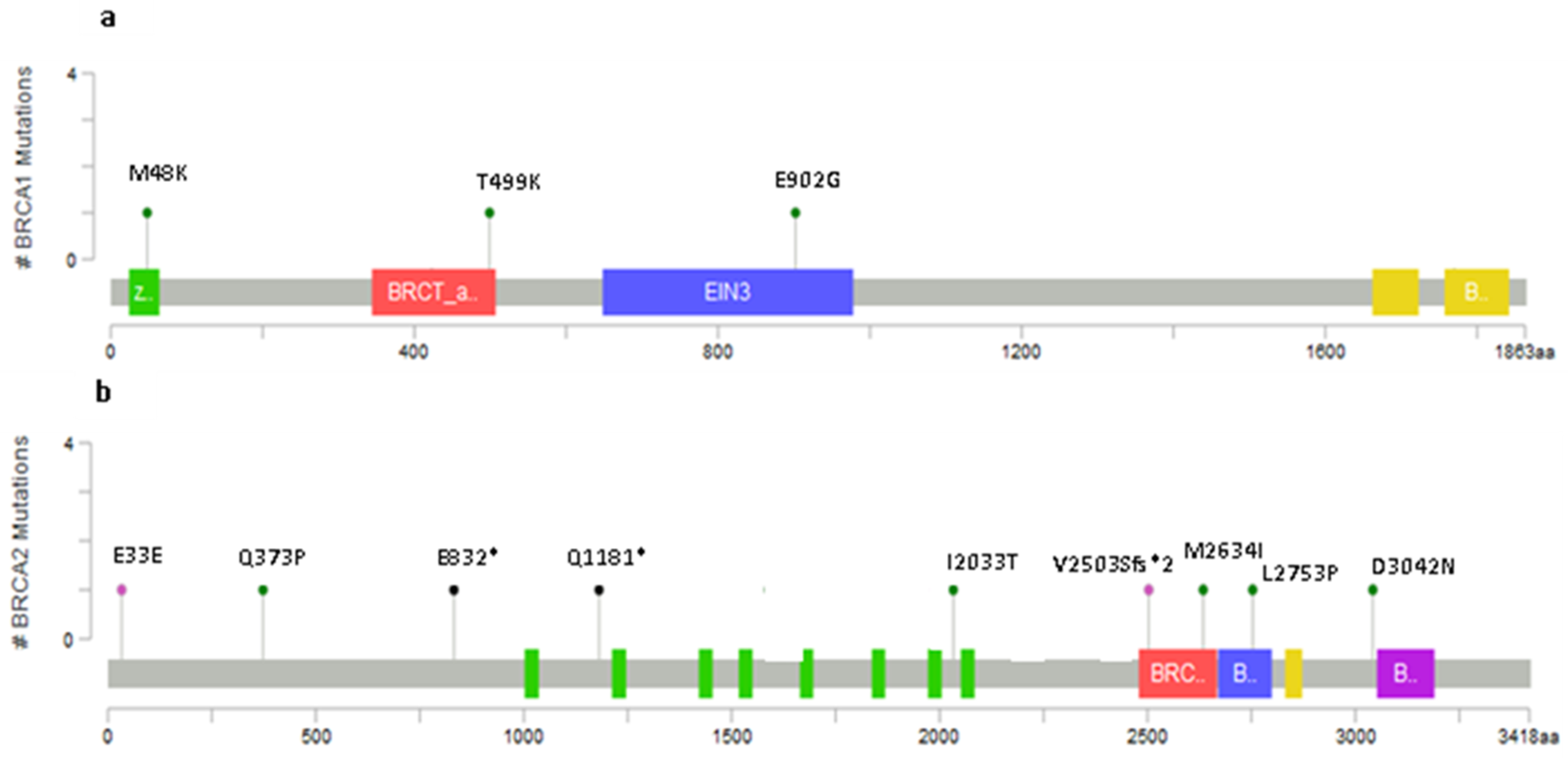
2.2. Novel Variants
2.3. Recurring Variants
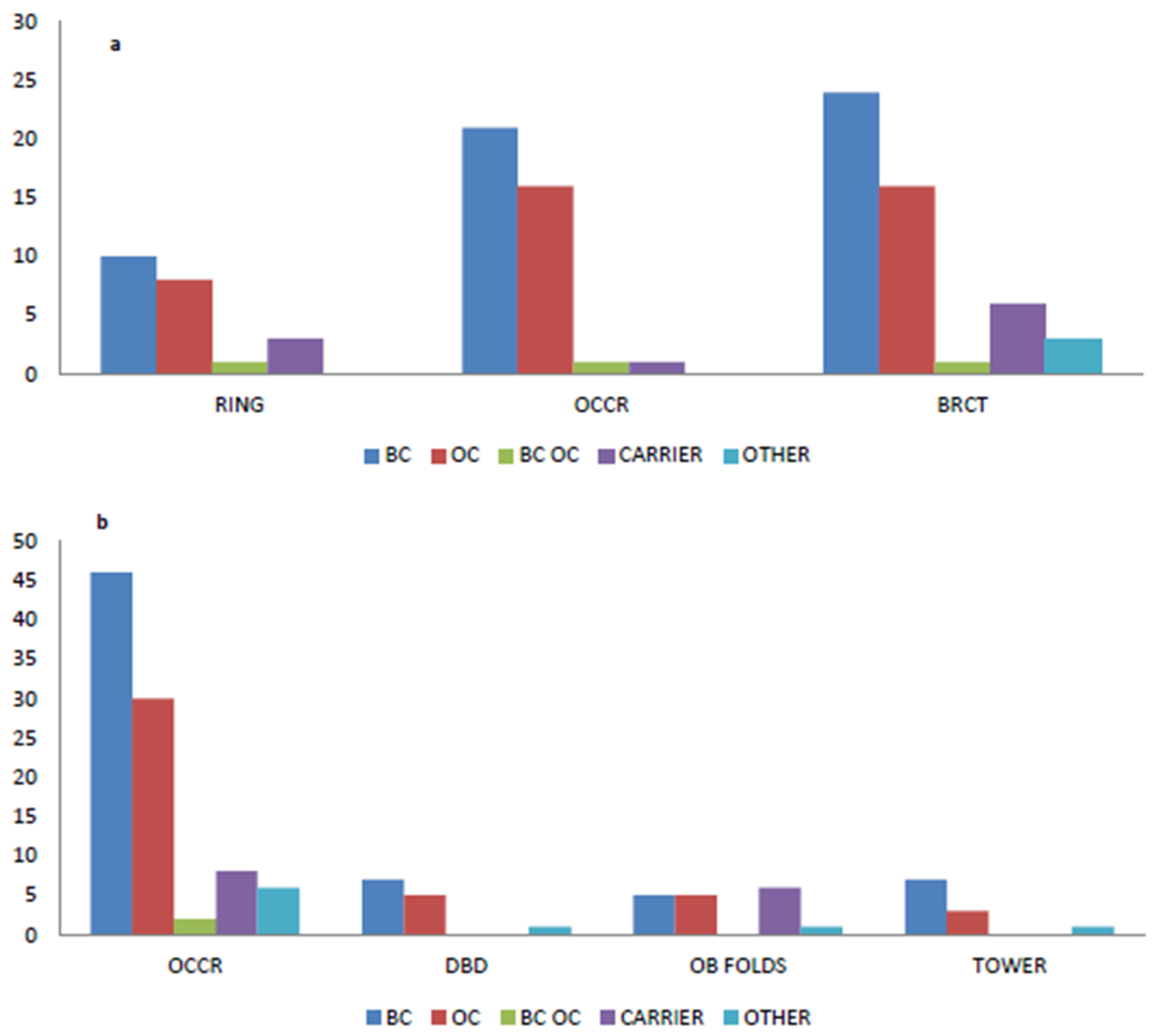
2.4. Variants Location and Type of Cancer
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Molecular Testing
DNA Extraction and Next-Generation Sequencing (NGS) Pipeline
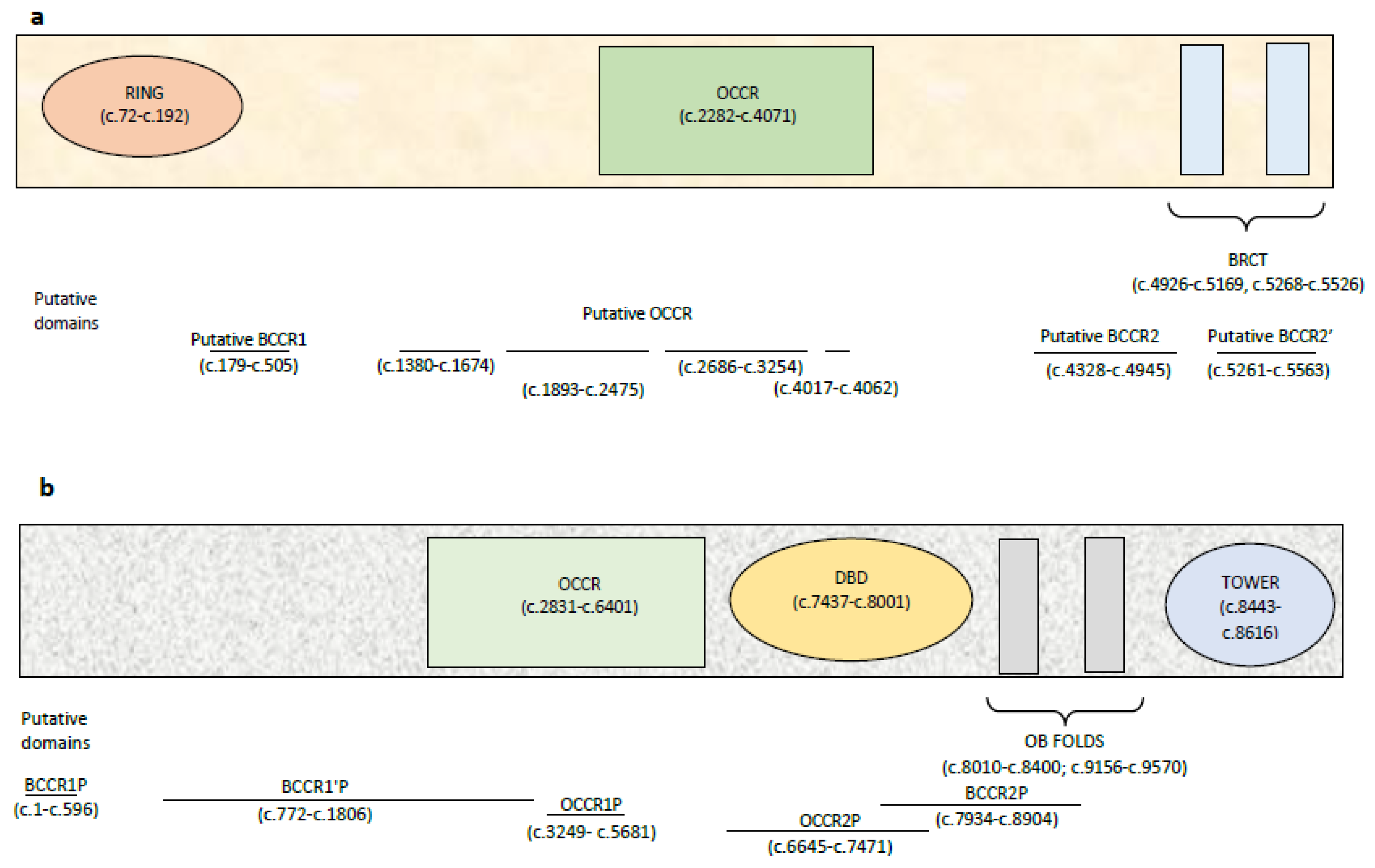
4.3. BRCA1 and BRCA2 Cluster Regions
4.4. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Tutt, A.; Ashworth, A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat. Rev. Cancer 2004, 4, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Venkitaraman, A.R. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002, 108, 171–182. [Google Scholar] [CrossRef]
- Lakhani, S.R.; Reis-Filho, J.S.; Fulford, L.; Penault-Llorca, F.; Van der Vijver, M.; Parry, S.; Bishop, T.; Benitez, J.; Rivas, C.; Chang-Claude, J.; et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005, 11, 5175–5180. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Marks, J.H.; Mandell, J.B. New York Breast Cancer Study Group Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; Van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. J. Am. Med. Assoc. 2017, 317, 2402–2416. [Google Scholar] [CrossRef]
- Edwards, S.M.; Evans, D.G.; Hope, Q.; Norman, A.R.; Barbachano, Y.; Bullock, S.; Kote-Jarai, Z.; Meitz, J.; Falconer, A.; Osin, P.; et al. Prostate cancer in BRCA2 germline mutation carriers is associated with poorer prognosis. Br. J. Cancer 2010, 103, 918–924. [Google Scholar] [CrossRef]
- Iqbal, J.; Ragone, A.; Lubinski, J.; Lynch, H.T.; Moller, P.; Ghadirian, P.; Foulkes, W.D.; Armel, S.; Eisen, A.; Senter, L.; et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012, 107, 2005–2009. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Voelkerding, K.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- ENIGMA (Evidence-Based Network for the Interpretation of Germline Mutant Alleles). Available online: https://enigmaconsortium.org/ (accessed on 27 March 2020).
- Parsons, M.T.; Tudini, E.; Li, H.; Hahnen, E.; Wappenschmidt, B.; Feliubadaló, L.; Aalfs, C.M.; Agata, S.; Aittomäki, K.; Alonso-Cerezo, M.C.; et al. Large scale multifactorial likelihood quantitative analysis of BRCA1 and BRCA2 variants: An ENIGMA resource to supportclinical variant classification. Hum. Mutat. 2019, 40, 1557–1578. [Google Scholar] [CrossRef]
- Santarosa, M. BRCA1 and BRCA2 genes: Role in hereditary breast and ovarian cancer in Italy. Int. J. Cancer 1999, 83, 5–9. [Google Scholar] [CrossRef]
- Ottini, L.; D’Amico, C.; Noviello, C.; Lauro, S.; Lalle, M.; Fornarini, G.; Colantuoni, O.A.; Pizzi, C.; Cortesi, E.; Carlini, S.; et al. BRCA1 and BRCA2 mutations in central and southern Italian patients. Breast Cancer Res. 2000, 2, 307. [Google Scholar] [CrossRef] [PubMed]
- Ottini, L.; Masala, G.; D’Amico, C.; Mancini, B.; Saieva, C.; Aceto, G.; Gestri, D.; Vezzosi, V.; Falchetti, M.; de Marco, M.; et al. BRCA1 and BRCA2 mutation status and tumor characteristics in male breast cancer: A population-based study in Italy. Cancer Res. 2003, 63, 342–347. [Google Scholar] [PubMed]
- Tommasi, S.; Crapolicchio, A.; Lacalamita, R.; Bruno, M.; Monaco, A.; Petroni, S.; Schittulli, F.; Longo, S.; Digennaro, M.; Calistri, D.; et al. BRCA1 mutations and polymorphisms in a hospital-based consecutive series of breast cancer patients from Apulia, Italy. Mutat. Res. 2005, 578, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Baudi, F.; Quaresima, B.; Grandinetti, C.; Cuda, G.; Faniello, C.; Tassone, P.; Barbieri, V.; Bisegna, R.; Ricevuto, E.; Conforti, S.; et al. Evidence of a founder mutation of BRCA1 in a highly homogeneous population from southern Italy with breast/ovarian cancer. Hum. Mutat. 2001, 18, 163–164. [Google Scholar] [CrossRef]
- Palomba, G.; Pisano, M.; Cossu, A.; Budroni, M.; Dedola, M.F.; Farris, A.; Contu, A.; Baldinu, P.; Tanda, F.; Palmieri, G. Spectrum and prevalence of BRCA1 and BRCA2 germline mutations in Sardinian patients with breast carcinoma through hospital-based screening. Cancer 2005, 104, 1172–1179. [Google Scholar] [CrossRef]
- Giannini, G.; Capalbo, C.; Ristori, E.; Ricevuto, E.; Sidoni, T.; Buffone, A.; Cortesi, E.; Marchetti, P.; Scambia, G.; Tomao, S.; et al. Novel BRCA1 and BRCA2 germline mutations and assessment of mutation spectrum and prevalence in Italian breast and/or ovarian cancer families. Breast Cancer Res. Treat. 2006, 100, 83–91. [Google Scholar] [CrossRef]
- Malacrida, S.; Agata, S.; Callegaro, M.; Casella, C.; Barana, D.; Scaini, M.C.; Oliani, C.; Radice, P.; Barile, M.; Menin, C.; et al. BRCA1 p.Val1688del is a deleterious mutation that recurs in breast and ovarian cancer families from Northeast Italy. J. Clin. Oncol. 2008, 26, 26–31. [Google Scholar] [CrossRef]
- Taylor, K.N.; Eskande, R.N. PARP inhibitors in epithelial ovarian cancer. Recent Pat. Anti Cancer Drug Discov. 2018, 13, 145–158. [Google Scholar] [CrossRef]
- Santonocito, C.; Scapaticci, M.; Guarino, D.; Bartolini, A.; Minucci, A.; Concolino, P.; Scambia, G.; Paris, I.; Capoluongo, E. Identification of twenty-nine novel germline unclassified variants of BRCA1 and BRCA2 genes in 1400 Italian individuals. Breast 2017, 36, 74–78. [Google Scholar] [CrossRef]
- Gori, S.; Barberis, M.; Bella, M.A.; Buttitta, F.; Capoluongo, E.; Carrera, P.; Colombo, N.; Cortesi, L.; Genuardi, M.; Gion, M.; et al. Recommendations for the implementation of BRCA testing in ovarian cancer patients and their relatives. Crit. Rev. Oncol. Hematol. 2019, 140, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Baer, R.; Lee, W.H. Functional domains of the BRCA1 and BRCA2 proteins. J. Mammary Gland Biol. Neoplasia 1998, 3, 403–412. [Google Scholar] [CrossRef] [PubMed]
- dbSNP Database. Available online: https://www.ncbi.nlm.nih.gov/snp (accessed on 27 March 2020).
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Nathanson, K.L.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.U.; Muhammad, N.; Naeemi, H.; Khan, F.A.; Hassan, M.; Faisal, S.; Gull, S.; Amin, A.; Loya, A.; Hamann, U. Spectrum and prevalence of BRCA1/2 germline mutations in Pakistani breast cancer patients: Results from a large comprehensive study. Hered. Cancer Clin. Pract. 2019, 17, 27. [Google Scholar] [CrossRef] [PubMed]
- Capalbo, C.; Ricevuto, E.; Vestri, A.; Ristori, E.; Sidoni, T.; Buffone, O.; Adamo, B.; Cortesi, E.; Marchetti, P.; Scambia, G.; et al. BRCA1 and BRCA2 genetic testing in Italian breast and/or ovarian cancer families: Mutation spectrum and prevalence and analysis of mutation prediction models. Ann. Oncol. 2006, 17 (Suppl. 7), vii34–vii40. [Google Scholar] [CrossRef] [PubMed]
- Hamel, N.; Feng, B.J.; Foretova, L.; Stoppa-Lyonnet, D.; Narod, S.A.; Imyanitov, E.; Sinilnikova, O.; Tihomirova, L.; Lubinski, J.; Gronwald, J.; et al. On the origin and diffusion of BRCA1 c.5266dupC (5382insC) in European populations. Eur. J. Hum. Genet. 2011, 19, 300–306. [Google Scholar] [CrossRef]
- Kaufman, B.; Laitman, Y.; Gronwald, J.; Lubinski, J.; Friedman, E. Haplotype of the C61G BRCA1 mutation in Polish and Jewish individuals. Genet. Test. Mol. Biomark. 2009, 13, 465–469. [Google Scholar] [CrossRef]
- Di Giacomo, D.; Calicchia, M.; Candria, S.; Bruera, G.; Ferella, S.; Lucci Cordisco, E.; Tosi, M.; Genuardi, M.; Ricevuto, E. Identification of the founder BRCA1 mutation c.4117G>T (p.Glu1373*) recurring in the Abruzzo and Lazio regions of Central Italy and predisposing to breast/ovarian and BRCA1-related cancers. Ann. Oncol. Abstr. 2019, 30, v767. [Google Scholar] [CrossRef]
- Nedelcu, R.; Liede, A.; Aubé, J.; Finch, A.; Kwan, E.; Jack, E.; Narod, S.A.; Randall, S.; Hugel, L.; Clark, K.; et al. BRCA mutations in Italian breast/ovarian cancer families. Eur. J. Hum. Genet. 2002, 10, 150–152. [Google Scholar] [CrossRef]
- Díez, O.; Osorio, A.; Durán, M.; Martinez-Ferrandis, J.I.; de la Hoya, M.; Salazar, R.; Vega, A.; Campos, B.; Rodríguez-López, R.; Velasco, E.; et al. Analysis of BRCA1 and BRCA2 genes in Spanish breast/ovarian cancer patients: A high proportion of mutations unique to Spain and evidence of founder effects. Hum. Mutat. 2003, 22, 301–312. [Google Scholar] [CrossRef]
- Shamoo, Y. Structural insights into BRCA2 function. Curr. Opin. Struct. Biol. 2003, 13, 206–211. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Janavičius, R. Founder BRCA1/2 mutations in the Europe: Implications for hereditary breast-ovarian cancer prevention and control. EPMA J. 2010, 1, 397–412. [Google Scholar] [CrossRef] [PubMed]
- National Comprehensive Cancer Network (NCCN). Available online: https://www.nccn.org/about/news/ebulletin/ebulletindetail.aspx?ebulletinid=535 (accessed on 27 March 2020).
- Concolino, P.; Gelli, G.; Rizza, R.; Costella, A.; Scambia, G.; Capoluongo, E. BRCA1 and BRCA2 testing through next-generation sequencing in a small cohort of Italian breast/ovarian cancer patients: Novel pathogenic and unknown clinical significance variants. Int. J. Mol. Sci. 2019, 20, 3442. [Google Scholar] [CrossRef] [PubMed]
- Concolino, P.; Capoluongo, E. Detection of BRCA1/2 large genomic rearrangements in breast and ovarian cancer patients: An overview of the current methods. Expert Rev. Mol. Diagn. 2019, 19, 795–802. [Google Scholar] [CrossRef]
- Rizza, R.; Hackmann, K.; Paris, I.; Minucci, A.; De Leo, R.; Schrock, E.; Urbani, A.; Capoluongo, E.; Gelli, G.; Concolino, P. Novel BRCA1 large genomic rearrangements in Italian breast/ovarian cancer patients. Mol. Diagn. Ther. 2019, 23, 121–126. [Google Scholar] [CrossRef]
- Human Genome Variation Sequence Systematic Nomenclature. Available online: http://www.hgvs.org/mutnomen/ (accessed on 27 March 2020).
- ClinVar. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 27 March 2020).
- LOVD v3.0. Available online: www.lovd.nl (accessed on 27 March 2020).
- HGMD. Available online: http://www.hgmd.cf.ac.uk/ac/ (accessed on 27 March 2020).
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Béroud, C.; Letovsky, S.I.; Braastad, C.D.; Caputo, S.M.; Beaudoux, O.; Bignon, Y.; Bressac-De Paillerets, B.; Bronner, M.; Buell, C.M.; Collod-Béroud, G.; et al. BRCA share: A collection of clinical BRCA gene variants. Hum. Mutat. 2016, 37, 1318–1328. [Google Scholar] [CrossRef]
- Glusman, G.; Caballero, J.; Mauldin, D.E.; Hood, L.; Roach, J.C. Kaviar: An accessible system for testing snv novelty. Bioinformatics 2011, 27, 3216–3217. [Google Scholar] [CrossRef]
- Hudson, T.J.; Anderson, W.; Aretz, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; Guttmacher, A.; et al. International network of cancer genome projects. Nature 2010, 464, 993–998. [Google Scholar]
- Quang, D.; Chen, Y.; Xie, X. Dann: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3. 0: A one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.D.; Nguyen, A.D.; Bear, K.A.; Wolfsberg, T.G. Clinical genomic database. Proc. Natl. Acad. Sci. USA 2013, 110, 9851–9855. [Google Scholar] [CrossRef]
- Köhler, S.; Vasilevsky, N.A.; Engelstad, M.; Foster, E.; McMurry, J.; Aymé, S.; Baynam, G.; Bello, S.M.; Boerkoel, C.F.; Brudno, M.; et al. The human phenotype ontology in 2017. Nucleic Acids Res. 2017, 45, D865–D876. [Google Scholar] [CrossRef]
- ALIGN-GVGD. Available online: https://agvgd.iarc.fr (accessed on 27 March 2020).
- Tavtigian, S.V.; Deffenbaugh, A.M.; Yin, L.; Judkins, T.; Scholl, T.; Samollow, P.B.; De Silva, D.; Zharkikh, A.; Thomas, A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J. Med. Genet. 2006, 43, 295–305. [Google Scholar] [CrossRef]
- Mathe, E.; Olivier, M.; Kato, S.; Ishioka, C.; Hainaut, P.; Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: A comparison of three sequence analysis based methods. Nucleic Acids Res. 2006, 34, 1317–1325. [Google Scholar] [CrossRef]
- microRNA.org. Available online: http://www.microrna.org/microrna/home.do (accessed on 27 March 2020).
- miRbase. Available online: http://www.mirbase.org/ (accessed on 27 March 2020).
- Microinspector. Available online: https://imbb.forth.gr (accessed on 27 March 2020).
- RegRNA 2.0 Software. Available online: http://regrna2.mbc.nctu.edu.tw/detection_output.php (accessed on 27 March 2020).
- Capoluongo, E.; De Matteis, E.; Cucinotto, I.; Ronzino, G.; Santonocito, C.; Tornesello, A.; De Giorgio, M.R.; Lucci Cordisco, E.; Minucci, A.; Genuardi, M. A new founder BRCA1 haplotype identified in the Puglia region is associated with a specific age-related cancer onset in three unrelated families. Clin. Chem. Lab. Med. 2020, 58. [Google Scholar] [CrossRef]
- Incorvaia, L.; Fanale, D.; Badalamenti, G.; Bono, M.; Calò, V.; Cancelliere, D.; Castiglia, M.; Fiorino, A.; Pivetti, A.; Barraco, N.; et al. Hereditary breast and ovarian cancer in families from Southern Italy (Sicily)-prevalence and geographic distribution of pathogenic variants in BRCA1/2 genes. Cancers 2020, 12, 1158. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Breast Cancer | Ovarian Cancer | Breast and Ovarian Cancer | Healthy Carriers | Other Cancer |
|---|---|---|---|---|---|
| Median Age | 49 years | 55 years | 57 years | 51 years | 58 years |
| (range) | (28–83) | (29–85) | (40–79) | (22–86) | (32–79) |
| Gender | |||||
| Female | 256 | 159 | 17 | 42 | 20 |
| Male | 7 | - | - | 15 | 2 |
| Variants Spectrum | |||||
| BRCA1 carriers (250) | 119 | 85 | 9 | 28 | 9 |
| BRCA2 carriers (260) | 140 | 70 | 8 | 29 | 13 |
| BRCA1/2 carriers (8) | 4 | 4 | - | - | - |
| BRCA 1 Gene Variants | |||||||
| Exon/Intron | HGVS Nucleotide | HGVS Protein | rs | Frequency | Variant Type | Class | |
| 2 | c.65T>C | p.(Leu22Ser) | rs80357438 | 2 | M | 5 | |
| IVS 2 | c.80+1G>A | - | rs80358010 | 1 | IVS | 5 | |
| IVS 2 | c.81-1G>C | - | rs80358018 | 2 | IVS | 5 | |
| 3 | c.134+2T>C | - | rs80358131 | 2 | IVS | 5 | |
| 5 | c.143T>A | p.(Met48Lys) | no rs | 2 | M | Novel 1,° | |
| 5 | c.181T>G* | p.(Cys61Gly) | rs28897672 | 13 | M | 5 | |
| 7 | c.398G>A | p.(Arg133His) | rs80357357 | 3 | M | CIP | |
| IVS 7 | c.441+5A>G | - | rs200358748 | 1 | IVS | 3 | |
| 8 | c.485_486delTG | p.(Val162GlufsTer19) | rs80357708 | 1 | F | 5 | |
| 8 | c.488G>C | p.(Arg163Thr) | rs1369043501 | 1 | M | 3 | |
| 8 | c.514delC | p.(Gln172AsnfsTer62) | rs80357872 | 5 | F | 5 | |
| IVS 8 | c.547+2T>A | - | rs80358047 | 3 | IVS | 5 | |
| 11 | c.755G>A | p.(Arg252His) | rs80357138 | 2 | M | CIP | |
| 11 | c.798_799delTT | p.(Ser267LysfsTer19) | no rs | 3 | F | 5 | |
| 11 | c.815_824dupAGCCATGTGG | p.(Thr276AlafsTer14) | rs387906563 | 1 | F | 5 | |
| 11 | c.843_846delCTCA | p.(Ser282TyrfsTer15) | rs80357919 | 1 | F | 5 | |
| 11 | c.850C>T | p.(Gln284Ter) | rs397509330 | 3 | NS | 5 | |
| 11 | c.946A>G | p.(Ser316Gly) | rs55874646 | 1 | M | 1 | |
| 11 | c.981_982delAT | p.(Cys328Ter) | rs80357772 | 1 | F | 5 | |
| 11 | c.997A>G | p.(Thr333Ala) | rs786201634 | 1 | M | 1 | |
| 11 | c.1016_1017insC | p.(Lys339AsnfsTer7) | rs1555592653 | 1 | F | 5 | |
| 11 | c.1063A>C | p.(Lys355Gln) | no rs | 1 | M | VUS | |
| 11 | c.1081T>C | p.(Ser361Pro) | rs80356946 | 1 | M | CIP | |
| 11 | c.1217dupA | p.(Asn406LysfsTer6) | rs397508846 | 2 | F | 5 | |
| 11 | c.1252G>T | p.(Glu418Ter) | rs80357083 | 1 | NS | 5 | |
| 11 | c.1268C>T | p.(Ser423Phe) | no rs | 1 | M | Novel2,# | |
| 11 | c.1297delG | p.(Ala433ProfsTer8) | rs80357794 | 1 | F | 5 | |
| 11 | c.1360_1361delAG* | p.(Ser454Ter) | rs80357969 | 4 | F | 5 | |
| 11 | c.1462dupA | p.(Thr488AsnfsTer2) | rs80357599 | 3 | F | 5 | |
| 11 | c.1496C>A | p.(Thr499Lys) | no rs | 1 | M | Novel# | |
| 11 | c.1513A>T | p.(Lys505Ter) | rs397508877 | 1 | NS | 5 | |
| 11 | c.1612C>T | p.(Gln538Ter) | rs80356893 | 2 | NS | 5 | |
| 11 | c.1687C>T | p.(Gln563Ter) | rs80356898 | 3 | NS | 5 | |
| 11 | c.1703C>T | p.(Pro568Leu) | rs80356910 | 3 | M | 1 | |
| 11 | c.1895G>A | p.(Ser632Asn) | rs80356983 | 2 | M | 3 | |
| 11 | c.1953dupG | p.(Lys652GlufsTer21) | rs80357753 | 2 | F | 5 | |
| 11 | c.2037delGinsCC | p.(Lys679AsnfsTer4) | rs397508932 | 1 | F | 5 | |
| 11 | c.2077delGinsATA | p.(Asp693ThrfsTer8) | rs886039991 | 1 | F | 5 | |
| 11 | c.2195_2196delAAinsG | p.(Glu732GlyfsTer4) | rs397508948 | 1 | F | 5 | |
| 11 | c.2281G>C | p.(Glu761Gln) | rs397507198 | 2 | M | 3 | |
| 11 | c.2296_2297delAG | p.(Ser766Ter) | rs80357780 | 3 | F | 5 | |
| 11 | c.2405_2406delTG | p.(Val802GlufsTer7) | rs80357706 | 3 | F | 5 | |
| 11 | c.2501G>A | p.(Gly834Glu) | rs757383244 | 1 | M | 3 | |
| 11 | c.2518A>T | p.(Ser840Cys) | rs377475866 | 1 | M | 3 | |
| 11 | c.2529_2530delAA | p.(Ser844HisfsTer7) | rs886040046 | 1 | F | 5 | |
| 11 | c.2705A>G | p.(Glu902Gly) | no rs | 1 | M | Novel# | |
| 11 | c.2760delA | p.(Gln921ArgfsTer79) | rs1064795769 | 1 | F | 5 | |
| 11 | c.3044dupG | p.(Asn1016LysfsTer2) | rs80357746 | 1 | F | 5 | |
| 11 | c.3082C>T | p.(Arg1028Cys) | rs80357049 | 1 | M | 1 | |
| 11 | c.3228_3229delAG * | p.(Gly1077AlafsTer8) | rs80357635 | 1 | F | 5 | |
| 11 | c.3285delA* | p.(Lys1095AsnfsTer14) | rs397509051 | 2 | F | 5 | |
| 11 | c.3331_3334delCAAG | p.(Gln1111AsnfsTer5) | rs80357701 | 1 | F | 5 | |
| 11 | c.3344_3346delAAG | p.(Glu1115del) | rs80358336 | 1 | IFDEL | 1 | |
| 11 | c.3454G>A | p.(Asp1152Asn) | rs80357175 | 1 | M | CIP | |
| 11 | c.3514G>T | p.(Glu1172Ter) | rs397509079 | 1 | NS | 5 | |
| 11 | c.3607C>T | p.(Arg1203Ter) | rs62625308 | 2 | NS | 5 | |
| 11 | c.3700_3704delGTAAA | p.(Val1234GlnfsTer8) | rs80357609 | 1 | F | 5 | |
| 11 | c.3756_3759delGTCT* | p.(Ser1253ArgfsTer10) | rs80357868 | 8 | F | 5 | |
| 11 | c.3868A>G | p.(Lys1290Glu) | rs80357254 | 1 | M | 3 | |
| 11 | c.3916_3917delTT | p.(Leu1306AspfsTer23) | rs80357678 | 1 | F | 5 | |
| 11 | c.3928dupA | p.(Thr1310AsnfsTer20) | rs886040176 | 1 | F | 5 | |
| 11 | c.3973delA | p.(Arg1325GlyfsTer11) | rs80357904 | 1 | F | 5 | |
| 11 | c.4054G>A | p.(Glu1352Lys) | rs80357202 | 1 | M | 3 | |
| 11 | c.4065_4068delTCAA | p.(Asn1355LysfsTer10) | rs80357508 | 1 | F | 5 | |
| IVS11 | c.4096+1G>A | - | rs80358178 | 2 | IVS | 3 | |
| 12 | c.4117G>T* | p.(Glu1373Ter) | rs80357259 | 23 | NS | 5 | |
| 12 | c.4132G>A | p.(Val1378Ile) | rs28897690 | 3 | M | 1 | |
| 12 | c.4162C>T | p.(Gln1388Ter) | rs876660601 | 1 | NS | 5 | |
| 12 | c.4183C>T | p.(Gln1395Ter) | rs80357260 | 1 | NS | 5 | |
| 13 | c.4213A>G | p.(Ile1405Val) | rs80357353 | 1 | M | CIP | |
| 13 | c.4327C>T | p.(Arg1443Ter) | rs41293455 | 1 | NS | 5 | |
| 13 | c.4357insT | Ala1453ValfsX9/Ala1453GlnfsX3 | no rs | 2 | F | 5 | |
| 14 | c.4361T>C | p.(Val1454Ala) | rs587782606 | 1 | M | CIP | |
| 14 | c.4484G>T | p.(Arg1495Met) | rs80357389 | 3 | M | 5 | |
| IVS 14 | c.4484+1G>T | - | rs80358063 | 1 | IVS | 5 | |
| IVS 15 | c.4675+3A>G | - | rs80358082 | 1 | IVS | 3 | |
| 16 | c.4739C>T | p.(Ser1580Phe) | rs80357411 | 1 | M | 3 | |
| 16 | c.4882A>G | p.(Met1628Val) | rs80357465 | 1 | M | CIP | |
| 16 | c.4964_4982del* | p.(Ser1655TyrfsTer16) | rs80359876 | 8 | F | 5 | |
| 17 | c.5030_5033delCTAA | p.(Thr1677IlefsTer2) | rs80357580 | 3 | F | 5 | |
| 17 | c.5035_5039delCTAAT | p.(Leu1679TyrfsTer2) | rs80357623 | 1 | F | 5 | |
| 17 | c.5062_5064delGTT* | p.(Val1688del) | rs80358344 | 2 | IFDEL | 5 | |
| 17 | c.5073A>T | p.(Thr1691=) | no rs | 5 | S | 5 | |
| IVS 17 | c.5074+6C>G | - | rs80358032 | 1 | IVS | 1 | |
| 18 | c.5095C>T | p.(Arg1699Trp) | rs55770810 | 1 | M | 5 | |
| 18 | c.5106delA | p.(Lys1702AsnfsTer4) | rs80357553 | 1 | F | 5 | |
| 18 | c.5123C>A* | p.(Ala1708Glu) | rs28897696 | 13 | M | 5 | |
| 18 | c.5150delT | p.(Phe1717SerfsTer3) | rs80357720 | 1 | F | 5 | |
| 20 | c.5239C>T | p.(Gln1747Ter) | rs80357367 | 1 | NS | 5 | |
| 20 | c.5266dupC* | p.(Gln1756ProfsTer74) | rs397507247 | 24 | F | 5 | |
| 21 | c.5308G>T | p.(Gly1770Trp) | no rs | 1 | M | Novel2,° | |
| 21 | c.5319dupC | p.(Asn1774GlnfsTer56) | rs80357823 | 1 | F | 5 | |
| 22 | c.5333A>G | p.(Asp1778Gly) | rs80357041 | 1 | M | 1/2 | |
| 22 | c.5353C>T | p.(Gln1785Ter) | rs80356969 | 4 | NS | 5 | |
| 23 | c.5431C>T | p.(Gln1811Ter) | rs397509283 | 1 | NS | 5 | |
| 23 | c.5434C>G | p.(Pro1812Ala) | rs1800751 | 1 | M | 4/5 | |
| 23 | c.5444G>A | p.(Trp1815Ter) | rs80356962 | 1 | NS | 5 | |
| IVS 23 | c.5468-1G>A | - | rs80358048 | 1 | IVS | 5 | |
| 24 | c.5504G>C | p.(Arg1835Pro) | rs273902776 | 1 | M | 3 | |
| 3-UTR | c.*85A>G | rs756518403 | 1 | M | Novel | ||
| c.(?_-1387-1)_(80+1_81-1)del | p.0? | 3 | LGR | 5 | |||
| c.(212+1_213-1)_(441+1_442-1)del | p.? | 1 | LGR | 5 | |||
| c.(4357+1_4358-1)_(4484+1_4485-1)del | p.? | 1 | LGR | 5 | |||
| c.(4675+1_4676-1)_(5074+1_5075-1)del | p.? | 1 | LGR | 5 | |||
| c.(5074+1_5075-1)_(5193+1_5192-1)del | p.? | 4 | LGR | 5 | |||
| c.(5193+1_5194-1)_(5277+1_5278-1)del | p.? | 1 | LGR | 5 | |||
| c.(5277+1_5277-1)_(5406+1_5407-1)del | p.? | 2 | LGR | 5 | |||
| BRCA2 Gene Variants | |||||||
| Exon/intron | HGVS Nucleotide | HGVS Protein | rs | Frequency | Variant Type | Class | |
| 2 | c.62A>G | p.(Lys21Arg) | rs397507367 | 2 | M | 3 | |
| IVS2 | c.67+1G>A | - | rs81002796 | 3 | IVS | 5 | |
| 3 | c.289G>T | p.(Glu97Ter) | no rs | 1 | NS | 5 | |
| 4 | c.353G>A | p.(Arg118His) | rs80358603 | 1 | M | CIP | |
| 4 | c.368_372delAAATG | p.(Lys123ArgfsTer5) | no rs | 1 | F | 5 | |
| IVS 4 | c.425+2T>C | - | rs876661045 | 1 | IVS | 4 | |
| IVS 6 | c.516+1G>C | - | rs397507762 | 2 | IVS | 5 | |
| 7 | c.599C>T | p.(Thr200Ile) | rs587781402 | 1 | M | 3 | |
| IVS 7 | c.632-2A>G | - | rs397507842 | 1 | IVS | 5 | |
| 7 | c.631G>A | p.(Val211Ile) | rs80358871 | 4 | M | 5 | |
| 8 | c.658_659delGT | p.(Val220IlefsTer4) | rs80359604 | 4 | F | 5 | |
| 10 | c.831T>G | p.(Asn277Lys) | rs28897705 | 1 | M | CIP | |
| 10 | c.1238delT | p.(Leu413HisfsTer17) | rs80359271 | 2 | F | 5 | |
| 10 | c.1244A>G | p.(His415Arg) | rs80358417 | 1 | M | CIP | |
| 10 | c.1247T>G | p.(Ile416Ser) | rs80358418 | 1 | M | 1/2 | |
| 10 | c.1257delT | p.(Cys419TrpfsTer11) | rs80359272 | 1 | F | 5 | |
| 10 | c.1259A>G | p.(Asp420Gly) | rs786201654 | 1 | M | 3 | |
| 10 | c.1296_1297delGA | p.8Asn433GlnfsTer18) | rs80359276 | 1 | F | 5 | |
| 10 | c.1322C>T | p.(Thr441Ile) | rs1064793062 | 1 | M | 3 | |
| 10 | c.1342C>T | p.(Arg448Cys) | rs80358422 | 1 | M | CIP | |
| 10 | c.1441A>G | p.(Ile481Val) | rs760559435 | 2 | M | 3 | |
| 10 | c.1514T>C | p.(Ile505Thr) | rs28897708 | 1 | M | 1 | |
| 10 | c.1550A>G | p.(Asn517Ser) | rs80358439 | 1 | M | CIP | |
| 10 | c.1670T>G | p.(Leu557Ter) | rs80358452 | 5 | NS | 5 | |
| 10 | c.1792A>G | p.(Thr598Ala) | rs28897710 | 1 | M | 1 | |
| 10 | c.1796_1800delCTTAT | p.(Ser599Ter) | rs276174813 | 3 | NS | 5 | |
| 10 | c.1813delA | p.(Ile605TyrfsTer9) | rs80359306 | 1 | F | 5 | |
| 10 | c.1820A>C | p.(Lys607Thr) | rs55962656 | 1 | M | CIP | |
| 11 | c.2014A>G | p.(Arg672Gly) | rs587781647 | 1 | M | 2 | |
| 11 | c.2094delA | p.(Gln699Serfs31) | rs80359323 | 1 | F | 5 | |
| 11 | c.2491_2492insT | p.(Glu832Ter) | no rs | 1 | NS | Novel° (Class 4) | |
| 11 | c.2494G>T | p.(Glu832Ter) | rs786202875 | 1 | NS | 5 | |
| 11 | c.2651C>G | p.(Ser884Ter) | rs777421358 | 1 | NS | 5 | |
| 11 | c.2684delC | p.(Ala895ValfsTer9) | rs80359342 | 1 | F | 5 | |
| 11 | c.2808_2811delACAA | p.(Ala938ProfsTer21) | rs80359351 | 4 | F | 5 | |
| 11 | c.2821G>A | p.(Val941Met) | rs863224586 | 1 | M | 3 | |
| 11 | c.2836delG | p.(Asp946IlefsTer14) | rs80359358 | 1 | F | 5 | |
| 11 | c.2905C>T | p.(Gln969Ter) | rs886038080 | 1 | NS | 5 | |
| 11 | c.2944A>C | p.(Ile982Leu) | rs28897717 | 1 | M | CIP | |
| 11 | c.3443A>G | p.(Gln1148Arg) | rs200808363 | 1 | M | 3 | |
| 11 | c.3499A>G | p.(Ile1167Val) | rs276174834 | 1 | M | 3 | |
| 11 | c.3541C>T | p.(Gln1181Ter) | no rs | 1 | NS | Novel° (Class 4) | |
| 11 | c.3551G>C | p.(Gly1184Ala) | rs431825309 | 1 | M | 3 | |
| 11 | c.3635delA | p.(Asn1212Metfs16) | no rs | 1 | F | 5 | |
| 11 | c.3680_3681delTG | p.(Leu1227GlnfsTer5) | rs80359395 | 2 | F | 5 | |
| 11 | c.3683A>G | p.(Asn1228Ser) | rs786202838 | 1 | M | 3 | |
| 11 | c.3723T>G | p.(Phe1241Leu) | rs587782723 | 1 | M | 3 | |
| 11 | c.3744_3747delTGAG | p.(Ser1248ArgfsTer10) | rs80359403 | 1 | F | 5 | |
| 11 | c.3847_3848delGT | p.(Val1283LysfsTer2) | rs80359405 | 1 | F | 5 | |
| 11 | c.3860delA | p.(Asn1287IlefsTer6) | rs80359406 | 1 | F | 5 | |
| 11 | c.3962A>G | p.(Asp1321Gly) | rs80358645 | 1 | M | 2 | |
| 11 | c.4131_4132insTGAGGA | p.(Thr1378Ter) | rs80359429 | 6 | IFINS | 5 | |
| 11 | c.4133_4136delCTCA | p.(Thr1378ArgfsTer9) | rs80359430 | 2 | F | 5 | |
| 11 | c.4284dupT | p.(Gln1429SerfsTer9) | rs80359439 | 4 | F | 5 | |
| 11 | c.4285_4286insT | p.(Gln1429LeufsTer9) | rs886040518 | 1 | F | 5 | |
| 11 | c.4325C>A | p.(Ser1442Ter) | rs80358670 | 1 | NS | 5 | |
| 11 | c.4334A>C | p.(Lys1445Thr) | no rs | 1 | M | 3 | |
| 11 | c.4419delC | p.(Asn1473LysfsTer6) | rs1064794337 | 1 | F | 5 | |
| 11 | c.4574A>G | p.(His1525Arg) | rs397507336 | 1 | M | 3 | |
| 11 | c.4647_4650delAGAG | p.(Lys1549AsnfsTer18) | rs397507734 | 1 | F | 5 | |
| 11 | c.4769A>G | p.(Lys1590Arg) | no rs | 1 | M | Novel1, # | |
| 11 | c.4803dupT | p.(Lys1602Ter) | no rs | 1 | NS | Novel1,° (Class 4) | |
| 11 | c.4899_4902delCTTT | p.(Phe1634Ter) | no rs | 1 | F | Novel2,° (Class 4) | |
| 11 | c.4936_4939delGAAA | p.(Glu1646GlnfsTer23) | rs80359473 | 1 | F | 5 | |
| 11 | c.5073dupA | p.(Trp1692MetfsTer3) | rs80359479 | 3 | F | 5 | |
| 11 | c.5158dupT | p.(Ser1720PhefsTer7) | rs80359489 | 2 | F | 5 | |
| 11 | c.5224_5229delAACAGT | p.(Asn1742_Ser1743del) | rs276174855 | 1 | IFDEL | 3 | |
| 11 | c.5239_5240insT | p.(Asn1747IlefsTer8) | rs80359500 | 1 | F | 5 | |
| 11 | c.5261A>G | p.(Asp1754Gly) | rs772772727 | 1 | M | 3 | |
| 11 | c.5345A>C | p.(Gln1782Pro) | rs758959174 | 1 | M | 3 | |
| 11 | c.5351_5352dupA | p.(Asn1784LysfsTer3) | rs80359507 | 4 | F | 5 | |
| 11 | c.5423T>C | p.(Ile1808Thr) | rs397507350 | 1 | M | CIP | |
| 11 | c.5428G>A | p.(Val1810Ile) | rs80358766 | 1 | M | 3 | |
| 11 | c.5492T>C | p.(Ile1831Thr) | rs587782007 | 1 | M | 3 | |
| 11 | c.5634C>G | p.(Asn1878Lys) | rs80358784 | 1 | M | 1 | |
| 11 | c.5722_5723delCT | p.(Leu1908ArgfsTer2) | rs80359530 | 2 | F | 5 | |
| 11 | c.5796_5797delTA | p.(His1932GlnfsTer12) | rs80359537 | 2 | F | 5 | |
| 11 | c.5851_5854delAGTT | p.(Ser1951TrpfsTer11) | rs80359543 | 1 | F | 5 | |
| 11 | c.5885T>C | p.(Ile1962Thr) | rs1060502377 | 1 | M | CIP | |
| 11 | c.5897A>G | p.(His1966Arg) | rs80358823 | 1 | M | CIP | |
| 11 | c.5946delT | p.(Ser1982ArgfsTer22) | rs80359550 | 2 | F | 5 | |
| 11 | c.5959C>T | p.(Gln1987Ter) | rs80358828 | 1 | NS | 5 | |
| 11 | c.5971G>A | p.(Ala1991Thr) | no rs | 1 | M | Novel2, $ | |
| 11 | c.5986G>A | p.(Ala1996Thr) | rs80358833 | 1 | M | CIP | |
| 11 | c.6024dupG | p.(Gln2009AlafsTer9) | rs80359554 | 1 | F | 5 | |
| 11 | c.6037A>T | p.(Lys2013Ter) | rs80358840 | 4 | NS | 5 | |
| 11 | c.6037A>G | p.(Lys2013Glu) | rs80358840 | 1 | M | 3 | |
| 11 | c.6039delA | p.(Val2014TyrfsTer26) | rs876660637 | 2 | F | 5 | |
| 11 | c.6078_6079delAA | p.(Glu2028ArgfsTer20) | rs80359557 | 1 | F | 5 | |
| 11 | c.6098T>C | p.(Ile2033Thr) | no rs | 1 | M | Novel$ | |
| 11 | c.6131G>C | p.(Gly2044Ala) | rs56191579 | 1 | M | CIP | |
| 11 | c.6267_6269delGCAinsC | p.(Glu2089AspfsTer2) | rs276174868 | 1 | F | 5 | |
| 11 | c.6322C>T | p.(Arg2108Cys) | rs55794205 | 1 | M | 1 | |
| 11 | c.6405_6409delCTTAA | p.(Asn2135LysfsTer3) | rs80359584 | 1 | F | 5 | |
| 11 | c.6468_6469delTC | p.(Gln2157IlefsTer18) | rs80359596 | 4 | F | 5 | |
| 11 | c.6486_6489delACAA | p.(Lys2162AsnfsTer5) | rs80359598 | 2 | F | 5 | |
| 11 | c.6496G>T | p.(Val2166Leu) | rs750084851 | 1 | M | 3 | |
| 11 | c.6590_6591insA | p.(Glu2198Ter) | no rs | 1 | F | Novel2,° (Class 4) | |
| 11 | c.6591_6592delTG | p.(Glu2198AsnfsTer4) | rs80359605 | 9 | F | 5 | |
| 11 | c.6650A>G | p.(Lys2217Arg) | rs1555284781 | 1 | M | 3 | |
| 11 | c.6761_6762delTT | p.(Phe2254TyrfsTer6) | rs80359624 | 1 | F | 5 | |
| IVS 11 | c.6841+1G>T | - | 1 | IVS | 3 | ||
| 12 | c.6875A>G | p.(Glu2292Gly) | rs397507378 | 1 | M | 3 | |
| 13 | c.7007G>A | p.(Arg2336His) | rs28897743 | 7 | M | 5 | |
| 13 | c.7007G>C | p.(Arg2336Pro) | rs28897743 | 4 | M | 5 | |
| IVS13 | c.7007+5G>A | - | rs81002816 | 1 | IVS | 3 | |
| 14 | c.7057G>C | p.(Gly2353Arg) | rs80358935 | 1 | M | 1 | |
| 14 | c.7072T>C | p.(Ser2358Pro) | rs80358937 | 1 | M | 3 | |
| 14 | c.7180A>T | p.(Arg2394Ter) | rs80358946 | 1 | NS | 5 | |
| 14 | c.7225C>T | p.(Pro2409Ser) | no rs | 1 | M | Novel2, # | |
| 14 | c.7435+10G>A | - | rs81002793 | 1 | IVS | CIP | |
| 15 | c.7505G>A | p.(Arg2502His) | rs56070345 | 1 | M | 1 | |
| 15 | c.7506delC | p.(Val2503SerfsTer21) | no rs | 1 | F | Novel° (Class 4) | |
| 15 | c.7561delA | p.(Ile2521SerfsTer3) | no rs | 1 | F | 5 | |
| 15 | c.7617+9_7617+12delTTGT | - | no rs | 1 | IVS | Novel# | |
| 16 | c.7636T>C | p.(Ser2546Pro) | rs1555286392 | 1 | M | 3 | |
| IVS 16 | c.7806-2A>G | - | rs81002836 | 1 | IVS | 5 | |
| 17 | c.7857G>A | p.(Trp2619Ter) | rs80359011 | 2 | NS | 5 | |
| 17 | c.7878G>C | p.(Trp2626Cys) | rs80359013 | 1 | M | 5 | |
| 18 | c.7994A>G | p.(Asp2665Gly) | rs28897745 | 2 | M | 1 | |
| 18 | c.8009C>T | p.(Ser2670Leu) | rs80359035 | 1 | M | CIP | |
| 18 | c.8245C>T | p.(Gln2749Ter) | rs1135401925 | 2 | NS | 5 | |
| 18 | c.8249_8251delAGA | p.(Lys2750del) | rs80359703 | 1 | IFDEL | 3 | |
| 18 | c.8258T>C | p.(Leu2753Pro) | rs786203357 | 1 | M | Novel2,# | |
| 18 | c.8299C>T | p.(Pro2767Ser) | rs587782619 | 2 | M | 3 | |
| 19 | c.8478C>A | p.(Tyr2826Ter) | rs776353983 | 2 | NS | 5 | |
| IVS 19 | c.8487+1G>A | - | rs81002798 | 6 | IVS | 5 | |
| 20 | c.8537_8538delAG* | p.(Glu2846GlyfsTer22) | rs80359714 | 1 | F | 5 | |
| 20 | c.8567A>C | p.(Glu2856Ala) | rs11571747 | 1 | M | CIP | |
| IVS 20 | c.8632+5A>G | - | rs763224070 | 1 | IVS | 2 | |
| IVS 21 | c.8754+4A>G | - | rs81002893 | 2 | IVS | 5 | |
| IVS 21 | c.8755-1G>A | - | rs81002812 | 4 | IVS | 5 | |
| 22 | c.8878C>T | p.(Gln2960Ter) | rs80359140 | 2 | NS | 5 | |
| 22 | c.8915T>G | p.(Leu2972Trp) | rs80359142 | 1 | M | 3 | |
| IVS 22 | c.8954-1_8955delGTTinsAA | - | rs276174916 | 1 | IVS | 5 | |
| 23 | c.9097delA | p.(Thr3033LeufsTer29) | rs397507419 | 3 | F | 5 | |
| 23 | c.9104A>C | p.(Tyr3035Ser) | rs80359165 | 1 | M | CIP | |
| 23 | c.9116C>T | p.(Pro3039Leu) | rs80359167 | 2 | M | CIP | |
| 24 | c.9124G>A | p.(Asp3042Asn) | no rs | 1 | M | Novel# | |
| 24 | c.9148C>T | p.(Gln3050Ter) | rs80359170 | 1 | NS | 5 | |
| 24 | c.9154C>T | p.(Arg3052Trp) | rs45580035 | 1 | M | 5 | |
| 24 | c.9171C>G | p.(Phe3057Leu) | rs747615055 | 1 | M | 3 | |
| 25 | c.9271G>A | p.(Val3091Ile) | rs80359194 | 1 | M | CIP | |
| 25 | c.9275A>G | p.(Tyr3092Cys) | rs80359195 | 1 | M | CIP | |
| 25 | c.9364G>A | p.(Ala3122Thr) | rs587782313 | 1 | M | CIP | |
| 25 | c.9382C>T | p.(Arg3128Ter) | rs80359212 | 2 | NS | 5 | |
| 25 | c.9413dupT | p.(Leu3138PhefsTer12) | rs876659435 | 1 | F | 5 | |
| IVS 25 | c.9501+3A>T | - | rs61757642 | 2 | IVS | 1 | |
| IVS 25 | c.9502-12T>G | - | rs81002803 | 1 | IVS | 1 | |
| 26 | c.9581C>A | p.(Pro3194Gln) | rs28897760 | 2 | M | CIP | |
| 26 | c.9583A>G | p.(Thr3195Ala) | rs80359227 | 1 | M | CIP | |
| 26 | c.9613_9614delGCinsCT | p.(Ala3205Leu) | rs276174926 | 3 | M | 3 | |
| 27 | c.9676delT | p.(Tyr3226IlefsTer23) | rs80359774 | 3 | F | 5 | |
| 27 | c.9959_9961delCTC | p.(Pro3320del) | rs745685382 | 2 | IFDEL | 3 | |
| 27 | c.10024G>A | p.(Glu3342Lys) | rs28897761 | 1 | M | CIP | |
| 27 | c.10040T>C | p.(Ile3347Thr) | rs587782373 | 1 | M | 3 | |
| Patients with Double or Triple Gene Variants | |||||||
|---|---|---|---|---|---|---|---|
| List of Patient | BC/OC | Gene | HGVS Nucleotide | HGVS Protein | rs | Variant Type | Class |
| 1 | BC | BRCA1 | c.3228_3229delAG; | p.(Gly1077AlafsTer8) | no rs | F | 5 |
| BRCA2 | c.464G>C | p.(Arg155Thr) | rs377639990 | M | 3 | ||
| 2 | BC | BRCA2 | c.1238delT | p.(Leu413HisfsTer17) | rs80359271 | F | 5 |
| BRCA1 | c.5095C>T | p.(Arg1699Trp) | rs55770810 | M | 5 | ||
| 3 | BC | BRCA1 | c.798_799delTT | p.(Ser267LysfsTer19) | no rs | F | 5 |
| BRCA2 | c.6290C>T | p.(Thr2097Met) | rs80358866 | M | 1 | ||
| 4 | OC | BRCA1 | c.5062_5064delGTT | p.(Val1688del) | rs80358344 | IFD | 5 |
| BRCA2 | c.4054G>T | p.(Asp1352Tyr) | rs80358655 | M | 3 | ||
| 5 | OC | BRCA2 | c.800G>A | p.(Gly267Glu) | rs80359036 | M | CIP |
| BRCA1 | c.4213A>G | p.(Ile1405Val) | rs80357353 | M | CIP | ||
| 6 | OC | BRCA2 | c.5796_5797delTA | p.(His1932GlnfsTer12) | rs80359537 | F | 5 |
| BRCA1 | c.(?_-232)_(4096+1_4097-1)del | p.0? | - | LGR | 5 | ||
| 7 | BC | BRCA1 | c.134+2T>C | - | rs80358131 | IVS | 5 |
| BRCA1 | c.2281G>C | p.(Glu761Gln) | rs397507198 | M | 3 | ||
| 8 | BC | BRCA1 | c.3756_3759delGTCT | p.(Ser1253ArgfsTer10) | rs80357868 | F | 5 |
| BRCA2 | c.3381delT | p.(Phe1127LeufsTer23) | no rs | F | 3 | ||
| 9 | OC | BRCA1 | c.4964_4982del | p.(Ser1655TyrfsTer16) | rs80359876 | F | 5 |
| BRCA1 | c.525G>A | p.(Lys175=) | rs1555594837 | S | 3 | ||
| 10 | OC | BRCA2 | c.2808_2811delACAA | p.(Ala938ProfsTer21) | rs80359351 | F | 5 |
| BRCA2 | c.9116C>T | p.(Pro3039Leu) | rs80359167 | M | CIP | ||
| 11 | BC | BRCA2 | c.2808_2811delACAA | p.(Ala938ProfsTer21) | rs80359351 | F | 5 |
| BRCA2 | c.9116C>T | p.(Pro3039Leu) | rs80359167 | M | CIP | ||
| 12 | OC | BRCA2 | c.865A>G | p.(Asn289Asp) | rs766173 | M | CIP |
| BRCA2 | c. 5126A>C | p.(Asp1709Ala) | rs786202836 | M | 3 | ||
| 13 | BC | BRCA2 | c.99A>G | p.(Glu33=) | no rs | S | Novel# |
| BRCA2 | c.9828A>T | p.(Arg3276Ser) | rs80359245 | M | 3 | ||
| BRCA2 | c.517-4C>G | - | rs81002804 | IVS | 3 | ||
| 14 | OC | BRCA2 | c.658_659delGT | p.(Val220IlefsTer4) | rs80359604 | F | 5 |
| BRCA2 | c.1118A>C | p.(Gln373Pro) | no rs | M | Novel$ | ||
| 15 | OC | BRCA2 | c.3635delA | p.(Asn1212Metfs16) | no rs | F | 5 |
| BRCA2 | c.7902G>A | p.(Met2634Ile) | M | Novel# | |||
| 16 | Other | BRCA2 | c.8452G>A | p.(Val2818Ile) | rs80359094 | M | 3 |
| BRCA2 | c.191C>T | p.(Thr64Ile) | rs397507615 | M | 3 | ||
| 17 | BC | BRCA2 | c.5959C>T | p.(Gln1987Ter) | rs80358828 | NS | 5 |
| BRCA2 | c.9038C>T | p.(Thr3013Ile) | rs28897755 | M | 1 | ||
| 18 | BC | BRCA2 | c.7008-2A>T | - | rs81002823 | IVS | 5 |
| BRCA2 | c.631G>A | p.(Val211Ile) | rs80358871 | M | 5 | ||
| 19 | OC | BRCA1 | c.5266dupC | p.(Gln1756ProfsTer74) | rs397507247 | F | 5 |
| BRCA2 | c.8262T>G | p.(His2754Gln) | rs587776472 | M | 3 | ||
| 20 | BC | BRCA2 | c.5202A>C | p.(Glu1734Asp) | rs1243093278 | M | 3 |
| BRCA2 | c.5867A>T | p.(Asp1956Val) | rs1309562690 | M | 3 | ||
| 21 | BC | BRCA2 | c.2049_2050delTC | p.(Ser683Argfs) | rs80359319 | F | 5 |
| BRCA2 | c.5191C>T | p.(His1731Tyr) | rs80358745 | M | 3 | ||
| 22 | OC | BRCA2 | c.631G>A | p.(Val211Ile) | rs80358871 | M | 5 |
| BRCA2 | c.7008-2A>T | - | rs81002823 | IVS | 5 | ||
| BRCA2 | c.79A>G | p.(Ile27Val) | rs80359034 | M | 3 | ||
| 23 | BC | BRCA2 | c.8567A>C | p.(Glu2856Ala) | rs11571747 | M | CIP |
| BRCA2 | c.7008-62A>G | - | rs76584943 | IVS | CIP | ||
| 24 | BC | BRCA2 | c.631G>A | p.(Val211Ile) | rs80358871 | M | 5 |
| BRCA2 | c.7008-2A>T | - | rs81002823 | IVS | 5 | ||
| 25 | BC | BRCA2 | c.2905C>T | p.(Gln969Ter) | rs886038080 | NS | 5 |
| BRCA2 | c.6447_6448dupTA | p.(Lys2150IlefsTer19) | rs397507858 | F | 5 | ||
| 26 | OC | BRCA2 | c.631G>A | p.(Val211Ile) | rs80358871 | M | 5 |
| BRCA2 | c.7008-2A>T | - | rs81002823 | IVS | 5 | ||
| Gene | HGVS Nucleotide | HGVS Protein | N (%) |
|---|---|---|---|
| BRCA1 | c.5266dupC | p.Gln1756Profs | 24 (9.6) |
| c.4117G>T | p.Glu1373Ter | 23 (9.2) | |
| 181T>G | p.Cys61Gly | 13 (5.2) | |
| c.5123C>A | p.Ala1708Glu | 13 (5.2) | |
| c.3756_3759delGTCT | p.Ser1253fs | 8 (3.2) | |
| c.4964_4982del | p.Ser1655fs | 8 (3.2) | |
| c.514delC | p.Gln172Asnfs | 5 (2) | |
| c.1360_1361delAG | p.Ser454Ter | 4 (1.6) | |
| DEL EXONS 18-19 | 4 (1.6) | ||
| BRCA2 | c.6591_6592delTG | p.Glu2198fs | 9 (3.6) |
| c.7007G>A | p.Arg2336His | 7 (2.8) | |
| c.4131_4132insTGAGGA | p.Thr1378Ter | 6 (2.4) | |
| c.8487+1G>A | IVS19+1G>A | 6 (2.4) | |
| c.631G>A | p.Val211Ile | 4 (1.6) | |
| c.7008-2A>T | IVS13-2A>T | 4 (1.6) | |
| c.658_659delGT | p.Val220fs | 4 (1.6) | |
| c.2808_2811delACAA | p.Ala938Profs | 4 (1.6) | |
| c.4284dupT | p.Gln1429fs | 4 (1.6) | |
| c.5351_5352dupA | p.Asn1784Lysfs | 4 (1.6) | |
| c.6037A>T | p.Lys2013Ter | 4 (1.6) | |
| c.6468_6469delTC | p.Gln2157fs | 4 (1.6) | |
| c.7007G>C | p.Arg2336Pro | 4 (1.6) | |
| c.8755-1G>A | IVS21-1G>A | 4 (1.6) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santonocito, C.; Rizza, R.; Paris, I.; De Marchis, L.; Paolillo, C.; Tiberi, G.; Scambia, G.; Capoluongo, E. Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers 2020, 12, 1286. https://doi.org/10.3390/cancers12051286
Santonocito C, Rizza R, Paris I, De Marchis L, Paolillo C, Tiberi G, Scambia G, Capoluongo E. Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers. 2020; 12(5):1286. https://doi.org/10.3390/cancers12051286
Chicago/Turabian StyleSantonocito, Concetta, Roberta Rizza, Ida Paris, Laura De Marchis, Carmela Paolillo, Giordana Tiberi, Giovanni Scambia, and Ettore Capoluongo. 2020. "Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome" Cancers 12, no. 5: 1286. https://doi.org/10.3390/cancers12051286
APA StyleSantonocito, C., Rizza, R., Paris, I., De Marchis, L., Paolillo, C., Tiberi, G., Scambia, G., & Capoluongo, E. (2020). Spectrum of Germline BRCA1 and BRCA2 Variants Identified in 2351 Ovarian and Breast Cancer Patients Referring to a Reference Cancer Hospital of Rome. Cancers, 12(5), 1286. https://doi.org/10.3390/cancers12051286