Proteomic Tissue-Based Classifier for Early Prediction of Prostate Cancer Progression

 ,
,  , , , ,
, , , ,  add
Show full author list
add
Show full author list
Abstract
1. Introduction
2. Results
2.1. Selection of Biomarker Candidates
2.2. Development of PRISM-SRM Assays and Targeted Proteomics Measurements
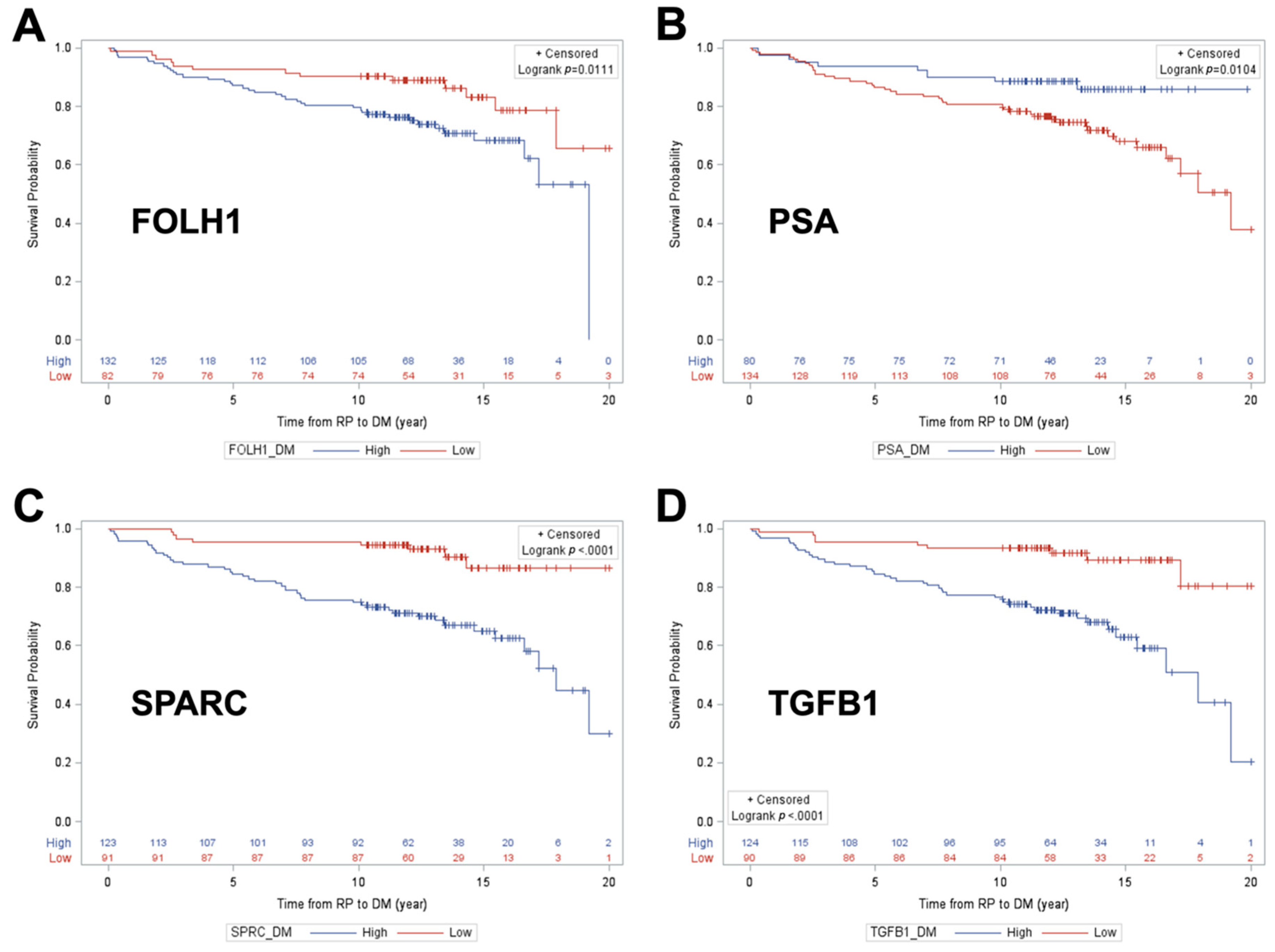
2.3. Determining Predictive Ability of Protein Biomarkers for Cancer Progression
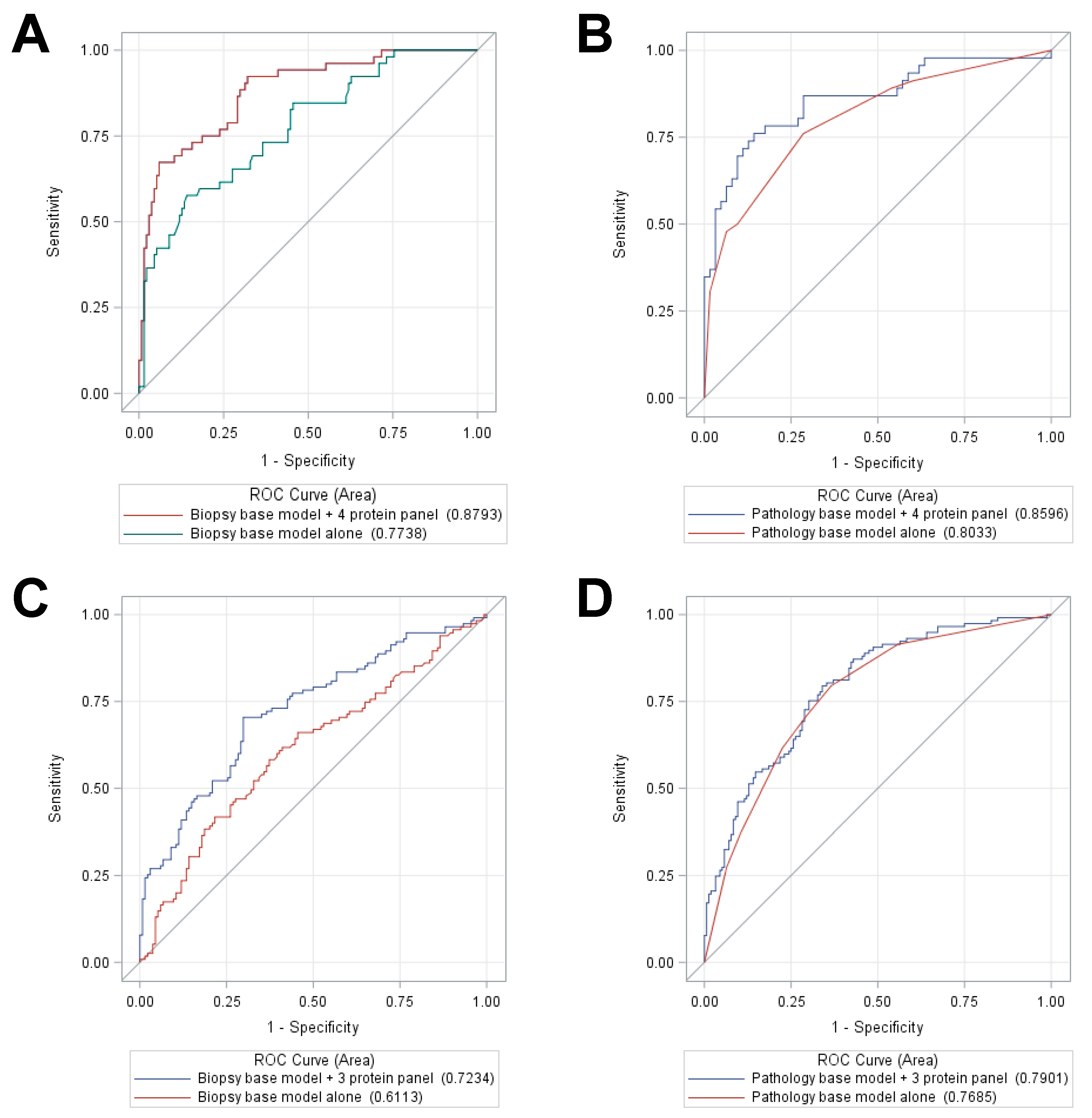
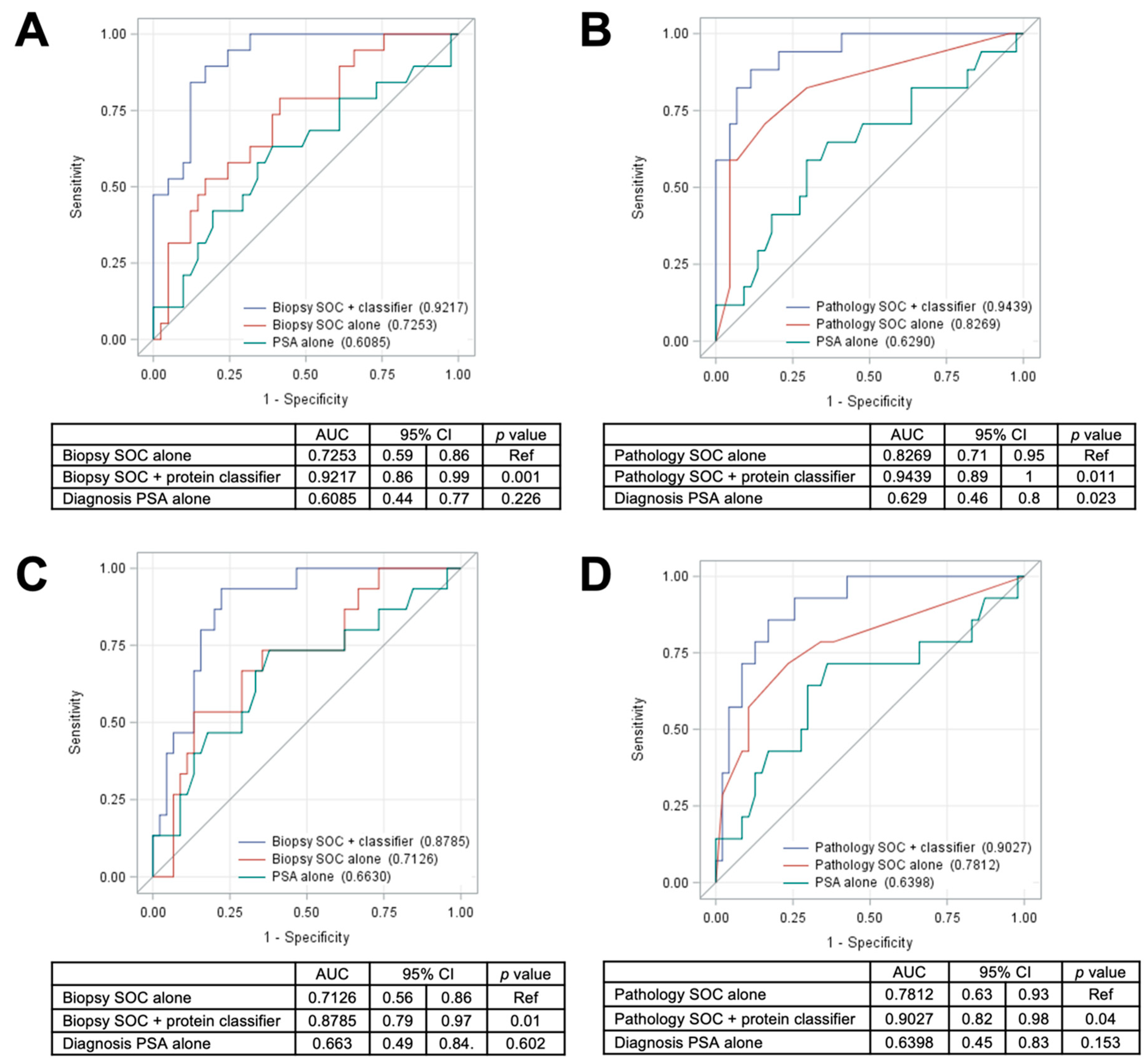
2.4. Training and Testing Set Analysis of a 5-Protein Classifier to Predict Distant Metastasis
3. Discussion
4. Materials and Methods
4.1. Study Cohort
4.2. Demographic, Clinical, and Treatment Variables
4.3. RP Specimen Processing and Pathologic Variable Measurement
4.4. Dependent Study Outcomes
4.5. Protein Digestion of FFPE Tissue Samples
4.6. PRISM-SRM Assay Configuration and Measurements
4.7. Response Curves for the PRISM-SRM Assays
4.8. SRM Data Analysis
4.9. Endogenous Concentration Calculation
4.10. Initial Evaluation of Performance of Protein Biomarker Panels
4.11. Protein Classifier Development and Evaluation
4.12. Data Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- American Cancer Society. Cancer Facts and Figures 2020. Available online: https://www.cancer.org/content/dam/cancer-org/research/cancer-facts-and-statistics/annual-cancer-facts-and-figures/2020/cancer-facts-and-figures-2020.pdf (accessed on 7 May 2020).
- Boutros, P.C.; Fraser, M.; Harding, N.J.; de Borja, R.; Trudel, D.; Lalonde, E.; Meng, A.; Hennings-Yeomans, P.H.; McPherson, A.; Sabelnykova, V.Y.; et al. Spatial genomic heterogeneity within localized, multifocal prostate cancer. Nat. Genet. 2015, 47, 736–745. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Erho, N.; Chan, J.M.; Feng, F.Y.; Fishbane, N.; Zhao, S.G.; Simko, J.P.; Cowan, J.E.; Lehrer, J.; Alshalalfa, M.; et al. The Diverse Genomic Landscape of Clinically Low-risk Prostate Cancer. Eur. Urol. 2018, 74, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Lovf, M.; Zhao, S.; Axcrona, U.; Johannessen, B.; Bakken, A.C.; Carm, K.T.; Hoff, A.M.; Myklebost, O.; Meza-Zepeda, L.A.; Lie, A.K.; et al. Multifocal Primary Prostate Cancer Exhibits High Degree of Genomic Heterogeneity. Eur. Urol. 2019, 75, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Salami, S.S.; Hovelson, D.H.; Kaplan, J.B.; Mathieu, R.; Udager, A.M.; Curci, N.E.; Lee, M.; Plouffe, K.R.; de la Vega, L.L.; Susani, M.; et al. Transcriptomic heterogeneity in multifocal prostate cancer. JCI Insight 2018, 3, e123468. [Google Scholar] [CrossRef] [PubMed]
- Fenton, J.J.; Weyrich, M.S.; Durbin, S.; Liu, Y.; Bang, H.; Melnikow, J. Prostate-Specific Antigen-Based Screening for Prostate Cancer: Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2018, 319, 1914–1931. [Google Scholar] [CrossRef]
- Kearns, J.T.; Lin, D.W. Improving the Specificity of PSA Screening with Serum and Urine Markers. Curr. Urol. Rep. 2018, 19, 80. [Google Scholar] [CrossRef]
- Ahdoot, M.; Wilbur, A.R.; Reese, S.E.; Lebastchi, A.H.; Mehralivand, S.; Gomella, P.T.; Bloom, J.; Gurram, S.; Siddiqui, M.; Pinsky, P.; et al. MRI-Targeted, Systematic, and Combined Biopsy for Prostate Cancer Diagnosis. N. Engl. J. Med. 2020, 382, 917–928. [Google Scholar] [CrossRef]
- Sarkar, D. The Role of Multi-Parametric MRI and Fusion Biopsy for the Diagnosis of Prostate Cancer—A Systematic Review of Current Literature. Adv. Exp. Med. Biol. 2018, 1095, 111–123. [Google Scholar]
- Marra, G.; Ploussard, G.; Futterer, J.; Valerio, M.; Party, E.-Y.P.C.W. Controversies in MR targeted biopsy: Alone or combined, cognitive versus software-based fusion, transrectal versus transperineal approach? World J. Urol. 2019, 37, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Alford, A.V.; Brito, J.M.; Yadav, K.K.; Yadav, S.S.; Tewari, A.K.; Renzulli, J. The Use of Biomarkers in Prostate Cancer Screening and Treatment. Rev. Urol. 2017, 19, 221–234. [Google Scholar] [PubMed]
- Lamy, P.J.; Allory, Y.; Gauchez, A.S.; Asselain, B.; Beuzeboc, P.; de Cremoux, P.; Fontugne, J.; Georges, A.; Hennequin, C.; Lehmann-Che, J.; et al. Prognostic Biomarkers Used for Localised Prostate Cancer Management: A Systematic Review. Eur. Urol. Focus 2018, 4, 790–803. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Jeon, J.; Mejia, S.; Yao, C.Q.; Ignatchenko, V.; Nyalwidhe, J.O.; Gramolini, A.O.; Lance, R.S.; Troyer, D.A.; Drake, R.R.; et al. Targeted proteomics identifies liquid-biopsy signatures for extracapsular prostate cancer. Nat. Commun. 2016, 7, 11906. [Google Scholar] [CrossRef]
- Shi, T.; Fillmore, T.L.; Sun, X.; Zhao, R.; Schepmoes, A.A.; Hossain, M.; Xie, F.; Wu, S.; Kim, J.S.; Jones, N.; et al. Antibody-free, targeted mass-spectrometric approach for quantification of proteins at low picogram per milliliter levels in human plasma/serum. Proc. Nat. Acad. Sci. USA 2012, 109, 15395–15400. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Jamal, M.; Tan, S.H.; Song, Y.; Young, D.; Chen, Y.; Katta, S.; Ying, K.; Ravindranath, L.; Woodle, T.; et al. Molecular profiling of radical prostatectomy tissue from patients with no sign of progression identifies ERG as the strongest independent predictor of recurrence. Oncotarget 2019, 10, 6466–6483. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.I.; Egevad, L.; Amin, M.B.; Delahunt, B.; Srigley, J.R.; Humphrey, P.A. The 2014 International Society of Urological Pathology (ISUP) Consensus Conference on Gleason Grading of Prostatic Carcinoma: Definition of Grading Patterns and Proposal for a New Grading System. Am. J. Surg. Pathol. 2016, 40, 244–252. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Dobbin, K.K.; Simon, R.M. Optimally splitting cases for training and testing high dimensional classifiers. BMC Med. Genom. 2011, 4, 31. [Google Scholar] [CrossRef]
- Simpraga, S.; Alvarez-Jimenez, R.; Mansvelder, H.D.; van Gerven, J.M.A.; Groeneveld, G.J.; Poil, S.S.; Linkenkaer-Hansen, K. EEG machine learning for accurate detection of cholinergic intervention and Alzheimer’s disease. Sci. Rep. 2017, 7, 5775. [Google Scholar] [CrossRef]
- Li, X.J.; Hayward, C.; Fong, P.Y.; Dominguez, M.; Hunsucker, S.W.; Lee, L.W.; McLean, M.; Law, S.; Butler, H.; Schirm, M.; et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Sci. Transl. Med. 2013, 5, 207ra142. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Pasta, D.J.; Elkin, E.P.; Litwin, M.S.; Latini, D.M.; Du Chane, J.; Carroll, P.R. The University of California, San Francisco Cancer of the Prostate Risk Assessment score: A straightforward and reliable preoperative predictor of disease recurrence after radical prostatectomy. J. Urol. 2005, 173, 1938–1942. [Google Scholar] [CrossRef] [PubMed]
- Memorial Sloan Kettering Cancer Center. Dynamic Prostate Cancer Nomogram: Coefficients. Available online: https://www.mskcc.org/nomograms/prostate/post_op/coefficients (accessed on 7 May 2020).
- Perner, S.; Hofer, M.D.; Kim, R.; Shah, R.B.; Li, H.; Moller, P.; Hautmann, R.E.; Gschwend, J.E.; Kuefer, R.; Rubin, M.A. Prostate-specific membrane antigen expression as a predictor of prostate cancer progression. Hum. Pathol. 2007, 38, 696–701. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Lawrentschuk, N.; Francis, R.J.; Tang, C.; Vela, I.; Thomas, P.; Rutherford, N.; Martin, J.M.; Frydenberg, M.; Shakher, R.; et al. Prostate-specific membrane antigen PET-CT in patients with high-risk prostate cancer before curative-intent surgery or radiotherapy (proPSMA): A prospective, randomised, multicentre study. Lancet 2020, 395, 1208–1216. [Google Scholar] [CrossRef]
- Thomas, R.; True, L.D.; Bassuk, J.A.; Lange, P.H.; Vessella, R.L. Differential expression of osteonectin/SPARC during human prostate cancer progression. Clin. Cancer Res. 2000, 6, 1140–1149. [Google Scholar] [PubMed]
- Derosa, C.A.; Furusato, B.; Shaheduzzaman, S.; Srikantan, V.; Wang, Z.; Chen, Y.; Seifert, M.; Ravindranath, L.; Young, D.; Nau, M.; et al. Elevated osteonectin/SPARC expression in primary prostate cancer predicts metastatic progression. Prostate Cancer Prostatic Dis. 2012, 15, 150–156. [Google Scholar] [CrossRef]
- Padua, D.; Massague, J. Roles of TGFbeta in metastasis. Cell Res. 2009, 19, 89–102. [Google Scholar] [CrossRef]
- Reis, S.T.; Pontes-Junior, J.; Antunes, A.A.; Sousa-Canavez, J.M.; Abe, D.K.; Cruz, J.A.; Dall’oglio, M.F.; Crippa, A.; Passerotti, C.C.; Ribeiro-Filho, L.A.; et al. Tgf-beta1 expression as a biomarker of poor prognosis in prostate cancer. Clinics (Sao Paulo) 2011, 66, 1143–1147. [Google Scholar]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef]
- He, J.; Sun, X.; Shi, T.; Schepmoes, A.A.; Fillmore, T.L.; Petyuk, V.A.; Xie, F.; Zhao, R.; Gritsenko, M.A.; Yang, F.; et al. Antibody-independent targeted quantification of TMPRSS2-ERG fusion protein products in prostate cancer. Mol. Oncol. 2014, 8, 1169–1180. [Google Scholar] [CrossRef]
- Wang, H.; Barbieri, C.E.; He, J.; Gao, Y.; Shi, T.; Wu, C.; Schepmoes, A.A.; Fillmore, T.L.; Chae, S.S.; Huang, D.; et al. Quantification of mutant SPOP proteins in prostate cancer using mass spectrometry-based targeted proteomics. J. Transl. Med. 2017, 15, 175. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Hossain, M.; Schepmoes, A.A.; Fillmore, T.L.; Sokoll, L.J.; Kronewitter, S.R.; Izmirlian, G.; Shi, T.; Qian, W.J.; Leach, R.J.; et al. Analysis of serum total and free PSA using immunoaffinity depletion coupled to SRM: Correlation with clinical immunoassay tests. J. Proteom. 2012, 75, 4747–4757. [Google Scholar] [CrossRef] [PubMed]
- Kohaar, I.; Petrovics, G.; Srivastava, S. A Rich Array of Prostate Cancer Molecular Biomarkers: Opportunities and Challenges. Int. J. Mol. Sci. 2019, 20, 1813. [Google Scholar] [CrossRef] [PubMed]
- Udager, A.M.; Tomlins, S.A. Molecular Biomarkers in the Clinical Management of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030601. [Google Scholar] [CrossRef]
- Carneiro, A.; Priante Kayano, P.; Gomes Barbosa, A.R.; Langer Wroclawski, M.; Ko Chen, C.; Cavlini, G.C.; Reche, G.J.; Sanchez-Salas, R.; Tobias-Machado, M.; Sowalsky, A.G.; et al. Are localized prostate cancer biomarkers useful in the clinical practice? Tumor Biol. 2018, 40, 1010428318799255. [Google Scholar] [CrossRef]
- Donovan, M.J.; Cordon-Cardo, C. Genomic analysis in active surveillance: Predicting high-risk disease using tissue biomarkers. Curr. Opin. Urol. 2014, 24, 303–310. [Google Scholar] [CrossRef]
- Cucchiara, V.; Cooperberg, M.R.; Dall’Era, M.; Lin, D.W.; Montorsi, F.; Schalken, J.A.; Evans, C.P. Genomic Markers in Prostate Cancer Decision Making. Eur. Urol. 2018, 73, 572–582. [Google Scholar] [CrossRef]
- Dani, H.; Loeb, S. The role of prostate cancer biomarkers in undiagnosed men. Curr. Opin. Urol. 2017, 27, 210–216. [Google Scholar] [CrossRef]
- Shoji, S. Magnetic resonance imaging-transrectal ultrasound fusion image-guided prostate biopsy: Current status of the cancer detection and the prospects of tailor-made medicine of the prostate cancer. Investig. Clin. Urol. 2019, 60, 4–13. [Google Scholar] [CrossRef]
- Elkhoury, F.F.; Simopoulos, D.N.; Marks, L.S. Targeted Prostate Biopsy in the Era of Active Surveillance. Urology 2018, 112, 12–19. [Google Scholar] [CrossRef]
- Johnson, L.M.; Choyke, P.L.; Figg, W.D.; Turkbey, B. The role of MRI in prostate cancer active surveillance. Biomed. Res. Int. 2014, 2014, 203906. [Google Scholar] [CrossRef] [PubMed]
- Litwin, M.S.; Tan, H.J. The Diagnosis and Treatment of Prostate Cancer: A Review. JAMA 2017, 317, 2532–2542. [Google Scholar] [CrossRef] [PubMed]
- Mottet, N.; De Santis, M.; Briers, E.; Bourke, L.; Gillessen, S.; Grummet, J.P.; Lam, T.B.; van der Poel, H.G.; Rouviere, O.; van den Bergh, R.C.N.; et al. Updated Guidelines for Metastatic Hormone-sensitive Prostate Cancer: Abiraterone Acetate Combined with Castration Is Another Standard. Eur. Urol. 2018, 73, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, I.A.; Vela, I.; Scardino, P.T. Molecular Profiles of Prostate Cancer: To Treat or Not to Treat. Annu. Rev. Med. 2016, 67, 119–135. [Google Scholar] [CrossRef]
- Sun, L.; Gancarczyk, K.; Paquette, E.L.; McLeod, D.; Kane, C.; Kusuda, L.; Lance, R.; Herring, J.; Foley, J.; Baldwin, D.; et al. Introduction to Department of Defense Center for Prostate Disease Research Multicenter National Prostate Cancer Database, and analysis of changes in the PSA-era. Urol. Oncol. 2001, 6, 203–209. [Google Scholar] [CrossRef]
- Furusato, B.; Gao, C.L.; Ravindranath, L.; Chen, Y.; Cullen, J.; McLeod, D.G.; Dobi, A.; Srivastava, S.; Petrovics, G.; Sesterhenn, I.A. Mapping of TMPRSS2-ERG fusions in the context of multi-focal prostate cancer. Mod. Pathol. 2008, 21, 67–75. [Google Scholar] [CrossRef]
- Furusato, B.; Tan, S.H.; Young, D.; Dobi, A.; Sun, C.; Mohamed, A.A.; Thangapazham, R.; Chen, Y.; McMaster, G.; Sreenath, T.; et al. ERG oncoprotein expression in prostate cancer: Clonal progression of ERG-positive tumor cells and potential for ERG-based stratification. Prostate Cancer Prostatic Dis. 2010, 13, 228–237. [Google Scholar] [CrossRef]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Total | Nonevent | BCR * | Metastasis | p Value |
|---|---|---|---|---|---|
| N | 338 | 161 | 124 | 53 | |
| Age at diagnosis (years) | |||||
| Mean (SD) | 59.5 (7.7) | 59.0 (8.1) | 59.2 (7.7) | 61.7 (5.9) | 0.0897 |
| Time from diagnosis to RP * (months) | |||||
| Median (range) | 2.3 (0.2–21) | 2.2 (0.2–21) | 2.5 (0.2–9) | 2.0 (0.7–10) | 0.4689 |
| Race | |||||
| AA * | 120 (35.6) | 55 (34.2) | 48 (39.0) | 17 (32.1) | |
| CA * and Other | 217 (64.4) | 106 (65.8) | 75 (61.0) | 36 (67.9) | 0.5882 |
| PSA * at diagnosis (ng/mL) | |||||
| <10 | 262 (78.0) | 133 (83.6) | 90 (72.6) | 39 (73.6) | |
| 10–20 | 59 (17.6) | 25 (15.7) | 25 (20.2) | 9 (17.0) | |
| >20 | 15 (4.5) | 1 (0.6) | 9 (7.3) | 5 (9.4) | 0.0062 |
| Clinical T stage | |||||
| T1-T2a | 274 (82.0) | 134 (85.4) | 107 (86.3) | 33 (62.3) | |
| T2b-T2c | 52 (15.6) | 22 (14.0) | 15 (12.1) | 15 (28.3) | |
| T3a-T4 | 8 (2.4) | 1 (0.6) | 2 (1.6) | 5 (9.4) | 0.0005 |
| Biopsy grade | |||||
| 6 or less | 182 (58.3) | 100 (70.9) | 68 (57.1) | 14 (26.9) | |
| 7 | 95 (30.4) | 35 (24.8) | 41 (34.4) | 19 (36.5) | |
| 8–10 | 35 (11.2) | 6 (4.3) | 10 (8.4) | 19 (36.5) | <0.0001 |
| NCCN * risk | |||||
| Low | 125 (40.6) | 69 (50.7) | 46 (38.3) | 10 (19.2) | |
| Intermediate | 134 (43.5) | 59 (43.4) | 55 (45.8) | 20 (38.5) | |
| High | 49 (15.9) | 8 (5.9) | 19 (15.8) | 22 (42.3) | <0.0001 |
| Pathological T stage | |||||
| pT2 | 174 (52.6) | 119 (74.4) | 46 (37.4) | 9 (18.8) | |
| pT3-4 | 157 (47.4) | 41 (25.6) | 77 (62.6) | 39 (81.2) | <0.0001 |
| GG * | |||||
| GG1 | 31 (9.3) | 18 (11.2) | 13 (10.6) | 0 | |
| GG2 | 105 (31.6) | 77 (48.1) | 27 (22.0) | 1 (2.0) | |
| GG3 | 6 (1.8) | 2 (1.2) | 4 (3.2) | 0 | |
| GG4 | 124 (37.4) | 54 (33.8) | 49 (39.8) | 21 (42.9) | |
| GG5 | 66 (19.9) | 9 (5.6) | 30 (24.4) | 27 (55.1) | <0.0001 |
| Surgical margin | |||||
| Negative | 209 (63.7) | 126 (79.2) | 62 (51.2) | 21 (43.8) | |
| Positive | 119 (36.3) | 33 (20.8) | 59 (48.8) | 27 (56.2) | <0.0001 |
| Post-RP Follow-up (months) | |||||
| Median (range) | 150 (18–253) | 156 (121–252) | 129 (18–229) | 124 (24–253) | <0.0001 |
| DM vs. Nonevent | BCR vs. Nonevent | GG (3–5 vs. 1–2) | ||||
|---|---|---|---|---|---|---|
| Protein | AUC | p Value | AUC | p Value | AUC | p Value |
| ANXA2 | 0.535 | 0.741 | 0.538 | 0.341 | 0.499 | 0.692 |
| CAMKK2 | 0.591 | 0.051 | 0.604 | 0.009 | 0.667 | <0.001 |
| CCND1 | 0.532 | 0.166 | 0.624 | 0.037 | 0.592 | 0.034 |
| EGFR | 0.628 | 0.012 | 0.578 | 0.035 | 0.653 | <0.001 |
| ERG | 0.543 | 0.668 | 0.546 | 0.830 | 0.482 | 0.708 |
| FOLH1 | 0.653 | 0.001 | 0.627 | <0.001 | 0.657 | <0.001 |
| MMP9 | 0.562 | 0.518 | 0.511 | 0.770 | 0.554 | 0.643 |
| MUC1 | 0.570 | 0.461 | 0.474 | 0.603 | 0.506 | 0.200 |
| NCOA2 | 0.637 | 0.095 | 0.613 | 0.225 | 0.670 | 0.001 |
| PSA | 0.730 | 0.001 | 0.529 | 0.955 | 0.608 | 0.005 |
| SMAD4 | 0.511 | 0.622 | 0.526 | 0.092 | 0.521 | 0.383 |
| SPINK1 | 0.486 | 0.207 | 0.548 | 0.535 | 0.547 | 0.470 |
| SPARC | 0.800 | <0.001 | 0.695 | <0.001 | 0.715 | <0.001 |
| TFF3 | 0.541 | 0.174 | 0.472 | 0.578 | 0.492 | 0.751 |
| TGFB1 | 0.788 | <0.001 | 0.649 | <0.001 | 0.705 | <0.001 |
| VEGFA | 0.528 | 0.168 | 0.601 | 0.040 | 0.573 | 0.009 |
| Protein | Cut-Point * | 95% CI ** | Sensitivity | Specificity | PPV *** | NPV |
|---|---|---|---|---|---|---|
| FOLH1 | −0.54 | −0.55, −0.53 | 0.731 | 0.419 | 0.325 | 0.803 |
| PSA | −0.12 | −0.15, −0.08 | 0.827 | 0.412 | 0.350 | 0.862 |
| SPARC | −0.53 | −0.55, −0.52 | 0.865 | 0.522 | 0.409 | 0.910 |
| TGFB1 | −0.50 | −0.52, −0.48 | 0.846 | 0.493 | 0.389 | 0.893 |
| Protein | Cut-Point * | 95% CI ** | Sensitivity | Specificity | PPV *** | NPV |
|---|---|---|---|---|---|---|
| SPARC | −0.74 | −0.75, −0.72 | 0.874 | 0.301 | 0.523 | 0.732 |
| TGFB1 | −0.71 | −0.73, −0.69 | 0.866 | 0.309 | 0.523 | 0.724 |
| Variable | Model 1 * | Model 2 ** | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p Value | HR | 95% CI | p Value | |
| Age at diagnosis | 1.00 | 0.93–1.07 | 0.898 | 1.03 | 0.96–1.11 | 0.407 |
| Race (AA vs. CA) | 0.94 | 0.33–2.74 | 0.916 | 1.59 | 0.54–4.64 | 0.396 |
| Risk (intermediate vs. low) | 2.31 | 0.69–7.76 | 0.176 | 1.49 | 0.41–5.47 | 0.545 |
| Risk (high vs. low) | 4.68 | 1.14–19.22 | 0.032 | 2.29 | 0.52–10.16 | 0.274 |
| 5-protein classifier (high vs. low) | 5.09 | 1.11–23.38 | 0.036 | 1.03 | 1.02–1.05 | <0.001 |
| Variable | Model 1 * | Model 2 ** | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | p Value | HR | 95% CI | p Value | |
| Pathology T (pT3 vs. pT2) | 2.54 | 0.78–8.27 | 0.122 | 1.94 | 0.52–7.15 | 0.321 |
| GG (GG5 vs. GG1-4) | 3.42 | 1.17–10.03 | 0.025 | 2.04 | 0.52–8.04 | 0.309 |
| Surgical margin (Pos vs. neg) | 1.31 | 0.47–3.68 | 0.603 | 1.23 | 0.42–3.57 | 0.705 |
| 5-protein classifier (high vs. low) | 3.71 | 0.82–16.88 | 0.089 | 1.02 | 1.01–1.05 | 0.018 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, Y.; Wang, Y.-T.; Chen, Y.; Wang, H.; Young, D.; Shi, T.; Song, Y.; Schepmoes, A.A.; Kuo, C.; Fillmore, T.L.; et al. Proteomic Tissue-Based Classifier for Early Prediction of Prostate Cancer Progression. Cancers 2020, 12, 1268. https://doi.org/10.3390/cancers12051268
Gao Y, Wang Y-T, Chen Y, Wang H, Young D, Shi T, Song Y, Schepmoes AA, Kuo C, Fillmore TL, et al. Proteomic Tissue-Based Classifier for Early Prediction of Prostate Cancer Progression. Cancers. 2020; 12(5):1268. https://doi.org/10.3390/cancers12051268
Chicago/Turabian StyleGao, Yuqian, Yi-Ting Wang, Yongmei Chen, Hui Wang, Denise Young, Tujin Shi, Yingjie Song, Athena A. Schepmoes, Claire Kuo, Thomas L. Fillmore, and et al. 2020. "Proteomic Tissue-Based Classifier for Early Prediction of Prostate Cancer Progression" Cancers 12, no. 5: 1268. https://doi.org/10.3390/cancers12051268
APA StyleGao, Y., Wang, Y.-T., Chen, Y., Wang, H., Young, D., Shi, T., Song, Y., Schepmoes, A. A., Kuo, C., Fillmore, T. L., Qian, W.-J., Smith, R. D., Srivastava, S., Kagan, J., Dobi, A., Sesterhenn, I. A., Rosner, I. L., Petrovics, G., Rodland, K. D., ... Liu, T. (2020). Proteomic Tissue-Based Classifier for Early Prediction of Prostate Cancer Progression. Cancers, 12(5), 1268. https://doi.org/10.3390/cancers12051268