Methylation-Based Signatures for Gastroesophageal Tumor Classification

Abstract
1. Introduction
2. Methods
2.1. Data Processing and Normalization
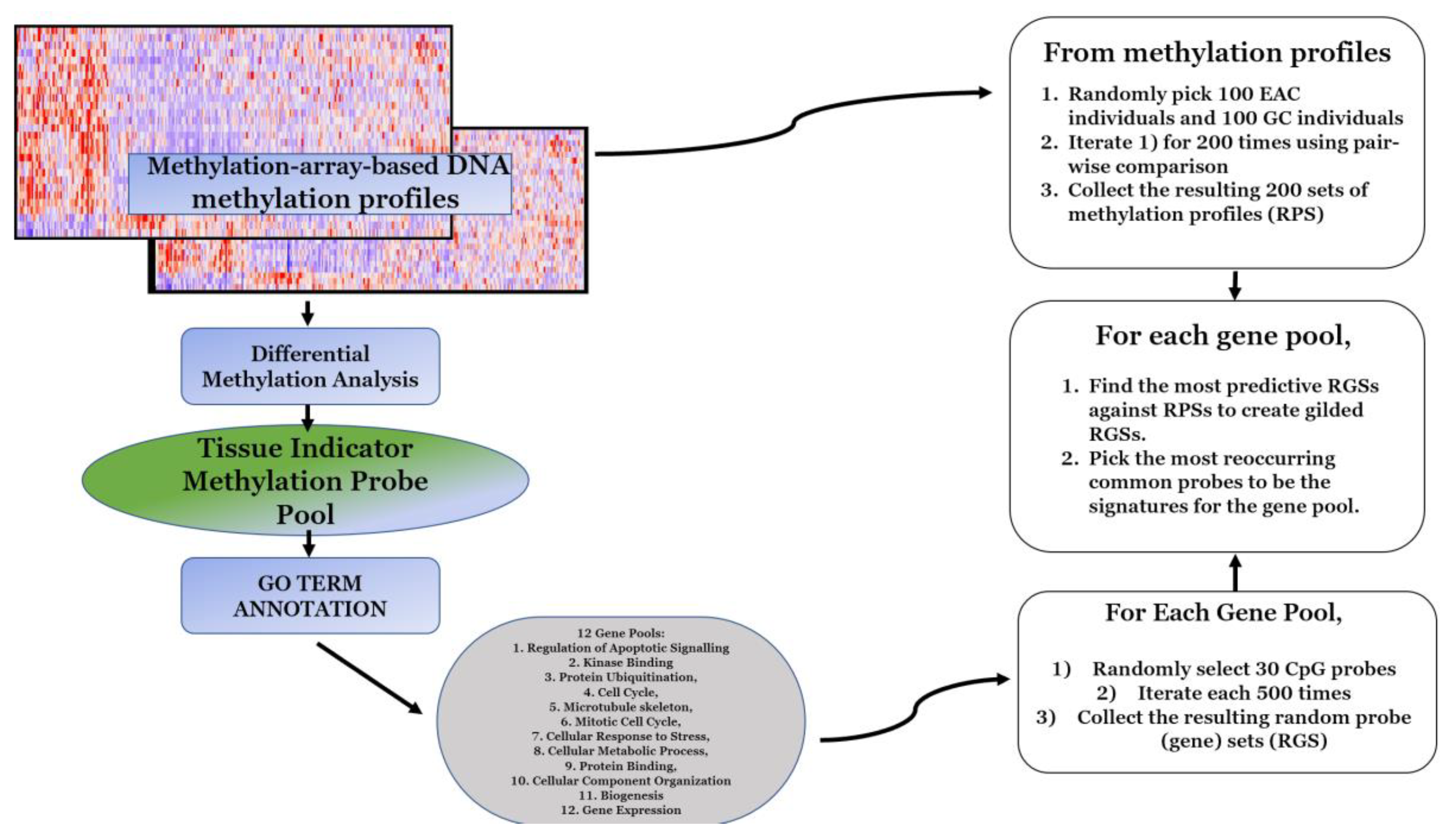
2.2. Multiple Survival Screening (MSS) Methodology and Optimization
- In the discovery dataset, probes that demonstrated significantly differential methylation profiles between the subgroup of esophageal carcinomas and the subgroup of stomach adenocarcinomas were selected to form a pool.
- Significance was first defined by a probe-wise analysis performed using the limma Bioconductor package pipeline to calculate moderated t-statistics.
- In original MSS methodology, statistical significance was determined by a p-value less than 0.05 and the application of other hyperparameters in downstream steps, while Feng et al. [8] suggested to optimize this step and choose the most significant 300–500 genes at this step. Thus, probes that had a false discovery rate (FDR)-corrected p-value of less than 0.0001 and a FC (fold change) greater than 3 were selected to form a gene pool of 536 unique probes.
- Gene pools were annotated for GO (gene ontology) terms by the Database for Annotation, Visualization and Integrated Discovery (DAVID) [24]. For the given probe pool, we partitioned genes with replacement into GO-defined subpools. In the subgroup of cellular response to DNA damage stimulus comprised of 54 genes, similarly, the numbers of genes were as the following for other subgroups: Regulation of apoptotic signaling (38), kinase binding (35), protein ubiquitination (47), cell cycle (87), microtubule skeleton (59), mitotic cell cycle (58), cellular response to stress (95), cellular metabolic process (377), protein binding (376), cellular component organization or biogenesis (252), and gene expression (210).
- Following the optimized MSS methodology proposed by Feng et al. [8], for a given GO-defined subpool, 500 random gene sets were created for each GO term. We then created 200 random patient sets (RPS) and to ensure an RPS was not dominated by a single subtype of stomach cancer we performed the following:
- We obtained assigned subtypes for samples in the TCGA STAD cohort: Epstein–Barr virus (EBV), microsatellite instability (MSI), genomic stability, and chromosomal instability (CIN).
- Twenty-five samples were randomly drawn from each subtype to obtain 100 samples of evenly distributed subtypes in each RPS.
- Then, 100 samples of esophageal carcinoma were drawn and added to each RPS to create a balanced 200 sample RPS ratio of 1:1 stomach adenocarcinoma to esophageal carcinoma.
- Then, 1,000,000 RPSs were created. A pairwise comparison was then performed to select the 200 most dissimilar RPSs. This was performed on a standard laptop in <8 hours.
- Each random gene set (RGS) was then tested against all 200 RPSs: Fisher’s tests were used to determine if the RGS enriched the RPSs. This took <3 hours on a standard laptop. The p-values yielded by the Fisher’s tests were recorded and the reciprocal of their average was considered as the enrichment score of the RGS. For each GO term, the top 50 most significant RGSs were selected to be “gilded RGSs” based on the enrichment score. According to Feng et al. [8], the threshold for choosing the genes in the gilded RGSs could be chosen freely as it did not affect the downstream results.
- The unique 30 most frequently appearing genes across the gilded RGSs of a GO term were then drawn as the set of signature genes for the corresponding GO term.
2.3. Gene Set Selection
- The sample in question would be removed from the dataset.
- For each GO term, we used the 30 signature genes to translate methylation profiles of patients in the training dataset into 1D vectors of shape (30, 1).
- Centroids of each cluster of samples (esophageal or stomach) were then calculated based on the GO term vector.
- The sample was then reintroduced to the dataset and assigned to a centroid based on the GO term vector.
- This was performed for each sample in each dataset to evaluate the prediction and recall accuracy of each signature set.
2.4. Probe Distance to Transcription Start Site Calculation
- The midpoint of each transcription start site was calculated and represented the TSS location.
- The distance from the midpoint of the TSS to the probe in question was calculated.
- The average distance from TSS to probe for each signature was then calculated.
3. Results
3.1. Methylation Signatures for Differentiation between Gastric and Esophageal Adenocarcinomas
3.2. Utilization of Optimized Methodology to Use Fewer Computational Resources
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Barra, W.F.; Moreira, F.C.; Cruz, A.M.P.; Khayat, A.S.; Calcagno, D.Q.; Dos Santos, N.P.C.; Junior, R.W.M.; Araújo, T.M.T.; Ishak, G.; Demachki, S.; et al. GEJ cancers: Gastric or esophageal tumors? Searching for the answer according to molecular identity. Oncotarget 2017, 8, 104286–104294. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Canto, M.I.; Piantadosi, S.; Montgomery, E.; Weinstein, W.M.; Herman, J.G.; Dannenberg, A.J.; Yang, V.W.; Shar, A.O.; Hawk, E.; et al. Secondary chemoprevention of Barrett’s esophagus with celecoxib: Results of a randomized trial. J. Natl. Cancer Inst. 2007, 99, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Peek, R.M., Jr.; Fiske, C.; Wilson, K.T. Role of innate immunity in Helicobacter pylori-induced gastric malignancy. Physiol. Rev. 2010, 90, 831–858. [Google Scholar] [CrossRef] [PubMed]
- Polk, D.B.; Peek, R.M., Jr. Helicobacter pylori: Gastric cancer and beyond. Nat. Rev. Cancer 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Shaheen, N.; Ransohoff, D.F. Gastroesophageal Reflux, Barrett Esophagus, and Esophageal Cancer. JAMA 2002, 287, 1972. [Google Scholar] [CrossRef]
- Li, J.; Lenferink, A.E.G.; Deng, Y.; Collins, C.; Cui, Q.; Purisima, E.O.; Wang, E. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat. Commun. 2010, 1. [Google Scholar] [CrossRef]
- Wang, J.; Wang, Q.; Liu, H.; Hu, B.; Zhou, W.; Cheng, Y. MicroRNA expression and its implication for the diagnosis and therapeutic strategies of gastric cancer. Cancer Lett. 2010, 297, 137–143. [Google Scholar] [CrossRef]
- Feng, X.; Wang, E.; Cui, Q. Gene Expression-Based Predictive Markers for Paclitaxel Treatment in ER+ and ER- Breast Cancer. Front. Genet. 2019, 10, 156. [Google Scholar] [CrossRef]
- Bédard, E.L.R.; Inculet, R.I.; Malthaner, R.A.; Brecevic, E.; Vincent, M.; Dar, R. The role of surgery and postoperative chemoradiation therapy in patients with lymph node positive esophageal carcinoma. Cancer 2001, 91, 2423–2430. [Google Scholar] [CrossRef]
- Rice, T.W.; Adelstein, D.J.; Chidel, M.A.; Rybicki, L.A.; Decamp, M.M.; Murthy, S.C.; Blackstone, E.H. Benefit of postoperative adjuvant chemoradiotherapy in locoregionally advanced esophageal carcinoma. J. Thorac. Cardiovasc. Surg. 2003, 126, 1590–1596. [Google Scholar] [CrossRef]
- Cunningham, D.; Allum, W.H.; Stenning, S.P.; Weeden, S. Perioperative chemotherapy in operable gastric and lower oesophageal cancer: Final results of a randomised, controlled trial (the MAGIC trial, ISRCTN 93793971). J. Clin. Oncol. 2005, 23, 4001. [Google Scholar] [CrossRef]
- Gallo, A.; Cha, C. Updates on esophageal and gastric cancers. World J. Gastroenterol. 2006, 12, 3237–3242. [Google Scholar] [CrossRef] [PubMed]
- Buas, M.F.; Vaughan, T.L. Epidemiology and risk factors for gastroesophageal junction tumors: Understanding the rising incidence of this disease. Semin. Radiat. Oncol. 2013, 23, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Ekstrom, A.M.; Signorello, L.B.; Hansson, L.-E.; Bergstrom, R.; Lindgren, A.; Nyren, O. Evaluating Gastric Cancer Misclassification: A Potential Explanation for the Rise in Cardia Cancer Incidence. JNCI J. Natl. Cancer Inst. 1999, 91, 786–790. [Google Scholar] [CrossRef]
- Zhu, J.; Zheng, Z.; Wang, J.; Sun, J.; Wang, P.; Cheng, X.; Fu, L.; Zhang, L.; Wang, Z.; Li, Z. Different miRNA expression profiles between human breast cancer tumors and serum. Front. Genet. 2014, 5, 149. [Google Scholar] [CrossRef]
- Xu, X.; Li, J.; Zou, J.; Feng, X.; Zhang, C.; Zheng, R.; Duanmu, W.; Saha-Mandal, A.; Ming, Z.; Wang, E. Association of Germline Variants in Natural Killer Cells With Tumor Immune Microenvironment Subtypes, Tumor-Infiltrating Lymphocytes, Immunotherapy Response, Clinical Outcomes, and Cancer Risk. JAMA Netw. Open 2019, 2, e199292. [Google Scholar] [CrossRef]
- Milanese, J.S.; Tibiche, C.; Zou, J.; Meng, Z.; Nantel, A.; Drouin, S.; Marcotte, R.; Wang, E. Germline variants associated with leukocyte genes predict tumor recurrence in breast cancer patients. NPJ Precis. Oncol. 2019, 3, 28. [Google Scholar] [CrossRef]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation Patterns Between DNA Methylation and Gene Expression in The Cancer Genome Atlas. Cancer Inform. 2019, 18, 1176935119828776. [Google Scholar] [CrossRef]
- Chen, Z.; Saad, R.; Jia, P.; Peng, D.; Zhu, S.; Washington, M.K.; Zhao, Z.; Xu, Z.; El-Rifai, W. Gastric adenocarcinoma has a unique microRNA signature not present in esophageal adenocarcinoma. Cancer 2013, 119, 1985–1993. [Google Scholar] [CrossRef]
- Mealy, K.; Feely, J.; Reid, I.; Mcsweeney, J.; Walsh, T.; Hennessy, T. Tumour marker detection in oesophageal carcinoma. Eur. J. Surg. Oncol. (EJSO) 1996, 22, 505–507. [Google Scholar] [CrossRef]
- Tan, C.; Qian, X.; Guan, Z.; Yang, B.; Ge, Y.; Wang, F.; Cai, J. Potential biomarkers for esophageal cancer. SpringerPlus 2016, 5, 467. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Cheng, C.; Ma, J.; Liew, C.C.; Geng, X. Gene expression signature for detection of gastric cancer in peripheral blood. Oncol. Lett. 2018, 15, 9802–9810. [Google Scholar] [CrossRef] [PubMed]
- Maksimovic, J.; Phipson, B.; Oshlack, A. A cross-package Bioconductor workflow for analysing methylation array data. F1000Research 2016, 5, 1281. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; A Lempicki, R. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Wang, E.; Zaman, N.; McGee, S.; Milanese, J.-S.; Masoudi-Nejad, A.; O’Connor-McCourt, M. Predictive genomics: A cancer hallmark network framework for predicting tumor clinical phenotypes using genome sequencing data. Semin. Cancer Boil. 2015, 30, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Oxnard, G.; Klein, E.; Swanton, C.; Seiden, M.; Cummings, S.R.; Absalan, F.; Alexander, G.; Allen, B.; Amini, H.; et al. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31. [Google Scholar] [CrossRef]
- Lhermitte, B.; Egele, C.; Weingertner, N.; Ambrosetti, D.; Dadone, B.; Kubiniek, V.; Bellocq, J. Adequately defining tumor cell proportion in tissue samples for molecular testing improves interobserver reproducibility of its assessment. Virchows Arch. 2017, 470, 21–27. [Google Scholar] [CrossRef]
- Van Krieken, J.H.; Rouleau, E.; Ligtenberg, M.J.; Normanno, N.; Patterson, S.D.; Jung, A. RAS testing in metastatic colorectal cancer: Advances in Europe. Virchow Arch. 2016, 468, 383–396. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Training Set (N = 628) | ||
|---|---|---|
| Sex | Male | 443 |
| Female | 185 | |
| Unknown | 0 | |
| Age (yrs.) | Range | 27–90 |
| Mean | 64.7 | |
| Unknown | 5 | |
| Stage | I | 54 |
| II | 136 | |
| III | 286 | |
| IV | 124 | |
| Unknown | 28 | |
| Validation Set (N = 548) | ||
|---|---|---|
| Sex | Male | 391 |
| Female | 139 | |
| Unknown | 18 | |
| Age (yrs.) | Range | 23–92 |
| Mean | 64.5 | |
| Unknown (No. of patients) | 43 | |
| Differentially Methylated Probes (DMPs) (FC >3, p-value < 10−4) | N = 81814 |
|---|---|
| Differentially Methylated Regions | N = 28054 |
| DMPs overlapping with 27k Array | N = 536 |
| Regulatory Feature Group | Percentage of DMPs |
| Promoter Associated | 81.3% |
| Gene Associated | 0.25% |
| Gene Associated Cell Specific | 0.55% |
| Relation to Island | Percentage of DMPs |
| OpenSea | 18.4% |
| Island | 55.0% |
| N_Shore | 11.5% |
| S_Shore | 9.7% |
| GO Term | Fold Enrichment | FDR | GO Accession Number |
|---|---|---|---|
| Single-multicellular organism process | 1.0508656015 | 0.82 × 10−7 | 0044707 |
| Anatomical structure morphogenesis | 1.083136144 | 3.45 × 10−6 | 0009653 |
| Single-organism developmental process | 1.050520832 | 6.97 × 10−6 | 0044767 |
| Anatomical structure development | 1.050309311 | 8.2 × 10−6 | 0048856 |
| Cell fate commitment | 1.281284221 | 2.60 × 10−5 | 0045165 |
| Epithelium development | 1.1318538 | 2.65 × 10−5 | 0060429 |
| Developmental process | 1.047949197 | 1.32 × 10−5 | 0032502 |
| Organ morphogenesis | 1.13549378 | 3.45 × 10−5 | 0009887 |
| Tissue development | 1.097778353 | 5.93 × 10−5 | 0009888 |
| Skeletal system development | 1.187655599 | 1.30 × 10−4 | 0001501 |
| Multicellular organism development | 1.050805467 | 1.45 × 10−4 | 0007275 |
| Tube development | 1.157944133 | 0.001406 | 0035295 |
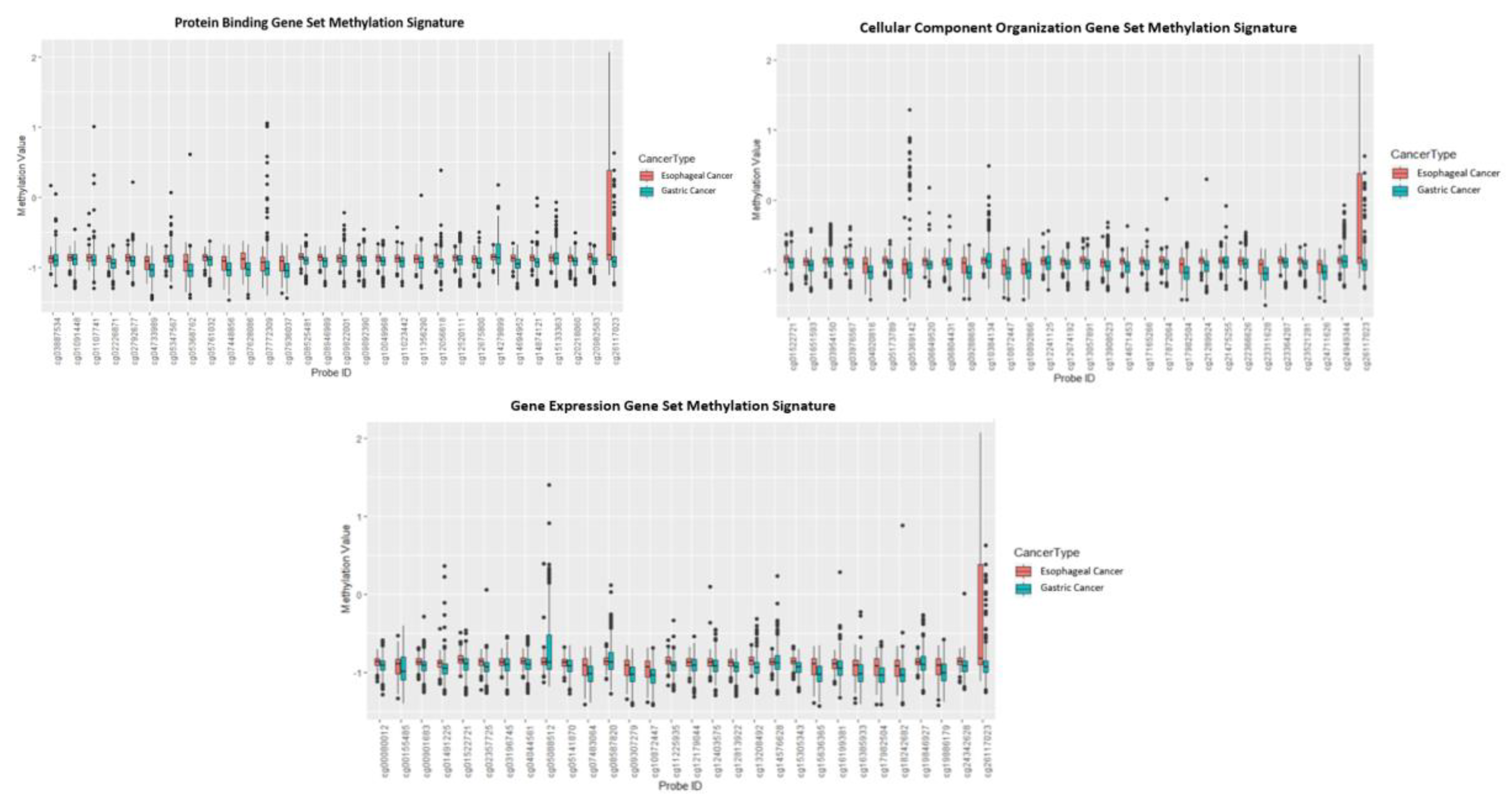
| Methylation Signature | Training Set (nEC = 185, nGC = 443) | Validation Set (nEC = 164, nGC = 383) |
|---|---|---|
| Protein Binding | Precision: 99.5% Recall: 96.6% | Precision: 94.7% Recall: 99.2% |
| Cellular Component Organization | Precision: 98.5% Recall: 98.0% | Precision: 96.8% Recall: 97.6% |
| Gene Expression | Precision: 98.0% Recall: 98.5% | Precision: 97.6% Recall: 96.8% |
| Cellular Component Biogenesis | Gene Expression | Protein Binding | ||||||
|---|---|---|---|---|---|---|---|---|
| Probe | Gene | Gene Description | Probe | Gene | Gene Description | Probe | Gene | Gene Description |
| cg26117023 | LOXL3 | Lysyl Oxidase Like 3 | cg00901683 | CPSF4 | Cleavage and Polyadenylation Specific Factor 4 | cg08946989 | TBC1D7 | TBC1 Domain Family Member 7 |
| cg04020816 | MAN2A1 | Mannosidase Alpha Class 2A Member 1 | cg01491225 | ZCCHC9 | Zinc Finger CCHC-Type Containing 9 | cg01091448 | AMACR | Alpha-Methylacyl-CoA Racemase |
| cg21475255 | DAG1 | Dystroglycan 1 | cg11225935 | KDM5A | Lysine Demethylase 5A | cg09892390 | ARHGAP21 | Rho GTPase Activating Protein 21 |
| cg23364287 | IP6K2 | Inositol Hexakisphosphate Kinase 2 | cg14576628 | PRMT1 | Protein Arginine Methyltransferase 1 | cg01107741 | CANT1 | Calcium Activated Nucleotidase 1 |
| cg01651593 | CDC20 | Cell Division Cycle 20 | cg00155485 | MED13L | Mediator Complex Subunit 13L | cg03887534 | BCL2L13 | BCL2 Like 13 |
| cg05173789 | RPLP0 | Ribosomal Protein Lateral Stalk Subunit P0 | cg08587820 | BHLHE40 | Basic Helix-Loop-Helix Family Member E40 | cg05368762 | TMBIM6 | Transmembrane BAX Inhibitor Motif Containing 6 |
| cg09288658 | ZAK | Mitogen-Activated Protein Kinase Kinase Kinase 20 | cg12403575 | TRADD | TNFRSF1A Associated Via Death Domain | cg26117023 | LOXL3 | Lysyl Oxidase Like 3 |
| cg06649520 | ARFIP1 | ADP Ribosylation Factor Interacting Protein 1 | cg12179044 | GCN1L1 | GCN1 Activator of EIF2AK4 | cg05761032 | CCPG1 | Cell Cycle Progression 1 |
| cg10384134 | RPS9 | Ribosomal Protein S9 | cg12813922 | RAB3GAP1 | RAB3 GTPase Activating Protein Catalytic Subunit 1 | cg07448856 | ZNF670 | Zinc Finger Protein 670 |
| cg10872447 | GTF2F2 | General Transcription Factor IIF Subunit 2 | cg17982504 | DDX28 | DEAD-Box Helicase 28 | cg07628086 | AP2B1 | Adaptor Related Protein Complex 2 Subunit Beta 1 |
| cg14671453 | STX4 | Syntaxin 4 | cg19846927 | MRPL44 | Mitochondrial Ribosomal Protein L44 | cg09822001 | APOA1BP | NAD(P)HX Epimerase |
| cg17982504 | DDX28 | DEAD-Box Helicase 28 | cg19886179 | PSMD14 | Proteasome 26S Subunit, Non-ATPase 14 | cg10049968 | FAM219A | Family with Sequence Similarity 219 Member A |
| cg21289924 | EIF3A | Eukaryotic Translation Initiation Factor 3 Subunit A | cg02357725 | IMP3 | IMP U3 Small Nucleolar Ribonucleoprotein 3 | cg11356290 | AZI2 | 5-Azacytidine Induced 2 |
| cg01522721 | MIR1181 | MicroRNA 1181 | cg05141870 | MIR423 | MicroRNA 423 | cg12520111 | PPIA | Peptidylprolyl Isomerase A |
| cg03954150 | C18orf55 | Translocase of Inner Mitochondrial Membrane 21 | cg07483064 | ENO1 | Enolase 1 | cg12675800 | TRAPPC6B | Trafficking Protein Particle Complex 6B |
| cg03976567 | AKD1 | Adenylate Kinase 9 | cg09307279 | GLT8D1 | Glycosyltransferase 8 Domain Containing 1 | cg14874121 | HSD17B4 | Hydroxysteroid 17-Beta Dehydrogenase 4 |
| cg05369142 | ALS2CL | ALS2 C-Terminal Like | cg10872447 | GTF2F2 | General Transcription Factor IIF Subunit 2 | cg20218060 | CLK1 | CDC Like Kinase 1 |
| cg06804431 | GNRHR2 | Gonadotropin Releasing Hormone Receptor 2 (Pseudogene) | cg13208492 | TSN | Translin | cg20982583 | POLR2F | RNA Polymerase II Subunit F |
| cg10892866 | PYGO2 | Pygopus Family PHD Finger 2 | cg15305343 | NSUN4 | NOP2/Sun RNA Methyltransferase 4 | cg02226871 | VPS28 | Vacuolar Protein Sorting-Associated Protein 28 Homolog |
| cg12241125 | EIF4H | Eukaryotic Translation Initiation Factor 4H | cg15636365 | PNPLA7 | Patatin Like Phospholipase Domain Containing 7 | cg02792677 | MRPL4 | Mitochondrial Ribosomal Protein L4 |
| cg12674192 | MAK16 | MAK16 Homolog | cg16199381 | TSTD2 | Thiosulfate Sulfurtransferase Like Domain Containing 2 | cg04733989 | NAGA | Alpha-N-Acetylgalactosaminidase |
| cg13057891 | ERCC5 | ERCC Excision Repair 5, Endonuclease | cg16385933 | PDCD4 | Programmed Cell Death 4 | cg05347567 | ZC3H10 | Zinc Finger CCCH-Type Containing 10 |
| cg13908523 | PRKCD | Protein Kinase C Delta | cg18242682 | FOXK2 | Forkhead Box K2 | cg07772309 | NELF | NMDA Receptor Synaptonuclear Signaling and Neuronal Migration Factor |
| cg17165266 | KRT18 | Keratin 18 | cg24342628 | KDM1B | Lysine Demethylase 1B | cg07936037 | SSR1 | Signal Sequence Receptor Subunit 1 |
| cg17872064 | NOP58 | NOP58 Ribonucleoprotein | cg26117023 | LOXL3 | Lysyl Oxidase Like 3 | cg08525481 | OGFR | Opioid Growth Factor Receptor |
| cg22366626 | ZFYVE20 | Rabenosyn, RAB Effector | cg00080012 | EED | Embryonic Ectoderm Development | cg11023442 | PITPNA-AS1 | PITPNA antisense RNA 1 |
| cg23311628 | RAB8B | RAB8B, Member RAS Oncogene Family | cg01522721 | CDC37 | Cell Division Cycle 37 | cg12056618 | KLF13 | Kruppel Like Factor 13 |
| cg23521281 | WDR75 | WD Repeat Domain 75 | cg03196745 | ISCU | Iron-Sulfur Cluster Assembly Enzyme | cg14279899 | IFNGR1 | Interferon Gamma Receptor 1 |
| cg24711626 | KIAA1012 | Trafficking Protein Particle Complex 8 | cg04044561 | POP7 | POP7 Homolog, Ribonuclease P/MRP Subunit | cg14694952 | HTT | Huntingtin |
| cg24949344 | ANO6 | Anoctamin 6 | cg05088512 | DKKL1 | Dickkopf Like Acrosomal Protein 1 | cg15133363 | HILPDA | Hypoxia Inducible Lipid Droplet Associated |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alabi, N.; Sheka, D.; Siddiqui, A.; Wang, E. Methylation-Based Signatures for Gastroesophageal Tumor Classification. Cancers 2020, 12, 1208. https://doi.org/10.3390/cancers12051208
Alabi N, Sheka D, Siddiqui A, Wang E. Methylation-Based Signatures for Gastroesophageal Tumor Classification. Cancers. 2020; 12(5):1208. https://doi.org/10.3390/cancers12051208
Chicago/Turabian StyleAlabi, Nikolay, Dropen Sheka, Ashar Siddiqui, and Edwin Wang. 2020. "Methylation-Based Signatures for Gastroesophageal Tumor Classification" Cancers 12, no. 5: 1208. https://doi.org/10.3390/cancers12051208
APA StyleAlabi, N., Sheka, D., Siddiqui, A., & Wang, E. (2020). Methylation-Based Signatures for Gastroesophageal Tumor Classification. Cancers, 12(5), 1208. https://doi.org/10.3390/cancers12051208