Opportunities for Conventional and In Situ Cancer Vaccine Strategies and Combination with Immunotherapy for Gastrointestinal Cancers, A Review



Abstract
1. Introduction
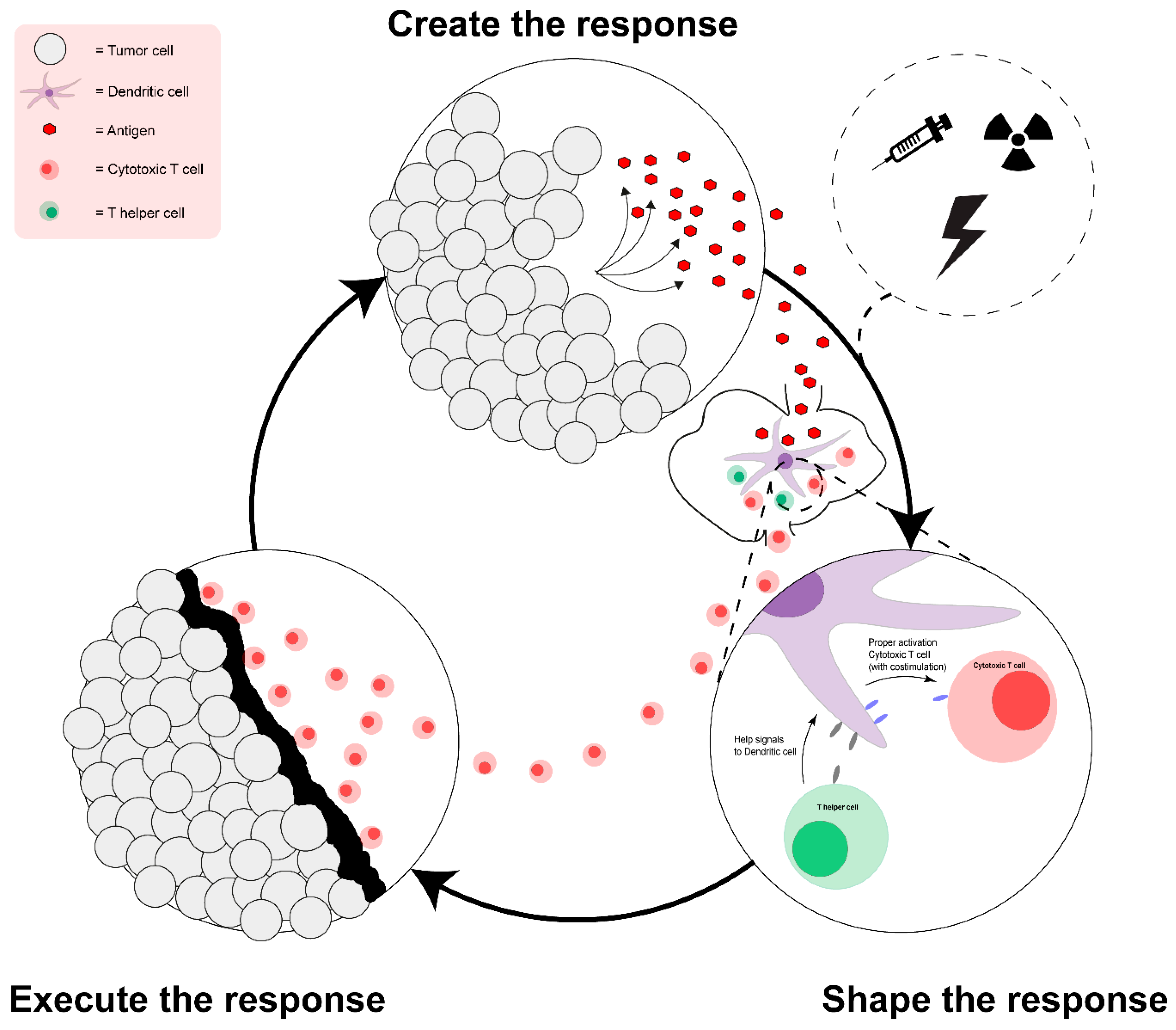
2. The Ideal Anti-Tumor Immune Response and the Limitation of Vaccination
3. Conventional Vaccines
3.1. Peptide Vaccine
3.2. Genetic Vaccine
3.3. Tumor Cell Vaccine
3.4. DC-Vaccine
4. In Situ Vaccines
4.1. Radiotherapy
4.2. Chemotherapy
4.3. Oncolytic Viruses
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rubin, G.; Walter, F.; Emery, J.; De Wit, N. Reimagining the diagnostic pathway for gastrointestinal cancer. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 181–188. [Google Scholar] [CrossRef]
- Kaiser, J.; Couzin-Frankel, J. Cancer immunotherapy sweeps Nobel for medicine. Science 2018, 362, 13. [Google Scholar] [CrossRef]
- Hodi, F.S. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Schachter, J.; Ribas, A.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus ipilimumab for advanced melanoma: Final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 2017, 390, 1853–1862. [Google Scholar] [CrossRef]
- Reck, M.; Rodríguez-Abreu, D.; Robinson, A.G.; Hui, R.; Csőszi, T.; Fülöp, A.; Gottfried, M.; Peled, N.; Tafreshi, A.; Cuffe, S.; et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2016, 375, 1823–1833. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Koemans, W.J.; Chalabi, M.; van Sandick, J.W.; van Dieren, J.M.; Kodach, L.L. Beyond the PD-L1 horizon: In search for a good biomarker to predict success of immunotherapy in gastric and esophageal adenocarcinoma. Cancer Lett. 2019, 442, 279–286. [Google Scholar] [CrossRef]
- Ahrends, T.; Busselaar, J.; Severson, T.M.; Bąbała, N.; de Vries, E.; Bovens, A.; Wessels, L.; van Leeuwen, F.; Borst, J. CD4(+) T cell help creates memory CD8(+) T cells with innate and help-independent recall capacities. Nat. Commun. 2019, 10, 5531. [Google Scholar] [CrossRef]
- Ahrends, T.; Spanjaard, A.; Pilzecker, B.; Bąbała, N.; Bovens, A.; Xiao, Y.; Jacobs, H.; Borst, J. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 2017, 47, 848–861.e5. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Mauriello, A.; Zeuli, R.; Cavalluzzo, B.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Ceccarelli, M.; Tagliamonte, M.; Buonaguro, L. High Somatic Mutation and Neoantigen Burden Do Not Correlate with Decreased Progression-Free Survival in HCC Patients not Undergoing Immunotherapy. Cancers 2019, 11, 1824. [Google Scholar] [CrossRef]
- Ito, Z.; Kan, S.; Bito, T.; Horiuchi, S.; Akasu, T.; Yoshida, S.; Kajihara, M.; Hokari, A.; Saruta, M.; Yoshida, N.; et al. Predicted Markers of Overall Survival in Pancreatic Cancer Patients Receiving Dendritic Cell Vaccinations Targeting WT1. Oncology 2019, 97, 135–148. [Google Scholar] [CrossRef]
- Balachandran, V.P.; Łuksza, M.; Zhao, J.N.; Makarov, V.; Moral, J.A.; Remark, R.; Herbst, B.; Askan, G.; Bhanot, U.; Senbabaoglu, Y.; et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 2017, 551, 512–516. [Google Scholar] [CrossRef]
- Stanley, M. Tumour virus vaccines: Hepatitis B virus and human papillomavirus. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160268. [Google Scholar] [CrossRef]
- Lavanchy, D. Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J. Viral Hepat. 2004, 11, 97–107. [Google Scholar] [CrossRef]
- Herzog, T.J.; Huh, W.K.; Downs, L.S.; Smith, J.S.; Monk, B.J. Initial lessons learned in HPV vaccination. Gynecol. Oncol. 2008, 109, S4–S11. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Ann. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Fridman, W.H.; Pagès, F.; Sautes-Fridman, C.; Galon, J. The immune contexture in human tumours: Impact on clinical outcome. Nat. Rev. Cancer 2012, 12, 298–306. [Google Scholar] [CrossRef]
- Pages, F.; Kirilovsky, A.; Mlecnik, B.; Asslaber, M.; Tosolini, M.; Bindea, G.; Lagorce, C.; Wind, P.; Marliot, F.; Bruneval, P.; et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. J. Clin. Oncol. 2009, 27, 5944–5951. [Google Scholar] [CrossRef]
- Łuksza, M.; Riaz, N.; Makarov, V.; Balachandran, V.P.; Hellmann, M.D.; Solovyov, A.; Rizvi, N.A.; Merghoub, T.; Levine, A.J.; Chan, T.A.; et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017, 551, 517–520. [Google Scholar] [CrossRef]
- Stromnes, I.M.; DelGiorno, K.E.; Greenberg, P.D.; Hingorani, S.R. Stromal reengineering to treat pancreas cancer. Carcinogenesis 2014, 35, 1451–1460. [Google Scholar] [CrossRef]
- Gunderson, A.J.; Kaneda, M.M.; Tsujikawa, T.; Nguyen, A.V.; Affara, N.I.; Ruffell, B.; Gorjestani, S.; Liudahl, S.M.; Truitt, M.; Olson, P.; et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016, 6, 270–285. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017, 27, 109–118. [Google Scholar] [CrossRef]
- Hirbod-Mobarakeh, A.; Mirghorbani, M.; Hajiju, F.; Marvi, M.; Bashiri, K.; Rezaei, N. Myeloid-derived suppressor cells in gastrointestinal cancers: A systematic review. J. Gastroenterol. Hepatol. 2016, 31, 1246–1256. [Google Scholar] [CrossRef]
- Bilgin, B.; Sendur, M.A.; Bülent Akıncı, M.; Şener Dede, D.; Yalçın, B. Targeting the PD-1 pathway: A new hope for gastrointestinal cancers. Curr. Med. Res. Opin. 2017, 33, 749–759. [Google Scholar] [CrossRef]
- Barriga, V. The Complex Interaction between the Tumor Micro-Environment and Immune Checkpoints in Breast Cancer. Cancers 2019, 11, 1205. [Google Scholar] [CrossRef]
- Barriga, V.; Kuol, N.; Nurgali, K.; Apostolopoulos, V. The mechanisms tumor cells utilize to evade the host’s immune system. Maturitas 2017, 105, 8–15. [Google Scholar]
- Shekarian, T.; Sivado, E.; Jallas, A.C.; Depil, S.; Kielbassa, J.; Janoueix-Lerosey, I. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci. Transl. Med. 2019, 11, eaat5025. [Google Scholar] [CrossRef]
- Aznar, M.A.; Molina, C.; Teijeira, A.; Rodriguez, I.; Azpilikueta, A.; Garasa, S. Repurposing the yellow fever vaccine for intratumoral immunotherapy. EMBO Mol. Med. 2020, 12, e10375. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar]
- Vanichapol, T.; Chutipongtanate, S.; Anurathapan, U.; Hongeng, S. Immune Escape Mechanisms and Future Prospects for Immunotherapy in Neuroblastoma. Biomed. Res. Int. 2018, 2018, 1812535. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 Challenges in Cancer Immunotherapy. Immunity 2020, 52, 17–35. [Google Scholar] [CrossRef]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.; Kastenmüller, W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Nimanong, S.; Ostroumov, D.; Wingerath, J.; Knocke, S.; Woller, N.; Gürlevik, E. CD40 Signaling Drives Potent Cellular Immune Responses in Heterologous Cancer Vaccinations. Cancer Res. 2017, 77, 1918–1926. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Umansky, V.; Blattner, C.; Gebhardt, C.; Utikal, J. The Role of Myeloid-Derived Suppressor Cells (MDSC) in Cancer Progression. Vaccines 2016, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Kraman, M.; Bambrough, P.J.; Arnold, J.N.; Roberts, E.W.; Magiera, L.; Jones, J.O. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science 2010, 330, 827–830. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Pachter, J.A. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Teichmann, S.A. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Patel, S.; Alam, A.; Pant, R.; Chattopadhyay, S. Wnt Signaling and Its Significance within the Tumor Microenvironment: Novel Therapeutic Insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef]
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef]
- Skwarczynski, M.; Toth, I. Peptide-based synthetic vaccines. Chem. Sci. 2016, 7, 842–854. [Google Scholar] [CrossRef]
- Bekaii-Saab, T.; Wesolowski, R.; Ahn, D.H.; Wu, C.; Mortazavi, A.; Lustberg, M.; Kaumaya, P. TPhase I Immunotherapy Trial with Two Chimeric HER-2 B-Cell Peptide Vaccines Emulsified in Montanide ISA 720VG and Nor-MDP Adjuvant in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2019, 25, 3495–3507. [Google Scholar] [CrossRef]
- Bezu, L.; Kepp, O.; Cerrato, G.; Pol, J.; Fucikova, J.; Spisek, R.; Galluzzi, L. Trial watch: Peptide-based vaccines in anticancer therapy. Oncoimmunology 2018, 7, e1511506. [Google Scholar] [CrossRef]
- Dou, Y.; van Montfoort, N.; van den Bosch, A.; De Man, R.A.; Zom, G.G.; Krebber, W.J.; Woltman, A.M. HBV-Derived Synthetic Long Peptide Can Boost CD4+ and CD8+ T-Cell Responses in Chronic HBV Patients Ex Vivo. J. Infect. Dis. 2018, 217, 827–839. [Google Scholar] [CrossRef]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Stachyra, A.; Gora-Sochacka, A.; Sirko, A. DNA vaccines against influenza. Acta Biochim. Pol. 2014, 61, 515–522. [Google Scholar] [CrossRef]
- Becker, J.T.; Olson, B.M.; Johnson, L.E.; Davies, J.G.; Dunphy, E.J.; McNeel, D.G. DNA vaccine encoding prostatic acid phosphatase (PAP) elicits long-term T-cell responses in patients with recurrent prostate cancer. J. Immunother. 2010, 33, 639–647. [Google Scholar] [CrossRef]
- Chudley, L.; McCann, K.; Mander, A.; Tjelle, T.; Campos-Perez, J.; Godeseth, R.; Heath, C. DNA fusion-gene vaccination in patients with prostate cancer induces high-frequency CD8(+) T-cell responses and increases PSA doubling time. Cancer Immunol. Immunother. 2012, 61, 2161–2170. [Google Scholar] [CrossRef]
- Leitner, W.W.; Ying, H.; Restifo, N.P. DNA and RNA-based vaccines: Principles, progress and prospects. Vaccine 1999, 18, 765–777. [Google Scholar] [CrossRef]
- Li, L.; Petrovsky, N. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert Rev. Vaccines 2016, 15, 313–329. [Google Scholar] [CrossRef]
- McNeel, D.G.; Dunphy, E.J.; Davies, J.G.; Frye, T.P.; Johnson, L.E.; Staab, M.J.; Liu, G. Safety and immunological efficacy of a DNA vaccine encoding prostatic acid phosphatase in patients with stage D0 prostate cancer. J. Clin. Oncol. 2009, 27, 4047–4054. [Google Scholar] [CrossRef]
- Hopkins, A.C.; Yarchoan, M.; Durham, J.N.; Yusko, E.C.; Rytlewski, J.A.; Robins, H.S.; Jaffee, E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Simons, J.W.; Sacks, N. Granulocyte-macrophage colony-stimulating factor-transduced allogeneic cancer cellular immunotherapy: The GVAX vaccine for prostate cancer. Urol. Oncol. 2006, 24, 419–424. [Google Scholar] [CrossRef]
- Van den Eertwegh, A.J.; Versluis, J.; Van den Berg, H.P.; Santegoets, S.J.; Van Moorselaar, R.J.A.; Van der Sluis, T.M.; Pinedo, H.M. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 509–517. [Google Scholar] [CrossRef]
- Yarchoan, M.; Ferguson, A.K.; Durham, J.N.; Rozich, N.; Rodriguez, C.; Huang, C.Y.; Donehower, R.C. A phase 2 study of GVAX colon vaccine with cyclophosphamide and pembrolizumab in patients with mismatch repair proficient advanced colorectal cancer. Cancer Med. 2020, 9, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Loveland, B.E.; Zhao, A.; White, S.; Gan, H.; Hamilton, K.; Xing, P.X.; Kyriakou, P. Mannan-MUC1-pulsed dendritic cell immunotherapy: A phase I trial in patients with adenocarcinoma. Clin. Cancer Res. 2006, 12, 869–877. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.L.; Quinn, M.A.; Grant, P.T.; Allen, D.G.; Jobling, T.W.; White, S.C.; McKenzie, I.F. A phase 2, single-arm study of an autologous dendritic cell treatment against mucin 1 in patients with advanced epithelial ovarian cancer. J. Immunother. Cancer 2014, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Bodey, B.; Bodey, B., Jr.; Siegel, S.E.; Kaiser, H.E. Failure of cancer vaccines: The significant limitations of this approach to immunotherapy. Anticancer. Res. 2000, 20, 2665–2676. [Google Scholar]
- Aerts, J.G.; de Goeje, P.L.; Cornelissen, R.; Kaijen-Lambers, M.E.; Bezemer, K.; van der Leest, C.H.; Braakman, E. Autologous Dendritic Cells Pulsed with Allogeneic Tumor Cell Lysate in Mesothelioma: From Mouse to Human. Clin. Cancer Res. 2018, 24, 766–776. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Gerritsen, W.R.; De Vries, I.J.M.; Figdor, C.G. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin. Cancer Res. 2016, 22, 1897–1906. [Google Scholar] [CrossRef]
- Bryant, C.E.; Sutherland, S.; Kong, B.; Papadimitrious, M.S.; Fromm, P.D.; Hart, D.N. Dendritic cells as cancer therapeutics. Semin. Cell Dev. Biol. 2019, 86, 77–88. [Google Scholar] [CrossRef]
- Rosalia, R.A.; Quakkelaar, E.D.; Redeker, A.; Khan, S.; Camps, M.; Drijfhout, J.W.; Janssen, G. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur. J. Immunol. 2013, 43, 2554–2565. [Google Scholar] [CrossRef]
- Sprooten, J.; Ceusters, J.; Coosemans, A.; Agostinis, P.; De Vleeschouwer, S.; Zitvogel, L.; Garg, A.D. Trial watch: Dendritic cell vaccination for cancer immunotherapy. Oncoimmunology 2019, 8, e1638212. [Google Scholar] [CrossRef]
- Van Willigen, W.W.; Bloemendal, M.; Gerritsen, W.R.; Schreibelt, G.; de Vries, I.J.M.; Bol, K.F. Dendritic Cell Cancer Therapy: Vaccinating the Right Patient at the Right Time. Front. Immunol. 2018, 9, 2265. [Google Scholar] [CrossRef]
- Frank, M.J.; Reagan, P.M.; Bartlett, N.L.; Gordon, L.I.; Friedberg, J.W.; Czerwinski, D.K.; Coffman, R.L. In Situ Vaccination with a TLR9 Agonist and Local Low-Dose Radiation Induces Systemic Responses in Untreated Indolent Lymphoma. Cancer Discov. 2018, 8, 1258–1269. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Radiation therapy to convert the tumor into an in situ vaccine. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 879–880. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Ng, B.; Devitt, M.L.; Babb, J.S.; Kawashima, N.; Liebes, L.; Formenti, S.C. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Goto, T. Radiation as an In Situ Auto-Vaccination: Current Perspectives and Challenges. Vaccines 2019, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, H.A.; Villar, R.C. Radiotherapy and immune response: The systemic effects of a local treatment. Clinics 2018, 73, e557s. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.; Rubner, Y.; Wunderlich, R.; Weiss, E.M.; Pockley, A.G.; Fietkau, R.; Gaipl, U.S. Induction of abscopal anti-tumor immunity and immunogenic tumor cell death by ionizing irradiation—Implications for cancer therapies. Curr. Med. Chem. 2012, 19, 1751–1764. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef]
- Bracci, L.; Schiavoni, G.; Sistigu, A.; Belardelli, F. Immune-based mechanisms of cytotoxic chemotherapy: Implications for the design of novel and rationale-based combined treatments against cancer. Cell Death Differ. 2014, 21, 15–25. [Google Scholar] [CrossRef]
- Casares, N.; Pequignot, M.O.; Tesniere, A.; Ghiringhelli, F.; Roux, S.; Chaput, N.; Coutant, F. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J. Exp. Med. 2005, 202, 1691–1701. [Google Scholar] [CrossRef]
- Cubas, R.; Moskalenko, M.; Cheung, J.; Yang, M.; McNamara, E.; Xiong, H.; Gould, S. Chemotherapy Combines Effectively with Anti-PD-L1 Treatment and Can Augment Antitumor Responses. J. Immunol. 2018, 201, 2273–2286. [Google Scholar] [CrossRef]
- Hannani, D.; Sistigu, A.; Kepp, O.; Galluzzi, L.; Kroemer, G.; Zitvogel, L. Prerequisites for the antitumor vaccine-like effect of chemotherapy and radiotherapy. Cancer J. 2011, 17, 351–358. [Google Scholar] [CrossRef]
- Haynes, N.M.; van der Most, R.G.; Lake, R.A.; Smyth, M.J. Immunogenic anti-cancer chemotherapy as an emerging concept. Curr. Opin. Immunol. 2008, 20, 545–557. [Google Scholar] [CrossRef]
- Ménard, C.; Martin, F.; Apetoh, L.; Bouyer, F.; Ghiringhelli, F. Cancer chemotherapy: Not only a direct cytotoxic effect, but also an adjuvant for antitumor immunity. Cancer Immunol. Immunother. 2008, 57, 1579–1587. [Google Scholar] [CrossRef]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Lin, Y.J. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef]
- Nguyen, T.; Avci, N.G.; Shin, D.H.; Martinez-Velez, N.; Jiang, H. Tune Up In Situ Autovaccination against Solid Tumors with Oncolytic Viruses. Cancers 2018, 10, 171. [Google Scholar] [CrossRef]
- Russell, S.J.; Barber, G.N. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell 2018, 33, 599–605. [Google Scholar] [CrossRef]
- Gatti-Mays, M.E.; Redman, J.M.; Donahue, R.N.; Palena, C.; Madan, R.A.; Karzai, F.; McMahon, S. A Phase I Trial Using a Multitargeted Recombinant Adenovirus 5 (CEA/MUC1/Brachyury)-Based Immunotherapy Vaccine Regimen in Patients with Advanced Cancer. Oncologist 2019. [Google Scholar] [CrossRef]
- Geary, S.M.; Lemke, C.D.; Lubaroff, D.M.; Salem, A.K. The combination of a low-dose chemotherapeutic agent, 5-fluorouracil, and an adenoviral tumor vaccine has a synergistic benefit on survival in a tumor model system. PLoS ONE 2013, 8, e67904. [Google Scholar] [CrossRef]
- Newman, J.H.; Chesson, C.B.; Herzog, N.L.; Bommareddy, P.K.; Aspromonte, S.M.; Pepe, R.; Lee, M. Intratumoral injection of the seasonal flu shot converts immunologically cold tumors to hot and serves as an immunotherapy for cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 1119–1128. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Kaufman, H.L. Talimogene Laherparepvec in Combination with Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef]
- Russell, L.; Peng, K.W. The emerging role of oncolytic virus therapy against cancer. Chin. Clin. Oncol. 2018, 7, 16. [Google Scholar] [CrossRef]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef]
- Graff, J.N.; Chamberlain, E.D. Sipuleucel-T in the treatment of prostate cancer: An evidence-based review of its place in therapy. Core Evid. 2015, 10, 1. [Google Scholar] [CrossRef]
- Jacobs, J.J.; Snackey, C.; Geldof, A.A.; Characiejus, D.; Van Moorselaar, R.J.A.; Den Otter, W. Inefficacy of therapeutic cancer vaccines and proposed improvements. Casus of prostate cancer. Anticancer. Res. 2014, 34, 2689–2700. [Google Scholar]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations with Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef]
- Robert, L.; Ribas, A.; Hu-Lieskovan, S. Combining targeted therapy with immunotherapy. Can 1+1 equal more than 2? Semin. Immunol. 2016, 28, 73–80. [Google Scholar] [CrossRef]
- Van der Burg, S.H. Correlates of immune and clinical activity of novel cancer vaccines. Semin. Immunol. 2018, 39, 119–136. [Google Scholar] [CrossRef]
- Ho, N.I.; Raaijmakers, T.K.; Adema, G.J. Adjuvants Enhancing Cross-Presentation by Dendritic Cells: The Key to More Effective Vaccines? Front. Immunol. 2018, 9, 2874. [Google Scholar] [CrossRef]
- Vermaelen, K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front. Immunol. 2019, 10, 8. [Google Scholar] [CrossRef]
- Bijker, M.S.; van den Eeden, S.J.; Franken, K.L.; Melief, C.J.; Offringa, R.; van der Burg, S.H. CD8+ CTL priming by exact peptide epitopes in incomplete Freund’s adjuvant induces a vanishing CTL response, whereas long peptides induce sustained CTL reactivity. J. Immunol. 2007, 179, 5033–5040. [Google Scholar] [CrossRef]
- Zom, G.G.; Filippov, D.V.; van der Marel, G.A.; Overkleeft, H.S.; Melief, C.J.; Ossendorp, F. Two in one: Improving synthetic long peptide vaccines by combining antigen and adjuvant in one molecule. Oncoimmunology 2014, 3, e947892. [Google Scholar] [CrossRef]
- Rajendra, S.; Wang, B.; Snow, E.T.; Sharma, P.; Pavey, D.; Merrett, N.; Robertson, I.K. Transcriptionally active human papillomavirus is strongly associated with Barrett’s dysplasia and esophageal adenocarcinoma. Am. J. Gastroenterol. 2013, 108, 1082–1093. [Google Scholar] [CrossRef]
- Kenter, G.G.; Welters, M.J.; Valentijn, A.R.P.; Lowik, M.J.; Berends-van der Meer, D.M.; Vloon, A.P.; Wafelman, A.R. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N. Engl. J. Med. 2009, 361, 1838–1847. [Google Scholar] [CrossRef]
- Van Poelgeest, M.I.; Welters, M.J.; van Esch, E.M.; Stynenbosch, L.F.; Kerpershoek, G.; van Meerten, E.L.V.P.; Valentijn, A.R.P. HPV16 synthetic long peptide (HPV16-SLP) vaccination therapy of patients with advanced or recurrent HPV16-induced gynecological carcinoma, a phase II trial. J. Transl. Med. 2013, 11, 88. [Google Scholar] [CrossRef]
- Welters, M.J.; van der Sluis, T.C.; van Meir, H.; Loof, N.M.; van Ham, V.J.; van Duikeren, S.; van Poelgeest, M.I. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci. Transl. Med. 2016, 8, 334ra52. [Google Scholar] [CrossRef]
- Melief, C.J.; Welters, M.J.; Vergote, I.; Kroep, J.R.; Kenter, G.G.; Ottevanger, P.B.; Reyners, A.K. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci. Transl. Med. 2020, 12, eaaz8235. [Google Scholar] [CrossRef]
- Massarelli, E.; William, W.; Johnson, F.; Kies, M.; Ferrarotto, R.; Guo, M.; Haymaker, C. Combining Immune Checkpoint Blockade and Tumor-Specific Vaccine for Patients with Incurable Human Papillomavirus 16-Related Cancer: A Phase 2 Clinical Trial. JAMA Oncol. 2019, 5, 67–73. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Chen, C. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Trimble, C.L.; Morrow, M.P.; Kraynyak, K.A.; Shen, X.; Dallas, M.; Yan, J.; Brown, A.S. Safety, efficacy, and immunogenicity of VGX-3100, a therapeutic synthetic DNA vaccine targeting human papillomavirus 16 and 18 E6 and E7 proteins for cervical intraepithelial neoplasia 2/3: A randomised, double-blind, placebo-controlled phase 2b trial. Lancet 2015, 386, 2078–2088. [Google Scholar] [CrossRef]
- Yuan, J.; Ku, G.Y.; Gallardo, H.F.; Orlandi, F.; Manukian, G.; Rasalan, T.S.; Chapman, P.B. Safety and immunogenicity of a human and mouse gp100 DNA vaccine in a phase I trial of patients with melanoma. Cancer Immun. 2009, 9, 5. [Google Scholar] [PubMed]
- Ginsberg, B.A.; Gallardo, H.F.; Rasalan, T.S.; Adamow, M.; Mu, Z.; Tandon, S.; Carvajal, R.D. Immunologic response to xenogeneic gp100 DNA in melanoma patients: Comparison of particle-mediated epidermal delivery with intramuscular injection. Clin. Cancer Res. 2010, 16, 4057–4065. [Google Scholar] [CrossRef] [PubMed]
- Norell, H.; Poschke, I.; Charo, J.; Wei, W.Z.; Erskine, C.; Piechocki, M.P.; Kiessling, R. Vaccination with a plasmid DNA encoding HER-2/neu together with low doses of GM-CSF and IL-2 in patients with metastatic breast carcinoma: A pilot clinical trial. J. Transl. Med. 2010, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Tiriveedhi, V.; Fleming, T.P.; Goedegebuure, P.S.; Naughton, M.; Ma, C.; Lockhart, C.; Mohanakumar, T. Mammaglobin-A cDNA vaccination of breast cancer patients induces antigen-specific cytotoxic CD4+ICOShi T cells. Breast Cancer Res. Treat. 2013, 138, 109–118. [Google Scholar] [CrossRef]
- Occhipinti, S.; Sponton, L.; Rolla, S.; Caorsi, C.; Novarino, A.; Donadio, M.; Amici, A. Chimeric rat/human HER2 efficiently circumvents HER2 tolerance in cancer patients. Clin. Cancer Res. 2014, 20, 2910–2921. [Google Scholar] [CrossRef]
- Staff, C.; Mozaffari, F.; Haller, B.K.; Wahren, B.; Liljefors, M. A Phase I safety study of plasmid DNA immunization targeting carcinoembryonic antigen in colorectal cancer patients. Vaccine 2011, 29, 6817–6822. [Google Scholar] [CrossRef]
- Bagarazzi, M.L.; Yan, J.; Morrow, M.P.; Shen, X.; Parker, R.L.; Lee, J.C.; Knott, C. Immunotherapy against HPV16/18 generates potent TH1 and cytotoxic cellular immune responses. Sci. Transl. Med. 2012, 4, 155ra138. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Omokoko, T. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Dranoff, G.; Jaffee, E.; Lazenby, A.; Golumbek, P.; Levitsky, H.; Brose, K.; Mulligan, R.C. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc. Natl. Acad. Sci. USA 1993, 90, 3539–3543. [Google Scholar] [CrossRef]
- Dranoff, G. GM-CSF-based cancer vaccines. Immunol. Rev. 2002, 188, 147–154. [Google Scholar] [CrossRef]
- Postow, M.A.; Manuel, M.; Wong, P.; Yuan, J.; Dong, Z.; Liu, C.; Pasqual, N. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J. Immunother. Cancer 2015, 3, 23. [Google Scholar] [CrossRef]
- Guermonprez, P.; Valladeau, J.; Zitvogel, L.; Théry, C.; Amigorena, S. Antigen presentation and T cell stimulation by dendritic cells. Ann. Rev. Immunol. 2002, 20, 621–667. [Google Scholar] [CrossRef]
- Vu Manh, T.P.; Bertho, N.; Hosmalin, A.; Schwartz-Cornil, I.; Dalod, M. Investigating Evolutionary Conservation of Dendritic Cell Subset Identity and Functions. Front. Immunol. 2015, 6, 260. [Google Scholar] [CrossRef]
- Crozat, K.; Guiton, R.; Contreras, V.; Feuillet, V.; Dutertre, C.A.; Ventre, E.; Boudinot, P. The XC chemokine receptor 1 is a conserved selective marker of mammalian cells homologous to mouse CD8alpha+ dendritic cells. J. Exp. Med. 2010, 207, 1283–1292. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Vulink, A.J. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef]
- Hor, J.L.; Whitney, P.G.; Zaid, A.; Brooks, A.G.; Heath, W.R.; Mueller, S.N. Spatiotemporally Distinct Interactions with Dendritic Cell Subsets Facilitates CD4+ and CD8+ T Cell Activation to Localized Viral Infection. Immunity 2015, 43, 554–565. [Google Scholar] [CrossRef]
- Qiao, G.; Wang, X.; Zhou, L.; Zhou, X.; Song, Y.; Wang, S.; Yi, X. Autologous Dendritic Cell-Cytokine Induced Killer Cell Immunotherapy Combined with S-1 Plus Cisplatin in Patients with Advanced Gastric Cancer: A Prospective Study. Clin. Cancer Res. 2019, 25, 1494–1504. [Google Scholar] [CrossRef]
- Katsuda, M.; Miyazawa, M.; Ojima, T.; Katanuma, A.; Hakamada, K.; Sudo, K.; Yamada, S. A double-blind randomized comparative clinical trial to evaluate the safety and efficacy of dendritic cell vaccine loaded with WT1 peptides (TLP0-001) in combination with S-1 in patients with advanced pancreatic cancer refractory to standard chemotherapy. Trials 2019, 20, 242. [Google Scholar] [CrossRef]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Liu, Q. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef]
- Bol, K.F.; Schreibelt, G.; Rabold, K.; Wculek, S.K.; Schwarze, J.K.; Dzionek, A.; Neyns, B. The clinical application of cancer immunotherapy based on naturally circulating dendritic cells. J. Immunother. Cancer 2019, 7, 109. [Google Scholar] [CrossRef]
- Belderbos, R.A.; Baas, P.; Berardi, R.; Cornelissen, R.; Fennell, D.A.; van Meerbeeck, J.P.; Aerts, J.G. A multicenter, randomized, phase II/III study of dendritic cells loaded with allogeneic tumor cell lysate (MesoPher) in subjects with mesothelioma as maintenance therapy after chemotherapy: DENdritic cell Immunotherapy for Mesothelioma (DENIM) trial. Transl. Lung Cancer Res. 2019, 8, 280–285. [Google Scholar] [CrossRef]
- Hammerich, L.; Bhardwaj, N.; Kohrt, H.E.; Brody, J.D. In situ vaccination for the treatment of cancer. Immunotherapy 2016, 8, 315–330. [Google Scholar] [CrossRef]
- Morris, Z.S.; Guy, E.I.; Werner, L.R.; Carlson, P.M.; Heinze, C.M.; Kler, J.S.; Loibner, H. Tumor-Specific Inhibition of In Situ Vaccination by Distant Untreated Tumor Sites. Cancer Immunol. Res. 2018, 6, 825–834. [Google Scholar] [CrossRef]
- Tsung, K.; Norton, J.A. In situ vaccine, immunological memory and cancer cure. Hum. Vaccines Immunother. 2016, 12, 117–119. [Google Scholar] [CrossRef]
- Hammerich, L.; Binder, A.; Brody, J.D. In situ vaccination: Cancer immunotherapy both personalized and off-the-shelf. Mol. Oncol. 2015, 9, 1966–1981. [Google Scholar] [CrossRef]
- Pierce, R.H.; Campbell, J.S.; Pai, S.I.; Brody, J.D.; Kohrt, H.E. In-situ tumor vaccination: Bringing the fight to the tumor. Hum. Vaccines Immunother. 2015, 11, 1901–1909. [Google Scholar] [CrossRef]
- Sheen, M.R.; Fiering, S. In situ vaccination: Harvesting low hanging fruit on the cancer immunotherapy tree. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11, e1524. [Google Scholar] [CrossRef]
- Ilson, D.H. The role of radiation therapy in upper gastrointestinal cancers. Clin. Adv. Hematol. Oncol. 2017, 15, 366–376. [Google Scholar]
- Mehta, S.R.; Suhag, V.; Semwal, M.; Sharma, N. Radiotherapy: Basic Concepts and Recent Advances. Med. J. Armed Forces India 2010, 66, 158–162. [Google Scholar] [CrossRef]
- Ko, E.C.; Formenti, S.C. Radiotherapy and checkpoint inhibitors: A winning new combination? Ther. Adv. Med. Oncol. 2018, 10, 1758835918768240. [Google Scholar] [CrossRef]
- Portella, L.; Scala, S. Ionizing radiation effects on the tumor microenvironment. Semin. Oncol. 2019, 46, 254–260. [Google Scholar] [CrossRef]
- Jiang, W.; Chan, C.K.; Weissman, I.L.; Kim, B.Y.; Hahn, S.M. Immune Priming of the Tumor Microenvironment by Radiation. Trends Cancer 2016, 2, 638–645. [Google Scholar] [CrossRef]
- Menon, H.; Ramapriyan, R.; Cushman, T.R.; Verma, V.; Kim, H.; Schoenhals, J.; Barsoumian, H. Role of Radiation Therapy in Modulation of the Tumor Stroma and Microenvironment. Front. Immunol. 2019, 10, 193. [Google Scholar] [CrossRef]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Demaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Yasmin-Karim, S.; Bruck, P.T.; Moreau, M.; Kunjachan, S.; Chen, G.Z.; Kumar, R.; Ngwa, W. Radiation and Local Anti-CD40 Generate an Effective in situ Vaccine in Preclinical Models of Pancreatic Cancer. Front. Immunol. 2018, 9, 2030. [Google Scholar] [CrossRef]
- Wang, D.Y.; Johnson, D.B.; Davis, E.J. Toxicities Associated With PD-1/PD-L1 Blockade. Cancer J. 2018, 24, 36–40. [Google Scholar] [CrossRef]
- Hiniker, S.M.; Reddy, S.A.; Maecker, H.T.; Subrahmanyam, P.B.; Rosenberg-Hasson, Y.; Swetter, S.M.; Knox, S.J. A Prospective Clinical Trial Combining Radiation Therapy with Systemic Immunotherapy in Metastatic Melanoma. Int. J. Radiat. Oncol. Biol. Phys. 2016, 96, 578–588. [Google Scholar] [CrossRef]
- Tang, C.; Welsh, J.W.; de Groot, P.; Massarelli, E.; Chang, J.Y.; Hess, K.R.; Basu, S.; Curran, M.A.; Cabanillas, M.E.; Subbiah, V.; et al. Ipilimumab with Stereotactic Ablative Radiation Therapy: Phase I Results and Immunologic Correlates from Peripheral T Cells. Clin. Cancer Res. 2017, 23, 1388–1396. [Google Scholar] [CrossRef]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef]
- Golden, E.B.; Demaria, S.; Schiff, P.B.; Chachoua, A.; Formenti, S.C. An abscopal response to radiation and ipilimumab in a patient with metastatic non-small cell lung cancer. Cancer Immunol. Res. 2013, 1, 365–372. [Google Scholar] [CrossRef]
- Shaverdian, N.; Lisberg, A.E.; Bornazyan, K.; Veruttipong, D.; Goldman, J.W.; Formenti, S.C.; Garon, E.B.; Lee, P. Previous radiotherapy and the clinical activity and toxicity of pembrolizumab in the treatment of non-small-cell lung cancer: A secondary analysis of the KEYNOTE-001 phase 1 trial. Lancet Oncol. 2017, 18, 895–903. [Google Scholar] [CrossRef]
- Shewach, D.S.; Kuchta, R.D. Introduction to cancer chemotherapeutics. Chem. Rev. 2009, 109, 2859–2861. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Ju, X.; Wang, J.; Fan, Y.; Ren, M.; Zhang, H. Immunogenic cell death in anticancer chemotherapy and its impact on clinical studies. Cancer Lett. 2018, 438, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Fletcher, R.; Yu, J.; Zhang, L. Immunogenic effects of chemotherapy-induced tumor cell death. Genes Dis. 2018, 5, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Wever, B.; Mazzaschi, G.; Assaraf, Y.G.; Rolfo, C.; Quaini, F.; Tiseo, M.; Giovannetti, E. Molecular basis and rationale for combining immune checkpoint inhibitors with chemotherapy in non-small cell lung cancer. Drug Resist. Updat. 2019, 46, 100644. [Google Scholar] [CrossRef]
- Shen, K.; Cui, J.; Wei, Y.; Chen, X.; Liu, G.; Gao, X.; Li, W.; Lu, H.; Zhan, P.; Lv, T.; et al. Effectiveness and safety of PD-1/PD-L1 or CTLA4 inhibitors combined with chemotherapy as a first-line treatment for lung cancer: A meta-analysis. J. Thorac. Dis. 2018, 10, 6636–6652. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Ma, J.; Wang, J.; Han, C.; Qian, Y.; Chen, G.; Li, X.; Zhang, J.; Cui, P.; Du, W.; et al. Anti-PD-1 therapy combined with chemotherapy in patients with advanced biliary tract cancer. Cancer Immunol. Immunother. 2019, 68, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Nakagawa, K. Combination therapy with PD-1 or PD-L1 inhibitors for cancer. Int. J. Clin. Oncol. 2019, 1–13. [Google Scholar] [CrossRef]
- Wang, C.; Kulkarni, P.; Salgia, R. Combined Checkpoint Inhibition and Chemotherapy: New Era of 1(st)-Line Treatment for Non-Small-Cell Lung Cancer. Mol. Ther. Oncolytics 2019, 13, 1–6. [Google Scholar] [CrossRef]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schutz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Zitvogel, L.; Kroemer, G. Clinical evidence that immunogenic cell death sensitizes to PD-1/PD-L1 blockade. Oncoimmunology 2019, 8, e1637188. [Google Scholar] [CrossRef] [PubMed]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanovic, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef]
- Mazzarella, L.; Duso, B.A.; Trapani, D.; Belli, C.; D’Amico, P.; Ferraro, E.; Viale, G.; Curigliano, G. The evolving landscape of ‘next-generation’ immune checkpoint inhibitors: A review. Eur. J. Cancer 2019, 117, 14–31. [Google Scholar] [CrossRef]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.K. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Cui, J.H.; Lin, K.R.; Yuan, S.H.; Jin, Y.B.; Chen, X.P.; Su, X.K.; Jiang, J.; Pan, Y.M.; Mao, S.L.; Mao, X.F.; et al. TCR Repertoire as a Novel Indicator for Immune Monitoring and Prognosis Assessment of Patients with Cervical Cancer. Front. Immunol. 2018, 9, 2729. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Therapy | Pros | Cons | References |
|---|---|---|---|
| Peptide vaccines |
|
| [47,48,49,50,51] |
| Genetic vaccines |
|
| [52,53,54,55,56,57] |
| Tumor cell vaccines |
|
| [58,59,60,61,62,63] |
| Dendritic cell vaccines |
|
| [64,65,66,67,68,69,70] |
| Therapy | Pros | Cons | References |
|---|---|---|---|
| Radiotherapy |
|
| [46,71,72,73,74,75,76,77] |
| Chemotherapy |
|
| [78,79,80,81,82,83,84] |
| Oncolytic virus |
|
| [32,85,86,87,88,89,90,91,92] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouzid, R.; Peppelenbosch, M.; Buschow, S.I. Opportunities for Conventional and In Situ Cancer Vaccine Strategies and Combination with Immunotherapy for Gastrointestinal Cancers, A Review. Cancers 2020, 12, 1121. https://doi.org/10.3390/cancers12051121
Bouzid R, Peppelenbosch M, Buschow SI. Opportunities for Conventional and In Situ Cancer Vaccine Strategies and Combination with Immunotherapy for Gastrointestinal Cancers, A Review. Cancers. 2020; 12(5):1121. https://doi.org/10.3390/cancers12051121
Chicago/Turabian StyleBouzid, Rachid, Maikel Peppelenbosch, and Sonja I. Buschow. 2020. "Opportunities for Conventional and In Situ Cancer Vaccine Strategies and Combination with Immunotherapy for Gastrointestinal Cancers, A Review" Cancers 12, no. 5: 1121. https://doi.org/10.3390/cancers12051121
APA StyleBouzid, R., Peppelenbosch, M., & Buschow, S. I. (2020). Opportunities for Conventional and In Situ Cancer Vaccine Strategies and Combination with Immunotherapy for Gastrointestinal Cancers, A Review. Cancers, 12(5), 1121. https://doi.org/10.3390/cancers12051121