MHC Class I Regulation: The Origin Perspective


Abstract
1. Introduction
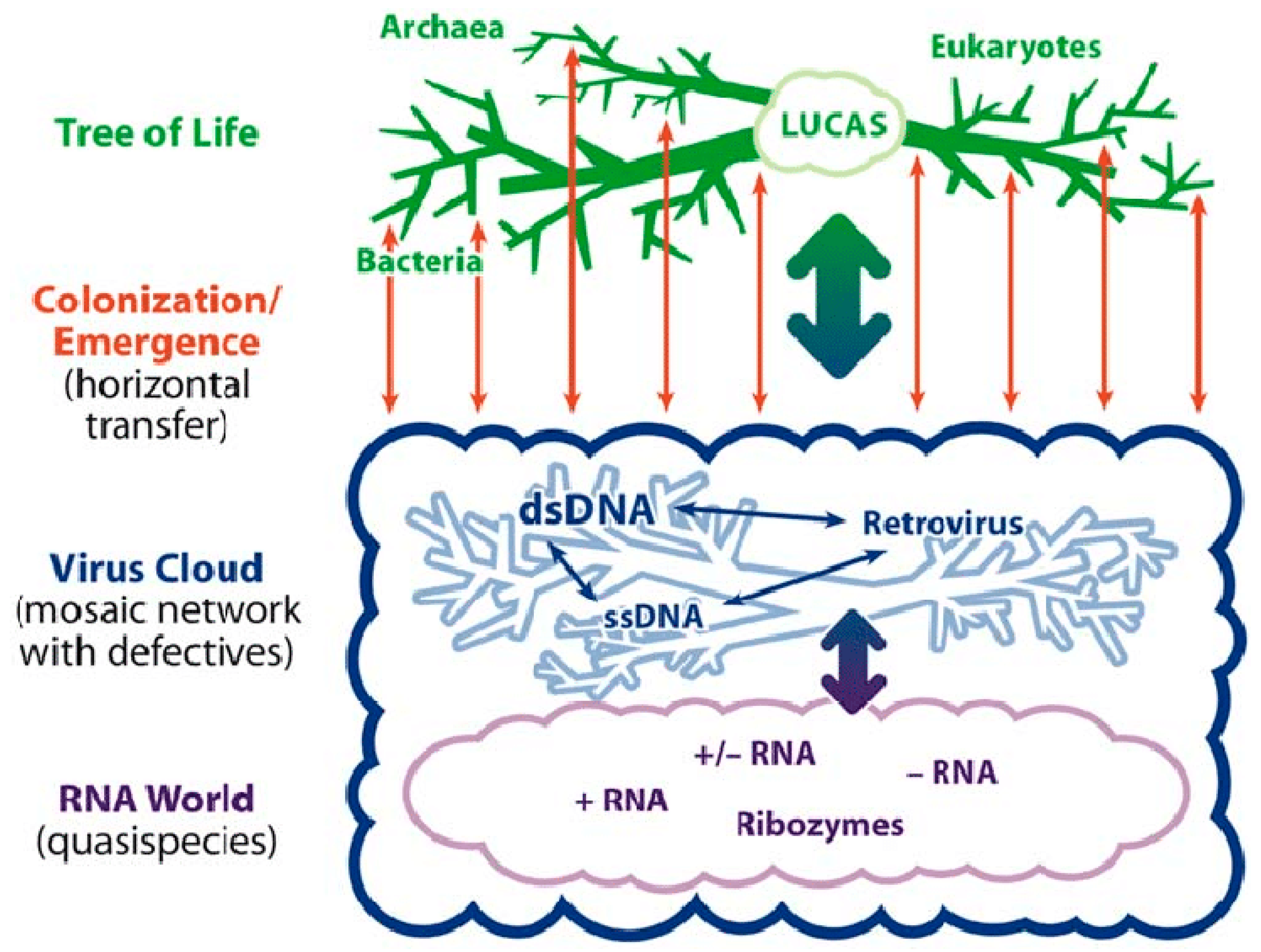
2. Living in the Virosphere
3. Survival in the Virosphere Thanks to Viral Addiction Modules
4. ‘Big Bang’ of an Adaptive Immune Response in Jawed Fish
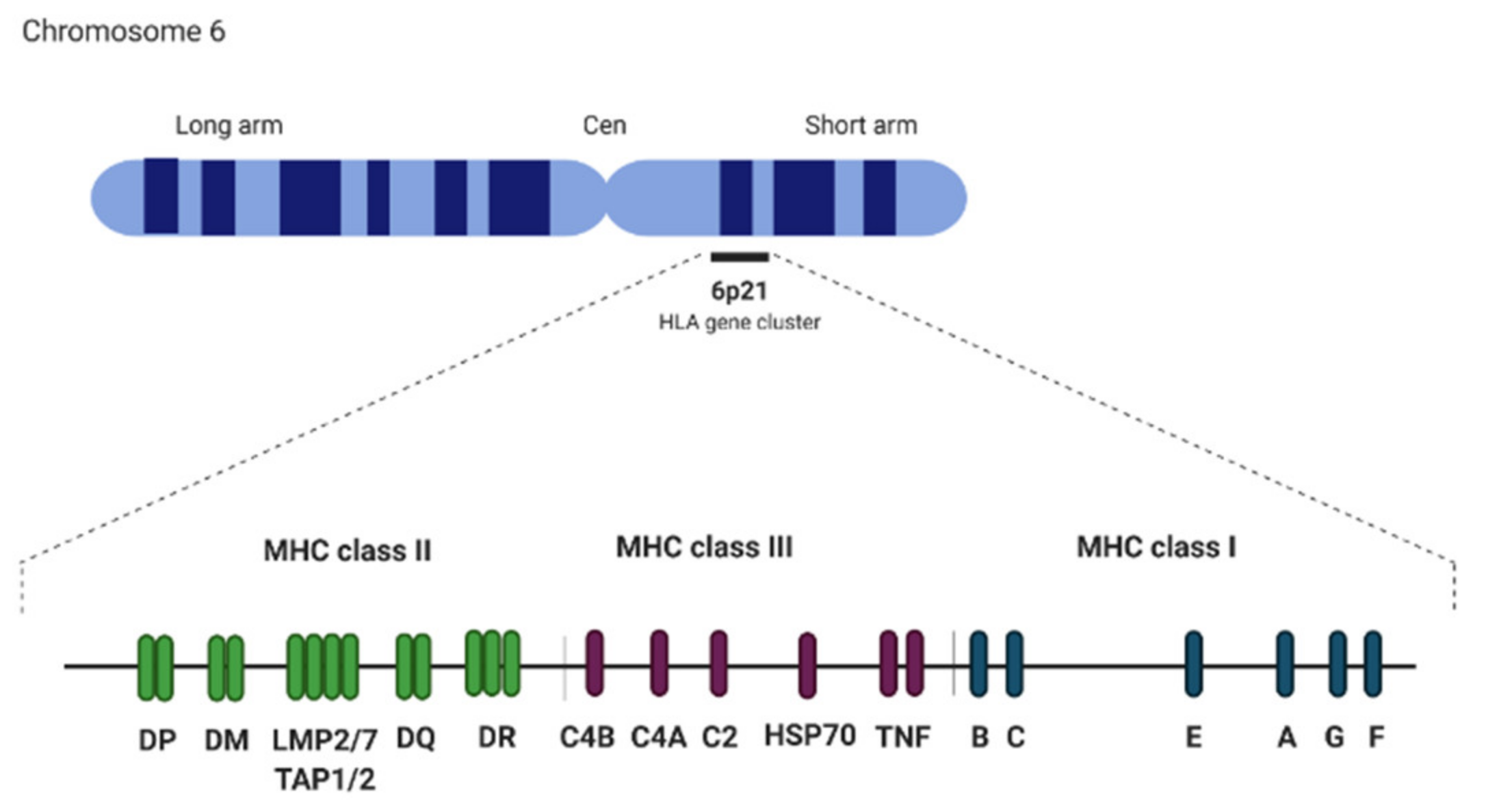
5. Major Histocompatibility Complex (MHC) Identity System
Major Histocompatibility Complex (MHC) Locus and Link to Odor Receptors
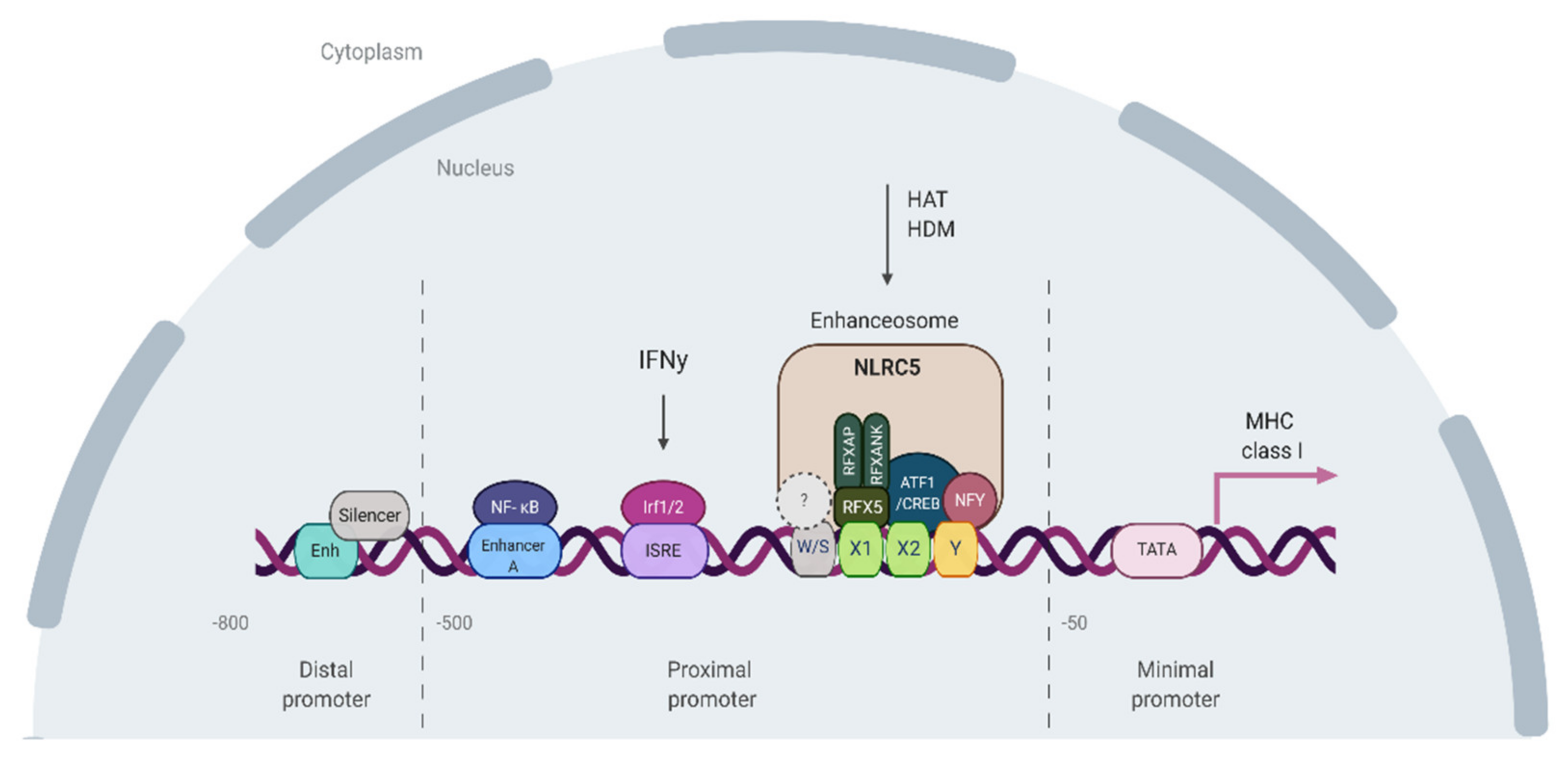
6. Viral Elements-Mediated Evolution and Regulation of MHCI Expression
7. MHC-Regulating Non-Coding RNAs in Cancer
8. MHC Class I Alterations in Tumors
8.1. HLA Class I Altered Phenotypes
8.2. Role of HLA Class I Alterations in Immunotherapy
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dávila-Ramos, S.; Castelán-Sánchez, H.G.; Martínez-ávila, L.; Sánchez-Carbente, M.D.R.; Peralta, R.; Hernández-Mendoza, A.; Dobson, A.D.W.; Gonzalez, R.A.; Pastor, N.; Batista-García, R.A. A review on viral metagenomics in extreme environments. Front. Microbiol. 2019, 10, e2403. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Meyerdierks, A.; Peña, A.; Rosselló-Mora, R.; Amann, R.; Antón, J. Metagenomic approach to the study of halophages: The environmental halophage 1. Environ. Microbiol. 2007, 9, 1711–1723. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Zayed, A.A.; Conceição-Neto, N.; Temperton, B.; Bolduc, B.; Alberti, A.; Ardyna, M.; Arkhipova, K.; Carmichael, M.; Cruaud, C.; et al. Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Haynes, M.; Kelley, S.; Angly, F.; Edwards, R.A.; Felts, B.; Mahaffy, J.M.; Mueller, J.; Nulton, J.; Rayhawk, S.; et al. Viral diversity and dynamics in an infant gut. Res. Microbiol. 2008, 159, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Lefeuvre, P.; Martin, D.P.; Elena, S.F.; Shepherd, D.N.; Roumagnac, P.; Varsani, A. Evolution and ecology of plant viruses. Nat. Rev. Microbiol. 2019, 17, 632–644. [Google Scholar] [CrossRef]
- Rivas-Carrillo, S.D.; Pettersson, M.E.; Rubin, C.-J.; Jern, P. Whole-genome comparison of endogenous retrovirus segregation across wild and domestic host species populations. Proc. Natl. Acad. Sci. USA 2018, 115, 11012–11017. [Google Scholar] [CrossRef]
- Stocking, C.; Kozak, C.A. Endogenous retroviruses: Murine endogenous retroviruses. Cell. Mol. Life Sci. 2008, 65, 3383–3398. [Google Scholar] [CrossRef]
- Tarlinton, R.E.; Meers, J.; Young, P.R. Retroviral invasion of the koala genome. Nature 2006, 442, 79–81. [Google Scholar] [CrossRef]
- Ryan, F.P. Human endogenous retroviruses in health and disease: A symbiotic perspective. J. R. Soc. Med. 2004, 97, 560–565. [Google Scholar] [CrossRef]
- Blond, J.-L.; Besème, F.; Duret, L.; Bouton, O.; Bedin, F.; Perron, H.; Mandrand, B.; Mallet, F. Molecular Characterization and Placental Expression of HERV-W, a New Human Endogenous Retrovirus Family. J. Virol. 1999, 73, 1175–1185. [Google Scholar] [CrossRef]
- Blond, J.-L.; Lavillette, D.; Cheynet, V.; Bouton, O.; Oriol, G.; Chapel-Fernandes, S.; Mandrand, B.; Mallet, F.; Cosset, F.-L. An Envelope Glycoprotein of the Human Endogenous Retrovirus HERV-W Is Expressed in the Human Placenta and Fuses Cells Expressing the Type D Mammalian Retrovirus Receptor. J. Virol. 2000, 74, 3321–3329. [Google Scholar] [CrossRef] [PubMed]
- Dupressoir, A.; Lavialle, C.; Heidmann, T. From ancestral infectious retroviruses to bona fide cellular genes: Role of the captured syncytins in placentation. Placenta 2012, 33, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Haig, D. Retroviruses and the placenta. Curr. Biol. 2012, 22, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P. Viruses and the placenta: The essential virus first view. APMIS 2016, 124, 20–30. [Google Scholar] [CrossRef]
- Buzdin, A.A. [Functional analysis of retroviral endogenous inserts in the human genome evolution]. Bioorg. Khim. 36, 38–46. [CrossRef]
- Buzdin, A. Human-specific endogenous retroviruses. Sci. World J. 2007, 7, 1848–1868. [Google Scholar] [CrossRef]
- Gogvadze, E.; Stukacheva, E.; Buzdin, A.; Sverdlov, E. Human-specific modulation of transcriptional activity provided by endogenous retroviral insertions. J. Virol. 2009, 83, 6098–6105. [Google Scholar] [CrossRef]
- Buzdin, A.A.; Prassolov, V.; Garazha, A.V. Friends-Enemies: Endogenous Retroviruses Are Major Transcriptional Regulators of Human DNA. Front. Chem. 2017, 5, e35. [Google Scholar] [CrossRef]
- Villarreal, L.P.; Witzany, G. The DNA habitat and its RNA inhabitants: At the dawn of RNA sociology. Genom. Insights 2013, 6, 1–12. [Google Scholar] [CrossRef]
- Villarreal, L.P.; Witzany, G. That is life: Communicating RNA networks from viruses and cells in continuous interaction. Ann. N. Y. Acad. Sci. 2019, 1447, 5–20. [Google Scholar] [CrossRef]
- Villarreal, L.P.; Witzany, G. Viruses are essential agents within the roots and stem of the tree of life. J. Theor. Biol. 2010, 262, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Villarreal, L.P. Persistent virus and addiction modules: An engine of symbiosis. Curr. Opin. Microbiol. 2016, 31, 70–79. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Villarreal, L.P. Viral ancestors of antiviral systems. Viruses 2011, 3, 1933–1958. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Makarova, K.S.; Aravind, L. Horizontal Gene Transfer in Prokaryotes: Quantification and Classification. Annu. Rev. Microbiol. 2001, 55, 709–742. [Google Scholar] [CrossRef]
- Puigb, P.; Wolf, Y.I.; Koonin, E.V. Search for a “tree of Life” in the thicket of the phylogenetic forest. J. Biol. 2009, 8, e59. [Google Scholar] [CrossRef]
- Villarreal, L.P. The source of self: Genetic parasites and the origin of adaptive immunity. Ann. N. Y. Acad. Sci. 2009, 1178, 194–232. [Google Scholar] [CrossRef]
- Villarreal, L.P.; Witzany, G. When Competing Viruses Unify: Evolution, Conservation, and Plasticity of Genetic Identities. J. Mol. Evol. 2015, 80, 305–318. [Google Scholar] [CrossRef]
- Villarreal, L.P. Viruses and the Evolution of Life; ASM Press: Washington, DC, USA, 2005; ISBN 1555813097. [Google Scholar]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory evolution of innate immunity through co-option of endogenous retroviruses. Science 2016, 351, 1083–1087. [Google Scholar] [CrossRef]
- Wang, T.; Zeng, J.; Lowe, C.B.; Sellers, R.G.; Salama, S.R.; Yang, M.; Burgess, S.M.; Brachmann, R.K.; Haussler, D. Species-specific endogenous retroviruses shape the transcriptional network of the human tumor suppressor protein p53. Proc. Natl. Acad. Sci. USA 2007, 104, 18613–18618. [Google Scholar] [CrossRef]
- Villarreal, L.P. Origin of Group Identity: Viruses, Addiction, and Cooperation; Springer: New York, NY, USA, 2009; ISBN 9780387779973. [Google Scholar]
- Lehnherr, H.; Maguin, E.; Jafri, S.; Yarmolinsky, M.B. Plasmid addiction genes of bacteriophage P1: Doc, which causes cell death on curing of prophage, and phd, which prevents host death when prophage is retained. J. Mol. Biol. 1993, 233, 414–428. [Google Scholar] [CrossRef]
- Mishra, R.; Kumar, A.; Ingle, H.; Kumar, H. The Interplay Between Viral-Derived miRNAs and Host Immunity During Infection. Front. Immunol. 2020, 10, e3079. [Google Scholar] [CrossRef] [PubMed]
- Withers, J.B.; Mondol, V.; Pawlica, P.; Rosa-Mercado, N.A.; Tycowski, K.T.; Ghasempur, S.; Torabi, S.F.; Steitz, J.A. Idiosyncrasies of Viral Noncoding RNAs Provide Insights into Host Cell Biology. Annu. Rev. Virol. 2019, 6, 297–317. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Sullivan, C.S. Lessons Learned from In Vivo Studies of a Viral Noncoding RNA. MSphere 2016, 1, e00026-16. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.E.; Sullivan, C.S. Balance and Stealth: The Role of Noncoding RNAs in the Regulation of Virus Gene Expression. Annu. Rev. Virol. 2014, 1, 89–109. [Google Scholar] [CrossRef] [PubMed]
- Tycowski, K.T.; Guo, Y.E.; Lee, N.; Moss, W.N.; Vallery, T.K.; Xie, M.; Steitz, J.A. Viral noncoding RNAs: More surprises. Genes Dev. 2015, 29, 567–584. [Google Scholar] [CrossRef] [PubMed]
- Witzany, G. Natural genome-editing competences of viruses. Acta Biotheor. 2006, 54, 235–253. [Google Scholar] [CrossRef]
- Witzany, G. Two genetic codes: Repetitive syntax for active non-coding RNAs; non-repetitive syntax for the DNA archives. Commun. Integr. Biol. 2017, 10, e1297352. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef]
- Flajnik, M.F. Primitive vertebrate immunity: What is the evolutionary derivative of molecules that define the adaptive immune system? Ciba Found. Symp. 1994, 186, 224–232. [Google Scholar]
- Flajnik, M.F.; Kasahara, M. Comparative genomics of the MHC: Glimpses into the evolution of the adaptive immune system. Immunity 2001, 15, 351–362. [Google Scholar] [CrossRef]
- Flajnik, M.F. Re-evaluation of the immunological big bang. Curr. Biol. 2014, 24, R1060–R1065. [Google Scholar] [CrossRef] [PubMed]
- Flajnik, M.F. A cold-blooded view of adaptive immunity. Nat. Rev. Immunol. 2018, 18, 438–453. [Google Scholar] [CrossRef] [PubMed]
- Bagasra, O.; Prilliman, K.R. RNA interference: The molecular immune system. J. Mol. Histol. 2004, 35, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Barstead, R. Genome-wide RNAi. Curr. Opin. Chem. Biol. 2001, 5, 63–66. [Google Scholar] [CrossRef]
- Wilkins, C.; Dishongh, R.; Moore, S.C.; Whitt, M.A.; Chow, M.; Machaca, K. RNA interference is an antiviral defence mechanism in Caenorhabditis elegans. Nature 2005, 436, 1044–1047. [Google Scholar] [CrossRef]
- Schott, D.H.; Cureton, D.K.; Whelan, S.P.; Hunter, C.P. An antiviral role for the RNA interference machinery in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2005, 102, 18420–18424. [Google Scholar] [CrossRef]
- Vastenhouw, N.L.; Plasterk, R.H.A. RNAi protects the Caenorhabditis elegans germline against transposition. Trends Genet. 2004, 20, 314–319. [Google Scholar] [CrossRef]
- Flint, S.J.; Enquist, L.W.; Racaniello, V.R.; Skalka, A.M. Principles of Virology: Molecular biology, Pathogenesis, and Control; ASM Press: Washington, DC, USA, 2000; ISBN 1555812591. [Google Scholar]
- You, E.-M.; Chiu, T.-S.; Liu, K.-F.; Tassanakajon, A.; Klinbunga, S.; Triwitayakorn, K.; de la Peña, L.D.; Li, Y.; Yu, H.-T. Microsatellite and mitochondrial haplotype diversity reveals population differentiation in the tiger shrimp (Penaeus monodon) in the Indo-Pacific region. Anim. Genet. 2008, 39, 267–277. [Google Scholar] [CrossRef]
- Xu, Z.; Dhar, A.K.; Wyrzykowski, J.; Alcivar-Warren, A. Identification of abundant and informative microsatellites from shrimp (Penaeus monodon) genome. Anim. Genet. 1999, 30, 150–156. [Google Scholar] [CrossRef]
- Williams, S.T.; Knowlton, N. Mitochondrial pseudogenes are pervasive and often insidious in the snapping shrimp genus Alpheus. Mol. Biol. Evol. 2001, 18, 1484–1493. [Google Scholar] [CrossRef]
- Corey, D.M.; Rosental, B.; Kowarsky, M.; Sinha, R.; Ishizuka, K.J.; Palmeri, K.J.; Quake, S.R.; Voskoboynik, A.; Weissman, I.L. Developmental cell death programs license cytotoxic cells to eliminate histocompatible partners. Proc. Natl. Acad. Sci. USA 2016, 113, 6520–6525. [Google Scholar] [CrossRef] [PubMed]
- Hibino, T.; Loza-Coll, M.; Messier, C.; Majeske, A.J.; Cohen, A.H.; Terwilliger, D.P.; Buckley, K.M.; Brockton, V.; Nair, S.V.; Berney, K.; et al. The immune gene repertoire encoded in the purple sea urchin genome. Dev. Biol. 2006, 300, 349–365. [Google Scholar] [CrossRef] [PubMed]
- Fugmann, S.D.; Messier, C.; Novack, L.A.; Andrew Cameron, R.; Rast, J.P. An ancient evolutionary origin of the Rag1/2 gene locus. Proc. Natl. Acad. Sci. USA 2006, 103, 3728–3733. [Google Scholar] [CrossRef] [PubMed]
- Delarbre, C.; Gallut, C.; Barriel, V.; Janvier, P.; Gachelin, G. Complete mitochondrial DNA of the hagfish, Eptatretus burgeri: The comparative analysis of mitochondrial DNA sequences strongly supports the cyclostome monophyly. Mol. Phylogenet. Evol. 2002, 22, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Furlong, R.F.; Holland, P.W.H. Bayesian Phylogenetic Analysis Supports Monophyly of Ambulacraria and of Cyclostomes. Zoolog. Sci. 2002, 19, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Acton, R.T.; Weinheimer, P.F.; Hildemann, W.H.; Evans, E.E. Bactericidal antibody response in the Pacific hagfish, Eptatretus stoutii. Infect. Immun. 1971, 4, 160–166. [Google Scholar] [CrossRef]
- Zapata, A.; Fänge, R.; Mattisson, A.; Villena, A. Plasma cells in adult Atlantic hagfish, Myxine glutinosa. Cell Tissue Res. 1984, 235, 691–693. [Google Scholar] [CrossRef]
- Newton, R.A.; Raftos, D.A.; Raison, R.L.; Geczy, C.L. Chemotactic responses of hagfish (Vertebrata, Agnatha) leucocytes. Dev. Comp. Immunol. 1994, 18, 295–303. [Google Scholar] [CrossRef]
- Raison, R.L.; Gilbertson, P.; Wotherspoon, J. Cellular requirements for mixed leucocyte reactivity in the cyclostome, Eptatretus stoutii. Immunol. Cell Biol. 1987, 65, 183–188. [Google Scholar] [CrossRef]
- Raison, R.L.; Coverley, J.; Hook, J.W.; Towns, P.; Weston, K.M.; Raftos, D.A. A cell-surface opsonic receptor on leucocytes from the phylogenetically primitive vertebrate, Eptatretus stouti. Immunol. Cell Biol. 1994, 72, 326–332. [Google Scholar] [CrossRef]
- Shintani, S.; Terzic, J.; Sato, A.; Saraga-Babic, M.; O’Huigin, C.; Tichy, H.; Klein, J. Do lampreys have lymphocytes? The Spi evidence. Proc. Natl. Acad. Sci. USA 2000, 97, 7417–7422. [Google Scholar] [CrossRef] [PubMed]
- Alder, M.N.; Rogozin, I.B.; Iyer, L.M.; Glazko, G.V.; Cooper, M.D.; Pancer, Z. Immunology: Diversity and function of adaptive immune receptors in a jawless vertebrate. Science 2005, 310, 1970–1973. [Google Scholar] [CrossRef] [PubMed]
- Pancer, Z.; Saha, N.R.; Kasamatsu, J.; Suzuki, T.; Amemiya, C.T.; Kasahara, M.; Cooper, M.D. Variable lymphocyte receptors in hagfish. Proc. Natl. Acad. Sci. USA 2005, 102, 9224–9229. [Google Scholar] [CrossRef] [PubMed]
- Kurosawa, Y.; Hashimoto, K. How did the primordial T cell receptor and MHC molecules function initially? Immunol. Cell Biol. 1997, 75, 193–196. [Google Scholar] [CrossRef]
- Marchalonis, J.J.; Schluter, S.F.; Bernstein, R.M.; Hohman, V.S. Antibodies of sharks: Revolution and evolution. Immunol. Rev. 1998, 166, 103–122. [Google Scholar] [CrossRef]
- Flajnik, M.; Ohta, Y.; Namikawa-Yomada, C.; Nonaka, M. Insight into the primordial MHC from studies in ectothermic vertebrates. Immunol. Rev. 1999, 167, 59–67. [Google Scholar] [CrossRef]
- Villarreal, L.P. Can Viruses Make Us Human? Proc. Am. Philos. Soc. 2004, 148, 296–323. [Google Scholar]
- Du Pasquier, L. Speculations on the origin of the vertebrate immune system. Immunol Lett. 2004, 92, 3–9. [Google Scholar] [CrossRef]
- Horton, R.; Gibson, R.; Coggill, P.; Miretti, M.; Allcock, R.J.; Almeida, J.; Forbes, S.; Gilbert, J.G.R.; Halls, K.; Harrow, J.L.; et al. Variation analysis and gene annotation of eight MHC haplotypes: The MHC Haplotype Project. Immunogenetics 2008, 60, 1–18. [Google Scholar] [CrossRef]
- Shiina, T.; Inoko, H.; Kulski, J.K. An update of the HLA genomic region, locus information and disease associations: 2004. Tissue Antigens 2004, 64, 631–649. [Google Scholar] [CrossRef]
- Danchin, E.; Vitiello, V.; Vienne, A.; Richard, O.; Gouret, P.; McDermott, M.F.; Pontarotti, P. The major histocompatibility complex origin. Immunol. Rev. 2004, 198, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA genomic loci map: Expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef] [PubMed]
- Jern, P.; Coffin, J.M. Effects of Retroviruses on Host Genome Function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef] [PubMed]
- Bannert, N.; Kurth, R. Retroelements and the human genome: New perspectives on an old relation. Proc. Natl. Acad. Sci. USA 2004, 101, 14572–14579. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- Robinson, J.; Barker, D.J.; Georgiou, X.; Cooper, M.A.; Flicek, P.; Marsh, S.G.E. IPD-IMGT/HLA Database. Nucleic Acids Res. 2020, 48, D948–D955. [Google Scholar]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, e76. [Google Scholar] [CrossRef]
- Liu, Y.; Helms, C.; Liao, W.; Zaba, L.C.; Duan, S.; Gardner, J.; Wise, C.; Miner, A.; Malloy, M.J.; Pullinger, C.R.; et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet. 2008, 4, e1000041. [Google Scholar] [CrossRef]
- Ichinokawa, K.; Nakanishi, Y.; Hida, Y.; Tsuchikawa, T.; Kato, T.; Itoh, T.; Kaji, M.; Kaga, K.; Hirano, S. Downregulated expression of human leukocyte antigen class I heavy chain is associated with poor prognosis in non-small-cell lung cancer. Oncol. Lett. 2019, 18, 117–126. [Google Scholar] [CrossRef]
- Yeung, J.T.; Hamilton, R.L.; Ohnishi, K.; Ikeura, M.; Douglas, M.; Nikiforova, M.N.; Ferrone, S.; Jakacki, R.I.; Pollack, I.F. LOH in the HLA class I region at 6p21 is associated with shorter survival in newly diagnosed adult glioblastoma. Clin. Cancer Res. 2013, 19, 1816–1826. [Google Scholar] [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018, 359, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Torvik, V.I. Alu elements within human mRNAs are probable microRNA targets. Trends Genet. 2006, 22, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Daskalova, E.; Baev, V.; Rusinov, V.; Minkov, I. 3’UTR-located ALU Elements: Donors of Potetial miRNA Target Sites and Mediators of Network miRNA-based Regulatory Interactions. Evol. Bioinform. 2006, 2. [Google Scholar] [CrossRef]
- Tokuyama, M.; Kong, Y.; Song, E.; Jayewickreme, T.; Kang, I.; Iwasaki, A. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc. Natl. Acad. Sci. USA 2018, 115, 12565–12572. [Google Scholar] [CrossRef]
- Kulski, J.K. Long Noncoding RNA HCP5, a Hybrid HLA Class I Endogenous Retroviral Gene: Structure, Expression, and Disease Associations. Cells 2019, 8, 480. [Google Scholar] [CrossRef]
- Liman, E.R.; Innan, H. Relaxed selective pressure on an essential component of pheromone transduction in primate evolution. Proc. Natl. Acad. Sci. USA 2003, 100, 3328–3332. [Google Scholar] [CrossRef]
- Giorgi, D.; Friedman, C.; Trask, B.J.; Rouquier, S. Characterization of nonfunctional V1R-like pheromone receptor sequences in human. Genome Res. 2000, 10, 1979–1985. [Google Scholar] [CrossRef][Green Version]
- Kouros-Mehr, H.; Pintchovski, S.; Melnyk, J.; Chen, Y.J.; Friedman, C.; Trask, B.; Shizuya, H. Identification of non-functional human VNO receptor genes provides evidence for vestigiality of the human VNO. Chem. Senses 2001, 26, 1167–1174. [Google Scholar] [CrossRef]
- Zhang, J.; Webb, D.M. Evolutionary deterioration of the vomeronasal pheromone transduction pathway in catarrhine primates. Proc. Natl. Acad. Sci. USA 2003, 100, 8337–8341. [Google Scholar] [CrossRef]
- Tönjes, R.R.; Löwer, R.; Boller, K.; Denner, J.; Hasenmaier, B.; Kirsch, H.; König, H.; Korbmacher, C.; Limbach, C.; Lugert, R.; et al. HERV-K: The biologically most active human endogenous retrovirus family. JAIDS J. Acquir. Immune Defic. Syndr. 1996, 13, S261–S267. [Google Scholar] [CrossRef] [PubMed]
- Vo√ate, P.A.; Lukashov, V.V.; Zs√≠ros, J.; Berkhout, B.; Jebbink, M.F. Evolutionary relationships within a subgroup of HERV-K-related human endogenous retroviruses. J. Gen. Virol. 1998, 79, 61–70. [Google Scholar]
- Garcia-Montojo, M.; Doucet-O’Hare, T.; Henderson, L.; Nath, A. Human endogenous retrovirus-K (HML-2): A comprehensive review. Crit. Rev. Microbiol. 2018, 44, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, M.Y.; Nonaka, M. Repetitive elements in the major histocompatibility complex (MHC) class I region of a teleost, medaka: Identification of novel transposable elements. Mech. Dev. 2004, 121, 771–777. [Google Scholar] [CrossRef]
- Ohta, Y.; Goetz, W.; Hossain, M.Z.; Nonaka, M.; Flajnik, M.F. Ancestral Organization of the MHC Revealed in the Amphibian Xenopus. J. Immunol. 2006, 176, 3674–3685. [Google Scholar] [CrossRef]
- Vogel, T.U.; Evans, D.T.; Urvater, J.A.; O’Connor, D.H.; Hughes, A.L.; Watkins, D.I. Major histocompatibility complex class I genes in primates: Co-evolution with pathogens. Immunol. Rev. 1999, 167, 327–337. [Google Scholar] [CrossRef]
- De Groot, N.G.; Otting, N.; Doxiadis, G.G.M.; Balla-Jhagjhoorsingh, S.S.; Heeney, J.L.; Van Rood, J.J.; Gagneux, P.; Bontrop, R.E. Evidence for an ancient selective sweep in the MHC class I gene repertoire of chimpanzees. Proc. Natl. Acad. Sci. USA 2002, 99, 11748–11753. [Google Scholar] [CrossRef]
- Andersson, G.; Svensson, A.C.; Setterblad, N.; Rask, L. Retroelements in the human MHC class II region. Trends Genet. 1998, 14, 109–114. [Google Scholar] [CrossRef]
- Kulski, J.K.; Gaudieri, S.; Inoko, H.; Dawkins, R.L. Comparison between two human endogenous retrovirus (HERV)-rich regions within the major histocompatibility complex. J. Mol. Evol. 1999, 48, 675–683. [Google Scholar] [CrossRef]
- Kulski, J.K.; Gaudieri, S.; Martin, A.; Dawkins, R.L. Coevolution of PERB11 (MIC) and HLA class I genes with HERV-16 and retroelements by extended genomic duplication. J. Mol. Evol. 1999, 49, 84–97. [Google Scholar] [CrossRef]
- Boegel, S.; Löwer, M.; Bukur, T.; Sorn, P.; Castle, J.C.; Sahin, U. HLA and proteasome expression body map. BMC Med. Genom. 2018, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Daar, A.S.; Fuggle, S.V.; Fabre, J.W.; Ting, A.; Morris, P.J. The detailed distribution of HLA–A, B, C antigens in normal human organs. Transplantation 1984, 38, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.H.; Liu, Y.J.; Bayir, E.; Iliopoulos, D.; Van Den Elsen, P.J.; Kobayashi, K.S. NLR family member NLRC5 is a transcriptional regulator of MHC class I genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef] [PubMed]
- Ludigs, K.; Seguín-Estévez, Q.; Lemeille, S.; Ferrero, I.; Rota, G.; Chelbi, S.; Mattmann, C.; MacDonald, H.R.; Reith, W.; Guarda, G. NLRC5 Exclusively Transactivates MHC Class I and Related Genes through a Distinctive SXY Module. PLoS Genet. 2015, 11, e1005088. [Google Scholar] [CrossRef]
- Robbins, G.R.; Truax, A.D.; Davis, B.K.; Zhang, L.; Brickey, W.J.; Ting, J.P.Y. Regulation of class I major histocompatibility complex (MHC) by nucleotide-binding domain, leucine-richrepeat-containing (NLR) Proteins. J. Biol. Chem. 2012, 287, 24294–24303. [Google Scholar] [CrossRef]
- Staehli, F.; Ludigs, K.; Heinz, L.X.; Seguín-Estévez, Q.; Ferrero, I.; Braun, M.; Schroder, K.; Rebsamen, M.; Tardivel, A.; Mattmann, C.; et al. NLRC5 Deficiency Selectively Impairs MHC Class I- Dependent Lymphocyte Killing by Cytotoxic T Cells. J. Immunol. 2012, 188, 3820–3828. [Google Scholar] [CrossRef]
- Chang, C.H.; Guerder, S.; Hong, S.C.; Van Ewijk, W.; Flavell, R.A. Mice lacking the MHC class II transactivator (CIITA) show tissue-specific impairment of MHC class II expression. Immunity 1996, 4, 167–178. [Google Scholar] [CrossRef]
- Collinge, M.; Pardi, R.; Bender, J.R. Class II transactivator-independent endothelial cell MHC class II gene activation induced by lymphocyte adhesion. J. Immunol. 1998, 161, 1589–1593. [Google Scholar]
- Weissman, J.D.; Singer, D.S. A complex regulatory DNA element associated with a major histocompatibility complex class I gene consists of both a silencer and an enhancer. Mol. Cell. Biol. 1991, 11, 4217–4227. [Google Scholar] [CrossRef]
- Howcroft, T.K.; Raval, A.; Weissman, J.D.; Gegonne, A.; Singer, D.S. Distinct Transcriptional Pathways Regulate Basal and Activated Major Histocompatibility Complex Class I Expression. Mol. Cell. Biol. 2003, 23, 3377–3391. [Google Scholar] [CrossRef]
- Van Den Elsen, P.J.; Gobin, S.J.P.; Van Eggermond, M.C.J.A.; Peijnenburg, A. Regulation of MHC class I and II gene transcription: Differences and similarities. Immunogenetics 1998, 48, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Gobin, S.J.; Keijsers, V.; van Zutphen, M.; van den Elsen, P.J. The role of enhancer A in the locus-specific transactivation of classical and nonclassical HLA class I genes by nuclear factor kappa B. J. Immunol. 1998, 161, 2276–2283. [Google Scholar] [PubMed]
- Van den Elsen, P.J. Expression regulation of major histocompatibility complex class I and class II encoding genes. Front. Immunol. 2011, 2, 1–9. [Google Scholar] [CrossRef]
- Gobin, S.J.; van Zutphen, M.; Woltman, A.M.; van den Elsen, P.J. Transactivation of classical and nonclassical HLA class I genes through the IFN-stimulated response element. J. Immunol. 1999, 163, 1428–1434. [Google Scholar] [PubMed]
- Lees, C.J.; Smorodinsky, N.; Horn, G.; Wreschner, D.H.; McKenzie, I.F.C.; Pietersz, G.; Stojanovska, L.; Apostolopoulos, V. MUC1 immunotherapy against a metastatic mammary adenocarcinoma model: Importance of IFN-gamma. Pril. Makedonska Akad. na Nauk. i Umet. Oddelenie za Med. Nauk. 2016, 37, 15–25. [Google Scholar] [CrossRef]
- Perkins, N.D.; Edwards, N.L.; Duckett, C.S.; Agranoff, A.B.; Schmid, R.M.; Nabel, G.J. A cooperative interaction between NF-kappa B and Sp1 is required for HIV-1 enhancer activation. EMBO J. 1993, 12, 3551–3558. [Google Scholar] [CrossRef]
- Harada, H.; Fujita, T.; Miyamoto, M.; Kimura, Y.; Maruyama, M.; Furia, A.; Miyata, T.; Taniguchi, T. Structurally similar but functionally distinct factors, IRF-1 and IRF-2, bind to the same regulatory elements of IFN and IFN-inducible genes. Cell 1989, 58, 729–739. [Google Scholar] [CrossRef]
- Hakem, R.; Bouteiller, P.L.; Harper, K.; Hakem, R.; Bouteiller, P.L.E.; Jezo-bremond, A.; Harper, K.; Campese, D.; Lemonnier, F.A. Differential regulation of HLA-A3 and HLA-B7 MHC class I genes by IFN is due to two nucleotide differences in their IFN response sequences. J. Immunol. 1991, 147, 2384–2390. [Google Scholar]
- Schmidt, H.; Gekeler, V.; Haas, H.; Engler-Blum, G.; Steiert, I.; Probst, H.; Müller, C.A. Differential regulation of HLA class I genes by interferon. Immunogenetics 1990, 31, 245–252. [Google Scholar] [CrossRef]
- Girdlestone, J.; Isamat, M.; Gewert, D.; Milstein, C. Transcriptional regulation of HLA-A and -B: Differential binding of members of the Rel and IRF families of transcription factors. Proc. Natl. Acad. Sci. USA 1993, 90, 11568–11572. [Google Scholar] [CrossRef]
- Girdlestone, J.; Milstein, C. Differential expression and interferon response of HLA class I genes in thymocyte lines and response variants. Eur. J. Immunol. 1988, 18, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J. Video Q&A: Non-coding RNAs and eukaryotic evolution-a personal view. BMC Biol. 2010, 8, e67. [Google Scholar]
- Qureshi, I.A.; Mattick, J.S.; Mehler, M.F. Long non-coding RNAs in nervous system function and disease. Brain Res. 2010, 1338, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A.; Wolvetang, E.J.; Mattick, J.S.; Rinn, J.L.; Barry, G. Mechanisms of Long Non-coding RNAs in Mammalian Nervous System Development, Plasticity, Disease, and Evolution. Neuron 2015, 88, 861–877. [Google Scholar] [CrossRef]
- Chen, Y.G.; Satpathy, A.T.; Chang, H.Y. Gene regulation in the immune system by long noncoding RNAs. Nat. Immunol. 2017, 18, 962–972. [Google Scholar] [CrossRef]
- Fitzgerald, K.A.; Caffrey, D.R. Long noncoding RNAs in innate and adaptive immunity. Curr. Opin. Immunol. 2014, 26, 140–146. [Google Scholar] [CrossRef]
- Kim, T.M.; Hong, S.J.; Rhyu, M.G. Periodic explosive expansion of human retroelements associated with the evolution of the hominoid primate. J. Korean Med. Sci. 2004, 19, 177–185. [Google Scholar] [CrossRef][Green Version]
- Smalheiser, N.R.; Torvik, V.I. Mammalian microRNAs derived from genomic repeats. Trends Genet. 2005, 21, 322–326. [Google Scholar] [CrossRef]
- Petri, R.; Brattås, P.L.; Sharma, Y.; Jonsson, M.E.; Pircs, K.; Bengzon, J.; Jakobsson, J. LINE-2 transposable elements are a source of functional human microRNAs and target sites. PLoS Genet. 2019, 15, e1008036. [Google Scholar] [CrossRef]
- Clark, P.M.; Chitnis, N.; Shieh, M.; Kamoun, M.; Johnson, F.B.; Monos, D. Novel and Haplotype Specific MicroRNAs Encoded by the Major Histocompatibility Complex. Sci. Rep. 2018, 8, 1–10. [Google Scholar] [CrossRef]
- Ladewig, E.; Okamura, K.; Flynt, A.S.; Westholm, J.O.; Lai, E.C. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res. 2012, 22, 1634–1645. [Google Scholar] [CrossRef] [PubMed]
- Chitnis, N.; Clark, P.M.; Kamoun, M.; Stolle, C.; Johnson, F.B.; Monos, D.S. An expanded role for HLA genes: HLA-B encodes a microRNA that regulates IgA and other immune response transcripts. Front. Immunol. 2017, 8, e583. [Google Scholar] [CrossRef] [PubMed]
- Vernet, C.; Ribouchon, M.T.; Chimini, G.; Jouanolle, A.M.; Sidibé, I.; Pontarotti, P. A novel coding sequence belonging to a new multicopy gene family mapping within the human MHC class I region. Immunogenetics 1993, 38, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.T.; Oberg, A.L.; Grill, D.E.; Ovsyannikova, I.G.; Haralambieva, I.H.; Kennedy, R.B.; Poland, G.A. System-wide associations between DNA-methylation, gene expression, and humoral immune response to influenza vaccination. PLoS ONE 2016, 11, e0152034. [Google Scholar] [CrossRef]
- Kulski, J.K.; Dawkins, R.L. The P5 multicopy gene family in the MHC is related in sequence to human endogenous retroviruses HERV-L and HERV-16. Immunogenetics 1999, 49, 404–412. [Google Scholar] [CrossRef]
- Yoon, W.; Ma, B.-J.; Fellay, J.; Huang, W.; Xia, S.-M.; Zhang, R.; Shianna, K.V.; Liao, H.-X.; Haynes, B.F.; Goldstein, D.B. A polymorphism in the HCP5 gene associated with HLA-B*5701 does not restrict HIV-1 in vitro. AIDS 2010, 24, 155–157. [Google Scholar] [CrossRef]
- Olgun, G.; Sahin, O.; Tastan, O. Discovering lncRNA mediated sponge interactions in breast cancer molecular subtypes. BMC Genom. 2018, 19, e650. [Google Scholar] [CrossRef]
- Teng, H.; Wang, P.; Xue, Y.; Liu, X.; Ma, J.; Cai, H.; Xi, Z.; Li, Z.; Liu, Y. Role of HCP5-miR-139-RUNX1 feedback loop in regulating malignant behavior of Glioma Cells. Mol. Ther. 2016, 24, 1806–1822. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, H.; Liu, Z.; Owzar, K.; Han, Y.; Su, L.; Wei, Y.; Hung, R.J.; McLaughlin, J.; Brhane, Y.; et al. A Novel Genetic Variant in Long Non-coding RNA Gene NEXN-AS1 is Associated with Risk of Lung Cancer. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Orvis, T.; Hepperla, A.; Walter, V.; Song, S.; Simon, J.; Parker, J.; Wilkerson, M.D.; Desai, N.; Major, M.B.; Hayes, D.N.; et al. BRG1/SMARCA4 inactivation promotes non-small cell lung cancer aggressiveness by altering chromatin organization. Cancer Res. 2014, 74, 6486–6498. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, R.; Fang, L.; Ge, X.; Chen, L.; Zhou, M.; Zhou, Y.; Xiong, W.; Hu, Y.; Tang, X.; et al. HCP5 is a SMAD3-responsive long non-coding RNA that promotes lung adenocarcinoma metastasis via miR-203/SNAI axis. Theranostics 2019, 9, 2460–2474. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Li, L. SP1-induced upregulation of long non-coding RNA HCP5 promotes the development of osteosarcoma. Pathol. Res. Pract. 2019, 215, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-J.; Kim, M.; Kim, H.-P. Epigenetic regulation of long noncoding RNA UCA1 by SATB1 in breast cancer. BMB Rep. 2016, 49, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Yan, Z.; Wu, C.; Zhang, Q.; Zhu, Y.; Li, K.; Xu, Y. Integrated analysis of dosage effect lncRNAs in lung adenocarcinoma based on comprehensive network. Oncotarget 2017, 8, e71430. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lu, B.; Sun, L.; Yan, X.; Xu, J. Identification of candidate genes or microRNAs associated with the lymph node metastasis of SCLC. Cancer Cell Int. 2018, 18, e161. [Google Scholar] [CrossRef]
- Yang, C.; Sun, J.; Liu, W.; Yang, Y.; Chu, Z.; Yang, T.; Gui, Y.; Wang, D. Long noncoding RNA HCP5 contributes to epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation and interacting with miR-139-5p. Am. J. Transl. Res. 2019, 11, 953–963. [Google Scholar]
- Hu, R.; Lu, Z. Long non-coding RNA HCP5 promotes prostate cancer cell proliferation by acting as the sponge of miR-4656 to modulate CEMIP expression. Oncol. Rep. 2020, 43, 328–336. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, Q.; Xu, J.; Guo, L.; Li, X. Clinical significance of serum miR-21 in breast cancer compared with CA153 and CEA. Chin. J. Cancer Res. 2013, 25, 743–748. [Google Scholar]
- Seliger, B. Role of microRNAs on HLA-G expression in human tumors. Hum. Immunol. 2016, 77, 760–763. [Google Scholar] [CrossRef]
- Kulkarni, S.; Savan, R.; Qi, Y.; Gao, X.; Yuki, Y.; Bass, S.E.; Martin, M.P.; Hunt, P.; Deeks, S.G.; Telenti, A.; et al. Differential microRNA regulation of HLA-C expression and its association with HIV control. Nature 2011, 472, 495–498. [Google Scholar] [CrossRef]
- O’Huigin, C.; Kulkarni, S.; Xu, Y.; Deng, Z.; Kidd, J.; Kidd, K.; Gao, X.; Carrington, M. The molecular origin and consequences of escape from miRNA regulation by HLA-C alleles. Am. J. Hum. Genet. 2011, 89, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Mari, L.; Hoefnagel, S.J.M.; Zito, D.; van de Meent, M.; van Endert, P.; Calpe, S.; del Sancho Serra, M.C.; Heemskerk, M.H.M.; van Laarhoven, H.W.M.; Hulshof, M.C.C.M.; et al. microRNA 125a Regulates MHC-I Expression on Esophageal Adenocarcinoma Cells, Associated with Suppression of Antitumor Immune Response and Poor Outcomes of Patients. Gastroenterology 2018, 155, 784–798. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Ruiz-Cabello, F.; Cabrera, T.; Pérez-Villar, J.J.; López-Botet, M.; Duggan-Keen, M.; Stern, P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today 1997, 18, 89–95. [Google Scholar] [CrossRef]
- Koopman, L.A.; Corver, W.E.; Van Der Slik, A.R.; Giphart, M.J.; Fleuren, G.J. Multiple genetic alterations cause frequent and heterogeneous human histocompatibility leukocyte antigen class I loss in cervical cancer. J. Exp. Med. 2000, 191, 961–975. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, P.; Cantón, J.; Collado, A.; Cabrera, T.; Serrano, A.; Real, L.M.; García, A.; Ruiz-Cabello, F.; Garrido, F. Chromosome loss is the most frequent mechanism contributing to HLA haplotype loss in human tumors. Int. J. Cancer 1999, 83, 91–97. [Google Scholar] [CrossRef]
- Feenstra, M.; Veltkamp, M.; van Kuik, J.; Wiertsema, S.; Slootweg, P.; van den Tweel, J.; de Weger, R.; Tilanus, M. HLA class I expression and chromosomal deletions at 6p and 15q in head and neck squamous cell carcinomas. Tissue Antigens 1999, 54, 235–245. [Google Scholar] [CrossRef]
- Versteeg, R.; Kruse-Wolters, K.M.; Plomp, A.C.; Van Leeuwen, A.; Stam, N.J.; Ploegh, H.L.; Ruiter, D.J.; Schrier, P.I. Suppression of class I human histocompatibility leukocyte antigen by c-myc is locus specific. J. Exp. Med. 1989, 170, 621–635. [Google Scholar] [CrossRef]
- Schrier, P.I.; Versteeg, R.; Peltenburg, L.T.; Plomp, A.C.; van ’t Veer, L.J.; Kruse-Wolters, K.M. Sensitivity of melanoma cell lines to natural killer cells: A role for oncogene-modulated HLA class I expression? Semin Cancer Biol. 1991, 2, 73–83. [Google Scholar]
- Soong, T.W.; Hui, K.M. Locus-specific transcriptional control of HLA genes. J Immunol. 1992, 149, 2008–2020. [Google Scholar]
- Browning, M.J.; Krausa, P.; Rowan, A.; Bicknell, D.C.; Bodmer, J.G.; Bodmer, W.F. Tissue typing the HLA-A locus from genomic DNA by sequence-specific PCR: Comparison of HLA genotype and surface expression on colorectal tumor cell lines. Proc. Natl. Acad. Sci. USA 1993, 90, 2842–2845. [Google Scholar] [CrossRef]
- Koopman, L.A.; Van Der Slik, A.R.; Giphart, M.J.; Fleuren, G.J. Human leukocyte antigen class I gene mutations in cervical cancer. J. Natl. Cancer Inst. 1999, 91, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Brady, C.S.; Bartholomew, J.S.; Burt, D.J.; Duggan-Keen, M.F.; Glenville, S.; Telford, N.; Little, A.M.; Davidson, J.A.; Jimenez, P.; Ruiz-Cabelto, F.; et al. Multiple mechanisms underlie HLA dysregulation in cervical cancer. Tissue Antigens 2000, 55, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Brady, C.S.; Jimenez, P.; Duggan-Keen, M.F.; Mendez, R.; Stern, P.; Garrido, F.; Ruiz-Cabello, F. A mutation determining the loss of HLA-A2 antigen expression in a cervical carcinoma reveals novel splicing of human MHC class I classical transcripts in both tumoral and normal cells. Immunogenetics 2000, 51, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Perea, F.; Bernal, M.; Sánchez-Palencia, A.; Carretero, J.; Torres, C.; Bayarri, C.; Gómez-Morales, M.; Garrido, F.; Ruiz-Cabello, F. The absence of HLA class I expression in non-small cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration. Int. J. Cancer 2017, 140, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Dovhey, S.E.; Ghosh, N.S.; Wright, K.L. Loss of interferon-γ inducibility of TAP1 and LMP2 in a renal cell carcinoma cell line. Cancer Res. 2000, 60, 5789–5796. [Google Scholar] [PubMed]
- Algarra, I.; García-Lora, A.; Cabrera, T.; Ruiz-Cabello, F.; Garrido, F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: Implications for tumor immune escape. Cancer Immunol. Immunother. 2004, 53, 904–910. [Google Scholar] [CrossRef] [PubMed]
- De Kruijf, E.M.; Sajet, A.; van Nes, J.G.H.; Natanov, R.; Putter, H.; Smit, V.T.H.B.M.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.H.; Kuppen, P.J.K. HLA-E and HLA-G Expression in Classical HLA Class I-Negative Tumors Is of Prognostic Value for Clinical Outcome of Early Breast Cancer Patients. J. Immunol. 2010, 185, 7452–7459. [Google Scholar] [CrossRef]
- Garrido, F.; Cabrera, T.; Aptsiauri, N. “Hard” and “soft” lesions underlying the HLA class I alterations in cancer cells: Implications for immunotherapy. Int. J. Cancer 2010, 127, 249–256. [Google Scholar] [CrossRef]
- Cabrera, T.; Fernandez, M.A.; Sierra, A.; Garrido, A.; Herruzo, A.; Escobedo, A.; Fabra, A.; Garrido, F. High frequency of altered HLA class I phenotypes in invasive breast carcinomas. Hum. Immunol. 1996, 50, 127–134. [Google Scholar] [CrossRef]
- Cabrera, T.; Collado, A.; Fernandez, M.A.; Ferron, A.; Sancho, J.; Ruiz-Cabello, F.; Garrido, F. High frequency of altered HLA class I phenotypes in invasive colorectal carcinomas. Tissue Antigens 1998, 52, 114–123. [Google Scholar] [CrossRef]
- Cabrera, T.; Salinero, J.; Fernandez, M.A.; Garrido, A.; Esquivias, J.; Garrido, F. High frequency of altered HLA class I phenotypes in laryngeal carcinomas. Hum. Immunol. 2000, 61, 499–506. [Google Scholar] [CrossRef]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F.; Aptsiauri, N.; Doorduijn, E.M.; Garcia Lora, A.M.; van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 2016, 39, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.R.; Barthen, C.; Williamson, D.J.; Davis, D.M. HLA-B and HLA-C differ in their nanoscale organization at cell surfaces. Front. Immunol. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Garrido, F. MHC/HLA Class I Loss in Cancer Cells. Adv. Exp. Med. Biol. 2019, 1151, 15–78. [Google Scholar] [PubMed]
- Aptsiauri, N.; Garcia-Lora, A.M.; Cabrera, T. MHC Class I Antigens in Malignant Cells: Immune Escape and Response to Immunotherapy; Springer Science & Business Media: Berlin, Germany, 2013; pp. 1–51. [Google Scholar]
- Lampen, M.H.; van Hall, T. Strategies to counteract MHC-I defects in tumors. Curr. Opin. Immunol. 2011, 23, 293–298. [Google Scholar] [CrossRef]
- Perea, F.; Sánchez-Palencia, A.; Gómez-Morales, M.; Bernal, M.; Concha, Á.; García, M.M.; González-Ramírez, A.R.; Kerick, M.; Martin, J.; Garrido, F.; et al. HLA class I loss and PD-L1 expression in lung cancer: Impact on T-cell infiltration and immune escape. Oncotarget 2018, 9, 4120–4133. [Google Scholar]
- Carretero, R.; Wang, E.; Rodriguez, A.I.; Reinboth, J.; Ascierto, M.L.; Engle, A.M.; Liu, H.; Camacho, F.M.; Marincola, F.M.; Garrido, F.; et al. Regression of melanoma metastases after immunotherapy is associated with activation of antigen presentation and interferon-mediated rejection genes. Int. J. Cancer 2012, 131, 387–395. [Google Scholar] [CrossRef]
- Carretero, R.; Cabrera, T.; Gil, H.; Saenz-Lopez, P.; Maleno, I.; Aptsiauri, N.; Cozar, J.M.; Garrido, F. Bacillus Calmette-Guerin immunotherapy of bladder cancer induces selection of human leukocyte antigen class I-deficient tumor cells. Int. J. Cancer 2011, 129, 839–846. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Phenotype | Characteristics | Description |
|---|---|---|
| I | Total loss of HLA class I molecules | Low frequency in laryngeal carcinomas (10%), colorectal carcinomas (18%), and melanomas (17%), and higher in breast (52%), prostate (40%), and bladder (35%) carcinomas [156]. |
| II | Loss of an HLA class I haplotype | Produced by loss of heterozygosity (LOH) associated with chromosome 6.Incidence of this altered phenotype is 46% in cervix carcinomas, 15–49% in head and neck, 17% in colorectal carcinomas, and 14% in breast carcinomas [157,158,159]. |
| III | Loss of an HLA class I locus | Found when both products of HLA-A, -B, or -C loci are coordinately downregulated [160,161].Since the levels of mRNA found in these tumor cell lines can frequently be upregulated in the presence of cytokines and low expression of transcription factors that bind to locus-specific DNA motifs can induce HLA-B locus downregulation, the assumption is that the mechanism of locus downregulation is often transcriptional [162]. |
| IV | HLA class I allelic loss | This molecular defect has been reported in colorectal carcinoma LS411, with a chromosomal break point in the HLA-A11 allele [163], or in the cervical cell lines CC11 and CSCC7 [164] or 808 and 778 [165,166]. |
| V | Compound phenotype | Requires a combination of at least two different alterations.Perea et al. recently reported a mechanism responsible for a total HLA class I loss in approximately 60% of studied small cell lung carcinoma samples. It is the combination of HLA haplotype loss together with a transcriptional downregulation of HLA-A, B and C genes [167]. |
| VI | Failure to respond to interferon (IFN) | This altered phenotype is found when tumor cells express basal levels of HLA class I antigens, but they do not respond to the stimulation of HLA class I expression with different cytokines, such as α and γ interferons (IFNs). For instance, the renal cell carcinoma Caki-2 does not have DNA-binding activity for IFN regulatory factor-1 or signal transducer and activator of transcription (STAT-1) [168]. |
| VII | Downregulation of classical HLA molecules (Ia) with aberrant expression of non-classical HLA molecules (Ib) | Based on unique HLA class I tissue distribution, that is used by the tumors to avoid both T and NK cell responses. It enables cancer cells to escape CTL responses by losing HLA-A, B, C. At the same time, these tumor cells, by engaging HLA-Ib molecules with NK inhibitory receptors, are resistant to NK lysis [156,169,170]. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sznarkowska, A.; Mikac, S.; Pilch, M. MHC Class I Regulation: The Origin Perspective. Cancers 2020, 12, 1155. https://doi.org/10.3390/cancers12051155
Sznarkowska A, Mikac S, Pilch M. MHC Class I Regulation: The Origin Perspective. Cancers. 2020; 12(5):1155. https://doi.org/10.3390/cancers12051155
Chicago/Turabian StyleSznarkowska, Alicja, Sara Mikac, and Magdalena Pilch. 2020. "MHC Class I Regulation: The Origin Perspective" Cancers 12, no. 5: 1155. https://doi.org/10.3390/cancers12051155
APA StyleSznarkowska, A., Mikac, S., & Pilch, M. (2020). MHC Class I Regulation: The Origin Perspective. Cancers, 12(5), 1155. https://doi.org/10.3390/cancers12051155