Mevalonate Pathway Enzyme HMGCS1 Contributes to Gastric Cancer Progression

 ,
,  , , ,
, , ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
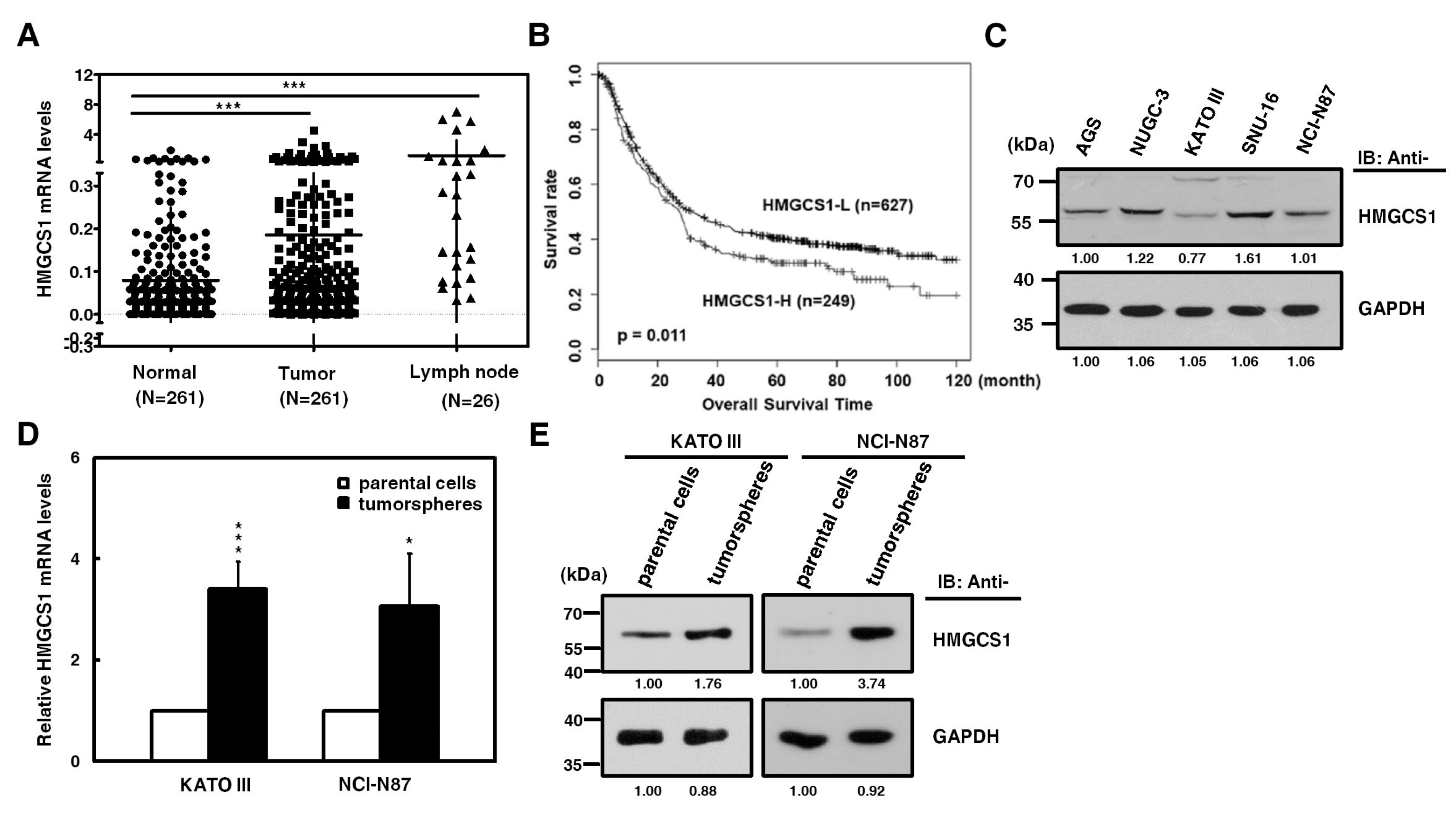
2.1. HMGCS1 Expression is Upregulated in Stomach Adenocarcinoma Samples of Patients and Tumorspheres of Gastric Cancer Cells
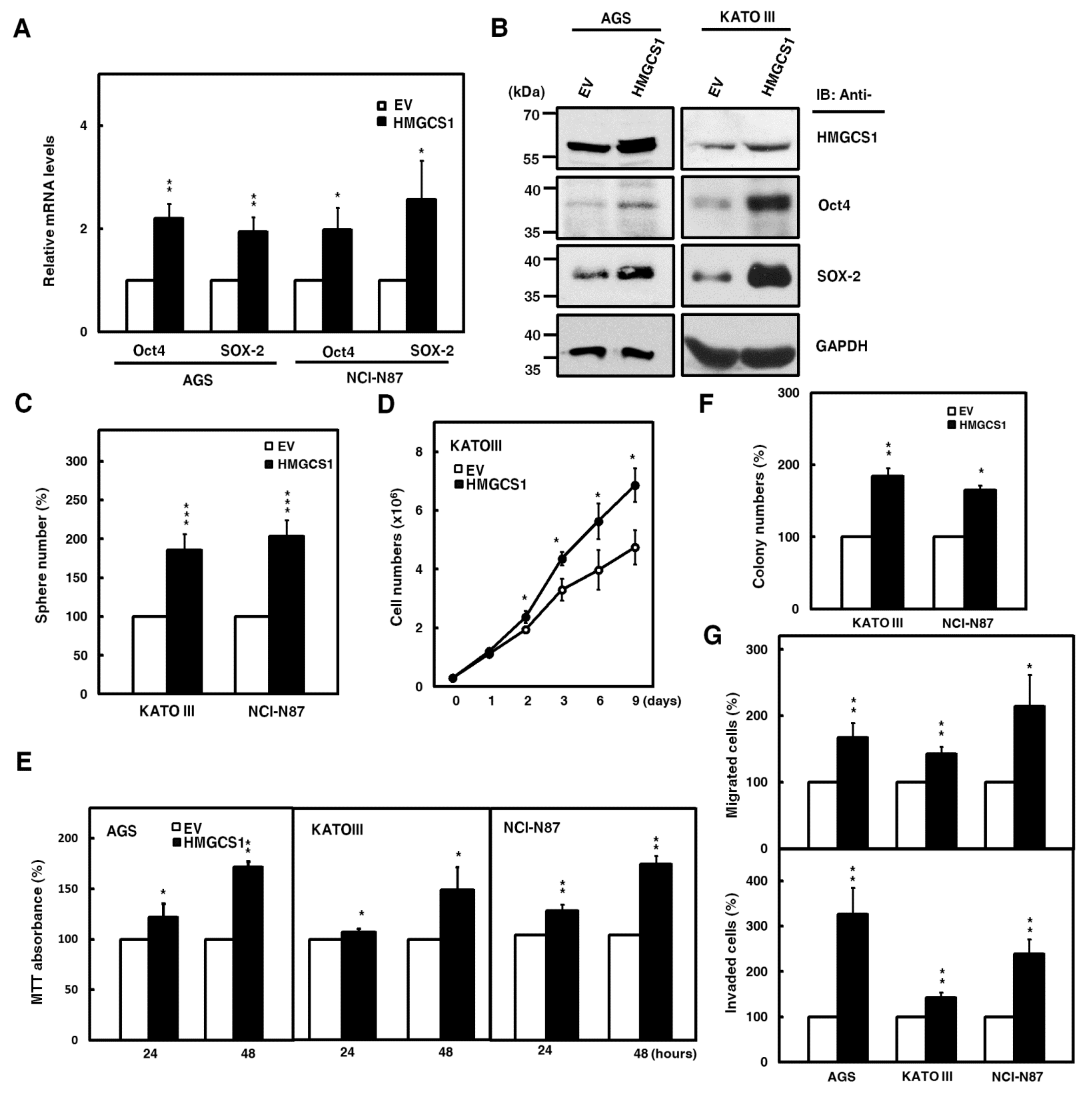
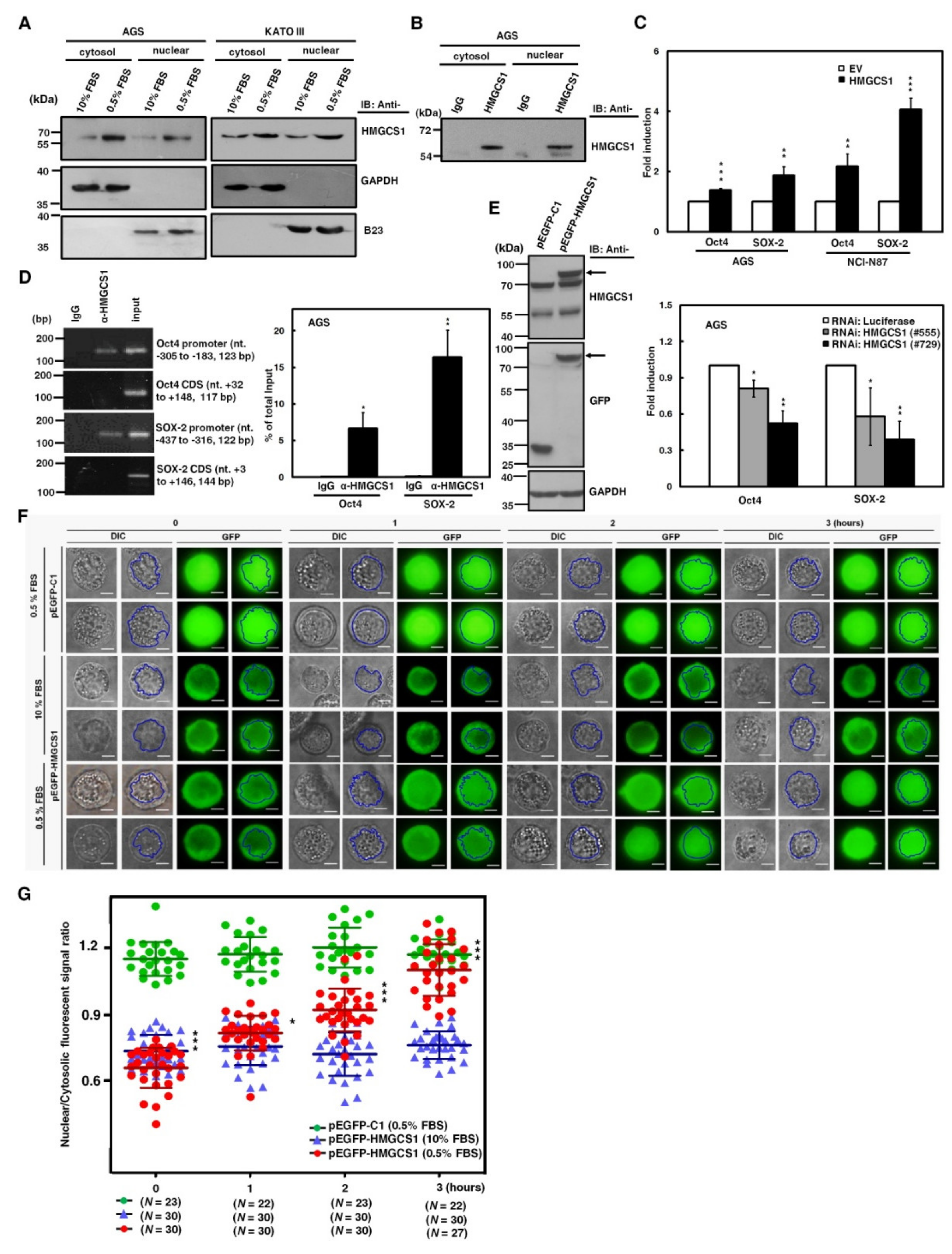
2.2. HMGCS1 Elevates Levels of Pluripotency Genes Oct4 and SRY (Sex Determining Region Y)-Box 2 (SOX-2) and Contributes to Progression in Gastric Cancer Cells
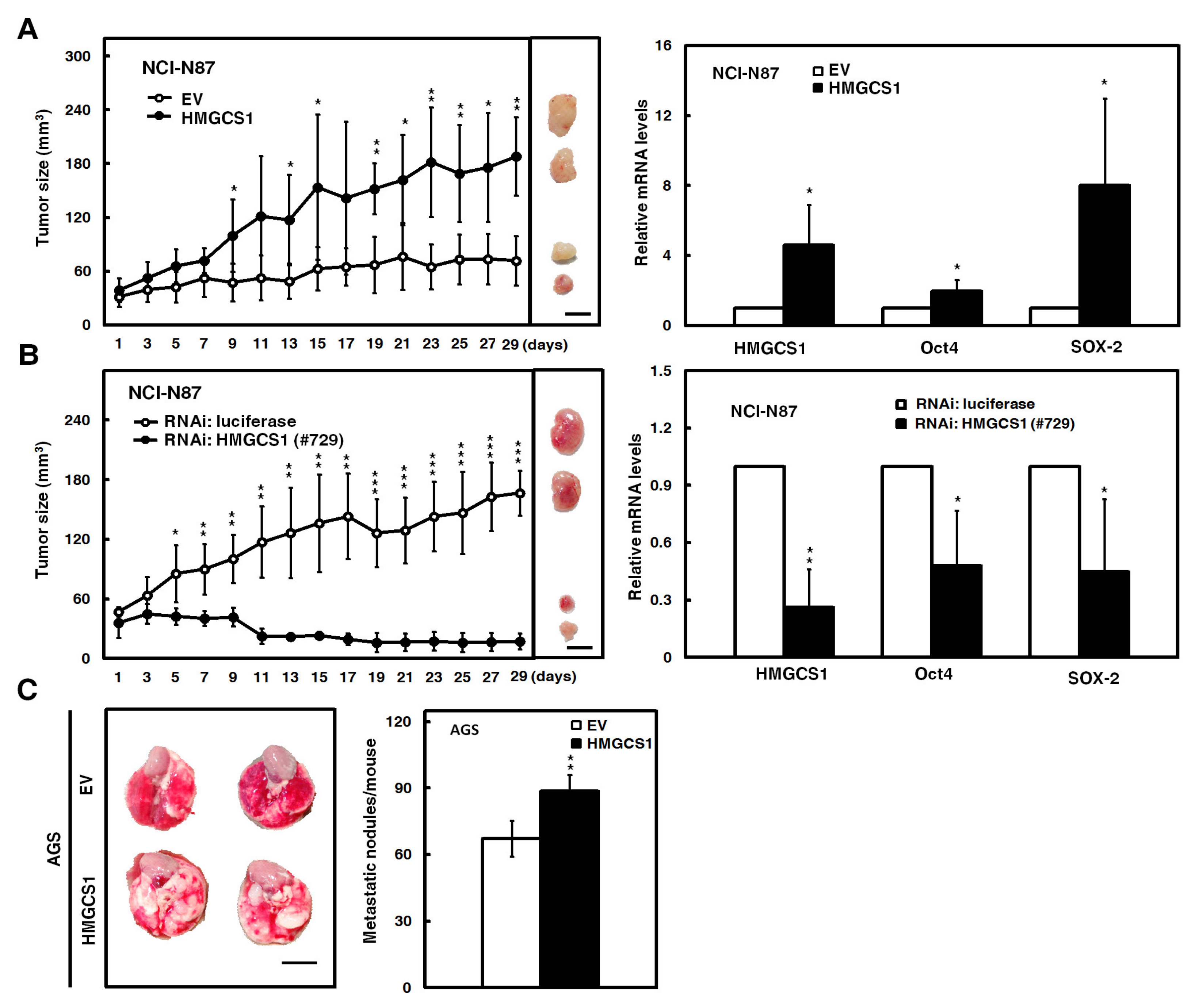
2.3. HMGCS1 Enhances Tumor Growth and Lung Metastasis of Gastric Cancer Cells
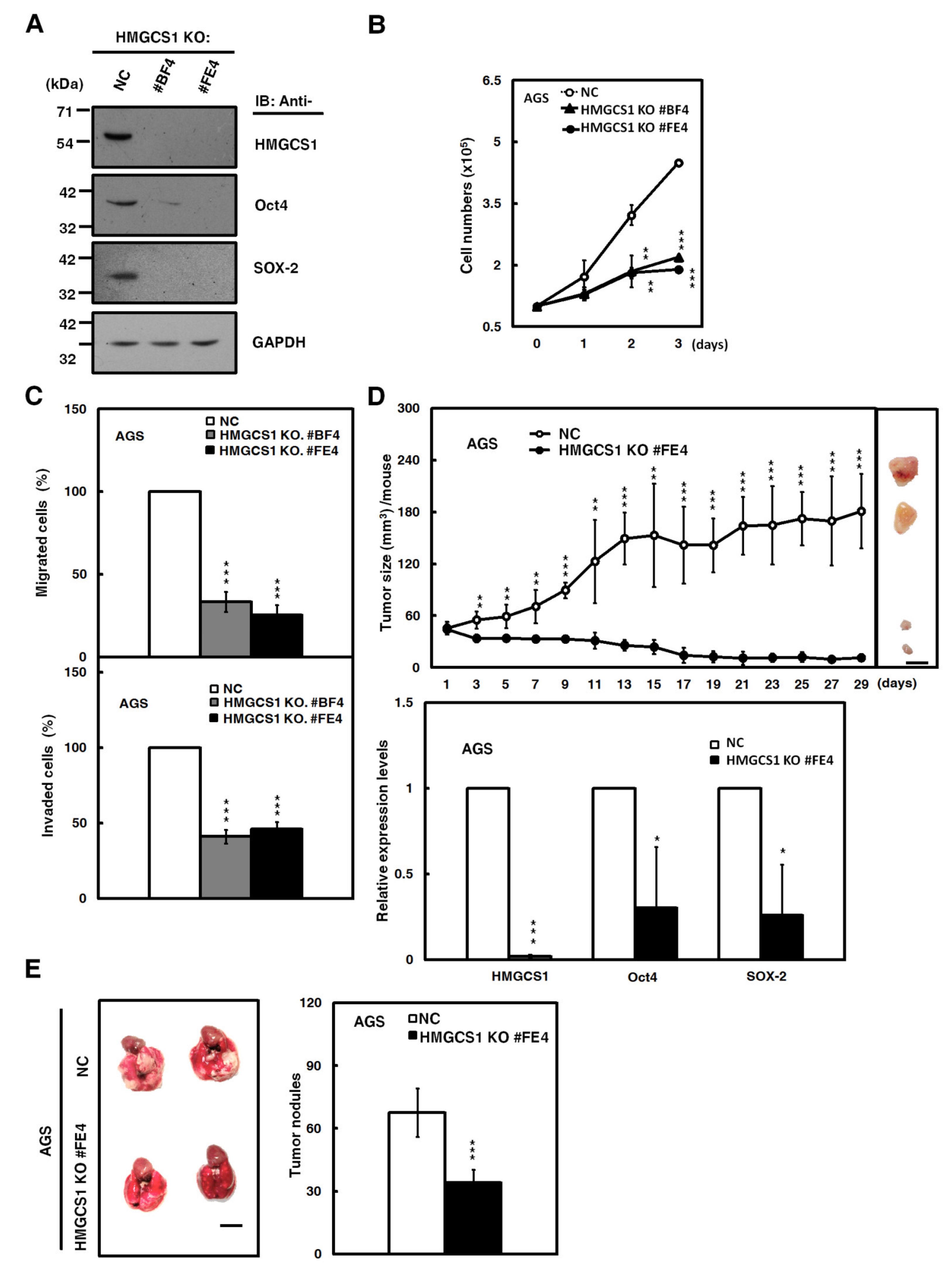
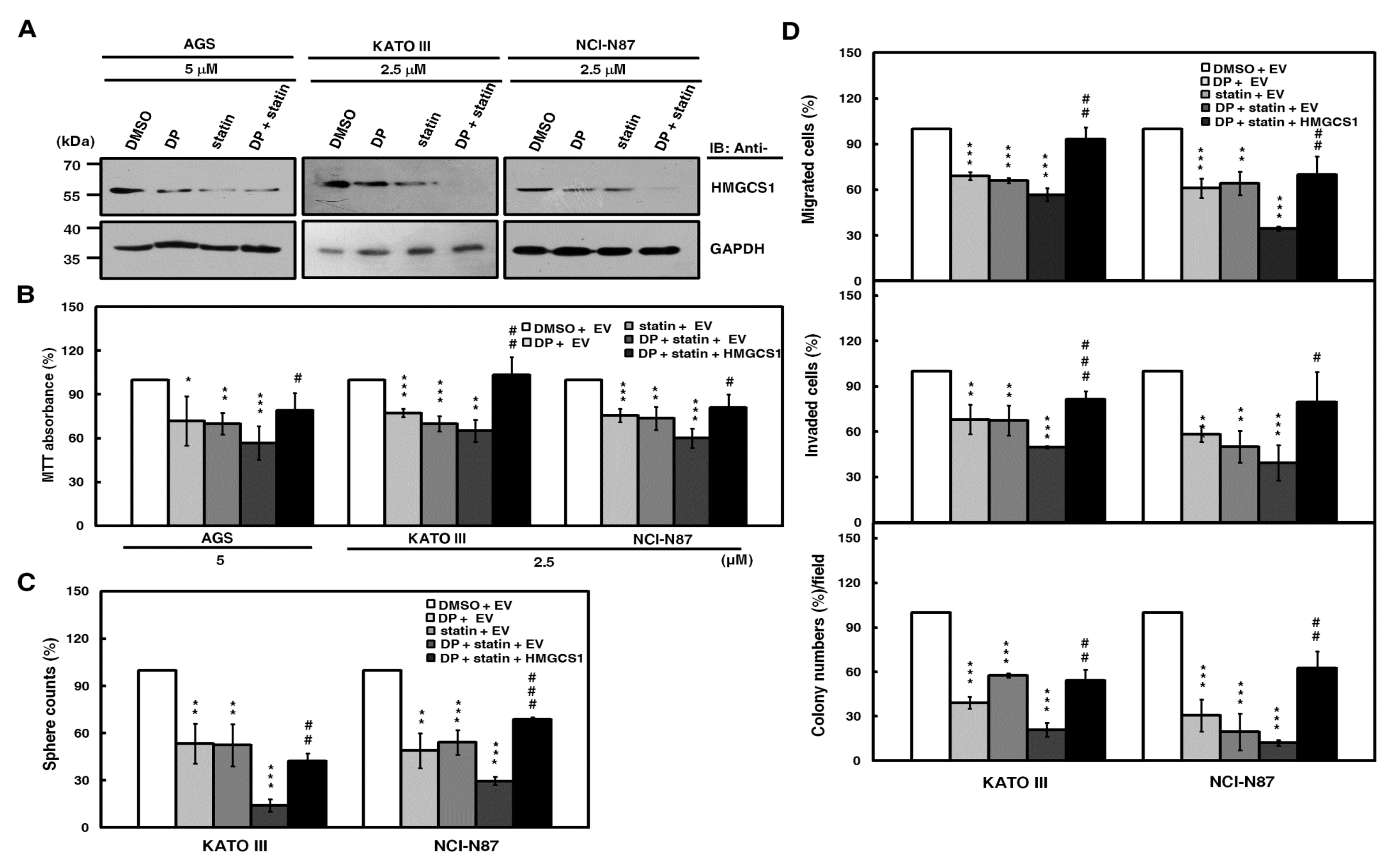
2.4. HMGCS1 Knockout Suppresses Growth and Progression of Gastric Cancer Cells
2.5. HMGCS1 Exerts Nonmetabolic Functions in Regulation of Gastric Cancer Progression
2.6. Nuclear HMGCS1 Translocation of Gastric Cancer Cells is Induced by Serum Deprivation
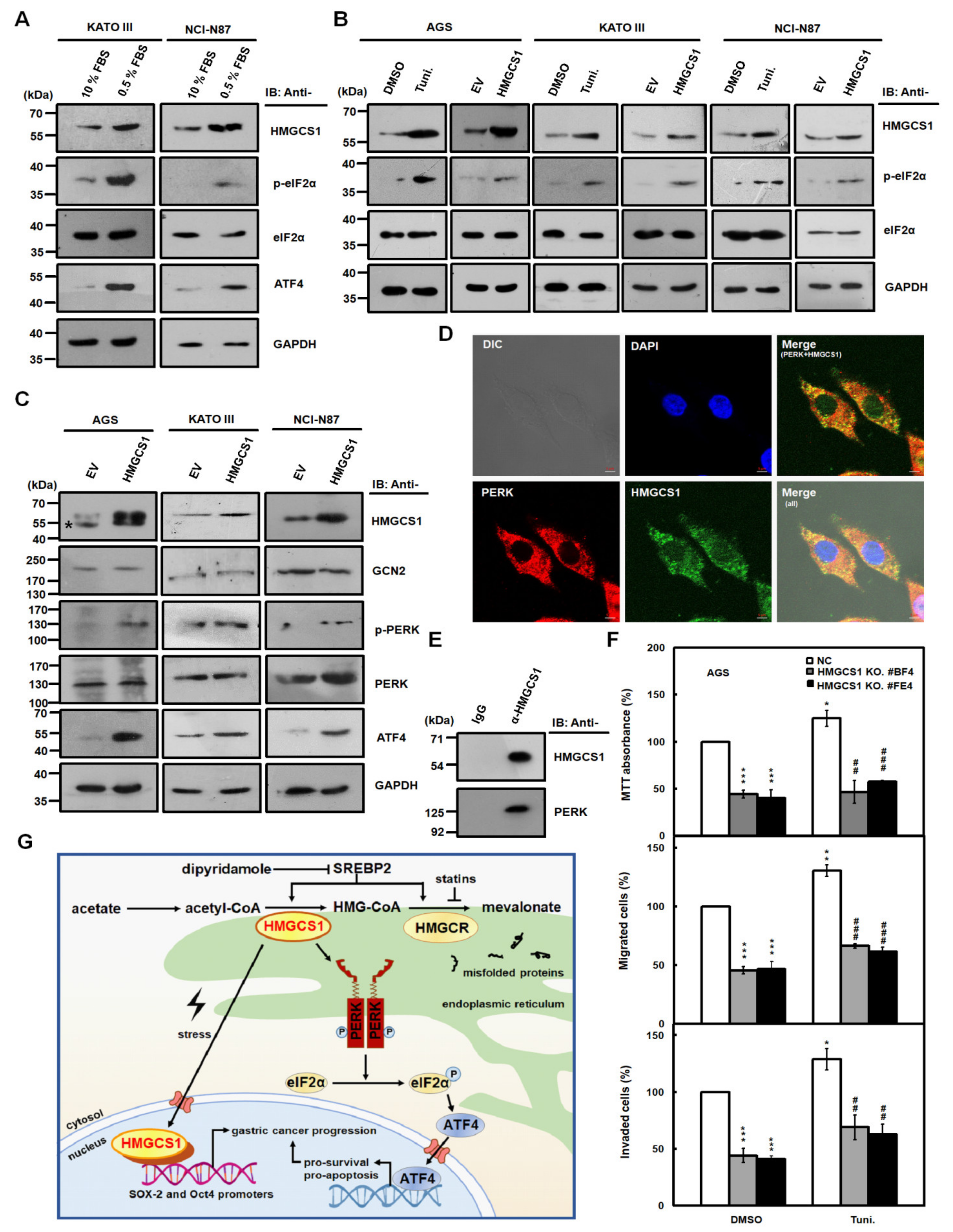
2.7. HMGCS1 Enhances the Integrated Stress Response (ISR) Pathway and Interacts with Protein Kinase RNA-Like Endoplasmic Reticulum (ER) Kinase (PERK), One of the Major ER Stress Transducers
3. Discussion
4. Materials and Methods
4.1. Plasmids and Plasmid Construction
4.2. Cell Culture and Transfection
4.3. The CRISPR-Cas9-Mediated HMGCS1 Knockout of AGS Gastric Cancer Cells
4.4. Quantitative Real-Time PCR Analysis
4.5. Western Blot Analysis
4.6. Subcellular Fractionation
4.7. Co-immunoprecipitation
4.8. Cell Growth and Viability Assays
4.9. Colony Formation, Migration, Invasion, and Tumorsphere Formation Assays
4.10. ChIP Assay
4.11. Time-Lapsed Epi-Fluorescence and DIC Microscopy Imaging
4.12. Immunofluorescence Staining and Confocal Microscopy
4.13. In vivo Xenografted Tumorigenicity and Tail Vein Metastasis Assays
4.14. Surgical Samples
4.15. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3-hydroxy-3-methylglutaryl-CoA synthase 1 | HMGCS1 |
| 3-hydroxy-3-methylglutaryl-CoA reductase | HMGCR |
| Clustered Regularly Interspaced Short Palindromic Repeats-associated protein-9 nuclease | CRISPR-Cas9 |
| eukaryotic translation initiation factor 2 alpha | eIF2α |
| integrated stress response | ISR |
| activating transcription factor 4 | ATF4 |
| endoplasmic reticulum | ER |
| protein kinase RNA-like ER kinase | PERK |
| general control nonderepressible 2 | GCN2 |
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Thurnher, M.; Gruenbacher, G.; Nussbaumer, O. Regulation of mevalonate metabolism in cancer and immune cells. Biochim. Biophys. Acta 2013, 1831, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Clendening, J.W.; Pandyra, A.; Boutros, P.C.; El Ghamrasni, S.; Khosravi, F.; Trentin, G.A.; Martirosyan, A.; Hakem, A.; Hakem, R.; Jurisica, I.; et al. Dysregulation of the mevalonate pathway promotes transformation. Proc. Natl. Acad. Sci. USA 2010, 107, 15051–15056. [Google Scholar] [CrossRef]
- Ginestier, C.; Monville, F.; Wicinski, J.; Cabaud, O.; Cervera, N.; Josselin, E.; Finetti, P.; Guille, A.; Larderet, G.; Viens, P.; et al. Mevalonate metabolism regulates Basal breast cancer stem cells and is a potential therapeutic target. Stem Cells 2012, 30, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef]
- Sharpe, L.J.; Brown, A.J. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J. Biol. Chem. 2013, 288, 18707–18715. [Google Scholar] [CrossRef]
- Sapir, A.; Tsur, A.; Koorman, T.; Ching, K.; Mishra, P.; Bardenheier, A.; Podolsky, L.; Bening-Abu-Shach, U.; Boxem, M.; Chou, T.F.; et al. Controlled sumoylation of the mevalonate pathway enzyme HMGS-1 regulates metabolism during aging. Proc. Natl. Acad. Sci. USA 2014, 111, E3880–E3889. [Google Scholar] [CrossRef]
- Nielsen, S.F.; Nordestgaard, B.G.; Bojesen, S.E. Statin use and reduced cancer-related mortality. N. Engl. J. Med. 2013, 368, 576–577. [Google Scholar] [CrossRef]
- Chiu, H.F.; Ho, S.C.; Chang, C.C.; Wu, T.N.; Yang, C.Y. Statins are associated with a reduced risk of gastric cancer: A population-based case-control study. Am. J. Gastroenterol. 2011, 106, 2098–2103. [Google Scholar] [CrossRef]
- Gobel, A.; Rauner, M.; Hofbauer, L.C.; Rachner, T.D. Cholesterol and beyond- The role of the mevalonate pathway in cancer biology. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188351. [Google Scholar] [CrossRef]
- Clendening, J.W.; Pandyra, A.; Li, Z.; Boutros, P.C.; Martirosyan, A.; Lehner, R.; Jurisica, I.; Trudel, S.; Penn, L.Z. Exploiting the mevalonate pathway to distinguish statin-sensitive multiple myeloma. Blood 2010, 115, 4787–4797. [Google Scholar] [CrossRef] [PubMed]
- Vock, C.; Doring, F.; Nitz, I. Transcriptional regulation of HMG-CoA synthase and HMG-CoA reductase genes by human ACBP. Cell. Physiol. Biochem. 2008, 22, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Pandyra, A.; Mullen, P.J.; Kalkat, M.; Yu, R.; Pong, J.T.; Li, Z.; Trudel, S.; Lang, K.S.; Minden, M.D.; Schimmer, A.D.; et al. Immediate utility of two approved agents to target both the metabolic mevalonate pathway and its restorative feedback loop. Cancer Res. 2014, 74, 4772–4782. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xu, H.; Tang, Q.; Xia, H.; Bi, F. Dipyridamole enhances the cytotoxicities of trametinib against colon cancer cells through combined targeting of HMGCS1 and MEK pathway. Mol. Cancer Ther. 2020, 19, 135–146. [Google Scholar] [CrossRef]
- Freed-Pastor, W.A.; Mizuno, H.; Zhao, X.; Langerod, A.; Moon, S.H.; Rodriguez-Barrueco, R.; Barsotti, A.; Chicas, A.; Li, W.; Polotskaia, A.; et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 2012, 148, 244–258. [Google Scholar] [CrossRef]
- Drynda, A.; Hoehn, R.; Peuster, M. Influence of Fe(II) and Fe(III) on the expression of genes related to cholesterol- and fatty acid metabolism in human vascular smooth muscle cells. J. Mater. Sci. Mater. Med. 2010, 21, 1655–1663. [Google Scholar] [CrossRef]
- Werstuck, G.H.; Lentz, S.R.; Dayal, S.; Hossain, G.S.; Sood, S.K.; Shi, Y.Y.; Zhou, J.; Maeda, N.; Krisans, S.K.; Malinow, M.R.; et al. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J. Clin. Investig. 2001, 107, 1263–1273. [Google Scholar] [CrossRef]
- Dubey, R.; Saini, N. STAT6 silencing up-regulates cholesterol synthesis via miR-197/FOXJ2 axis and induces ER stress-mediated apoptosis in lung cancer cells. Biochim. Biophys. Acta 2015, 1849, 32–43. [Google Scholar] [CrossRef]
- Pandyra, A.A.; Mullen, P.J.; Goard, C.A.; Ericson, E.; Sharma, P.; Kalkat, M.; Yu, R.; Pong, J.T.; Brown, K.R.; Hart, T.; et al. Genome-wide RNAi analysis reveals that simultaneous inhibition of specific mevalonate pathway genes potentiates tumor cell death. Oncotarget 2015, 6, 26909–26921. [Google Scholar] [CrossRef]
- Lee, W.J.; Kim, S.C.; Yoon, J.H.; Yoon, S.J.; Lim, J.; Kim, Y.S.; Kwon, S.W.; Park, J.H. Meta-analysis of tumor stem-like breast cancer cells using gene set and network analysis. PLoS ONE 2016, 11, e0148818. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Lincet, H.; Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 2015, 34, 3751–3759. [Google Scholar] [CrossRef] [PubMed]
- Tamada, M.; Suematsu, M.; Saya, H. Pyruvate kinase M2: Multiple faces for conferring benefits on cancer cells. Clin. Cancer Res. 2012, 18, 5554–5561. [Google Scholar] [CrossRef] [PubMed]
- Hartman, I.Z.; Liu, P.; Zehmer, J.K.; Luby-Phelps, K.; Jo, Y.; Anderson, R.G.; DeBose-Boyd, R.A. Sterol-induced dislocation of 3-hydroxy-3-methylglutaryl coenzyme A reductase from endoplasmic reticulum membranes into the cytosol through a subcellular compartment resembling lipid droplets. J. Biol. Chem. 2010, 285, 19288–19298. [Google Scholar] [CrossRef]
- Ohashi, M.; Mizushima, N.; Kabeya, Y.; Yoshimori, T. Localization of mammalian NAD(P)H steroid dehydrogenase-like protein on lipid droplets. J. Biol. Chem. 2003, 278, 36819–36829. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef]
- Hetz, C.; Papa, F.R. The unfolded protein response and cell fate control. Mol. Cell. 2018, 69, 169–181. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Arnold, S.M.; Miller, C.N.; Wu, J.; Li, J.; Gunnison, K.M.; Mori, K.; Sadighi Akha, A.A.; Raden, D.; Kaufman, R.J. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006, 4, e374. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef] [PubMed]
- Denoyelle, C.; Abou-Rjaily, G.; Bezrookove, V.; Verhaegen, M.; Johnson, T.M.; Fullen, D.R.; Pointer, J.N.; Gruber, S.B.; Su, L.D.; Nikiforov, M.A.; et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat. Cell Biol. 2006, 8, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- Tameire, F.; Verginadis, I.I.; Leli, N.M.; Polte, C.; Conn, C.S.; Ojha, R.; Salas Salinas, C.; Chinga, F.; Monroy, A.M.; Fu, W.; et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat. Cell Biol. 2019, 21, 889–899. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Zheng, Z.; Mendez, R.; Ha, S.W.; Xie, Y.; Zhang, K. Pharmacologic ER stress induces non-alcoholic steatohepatitis in an animal model. Toxicol. Lett. 2012, 211, 29–38. [Google Scholar] [CrossRef]
- Van Hoof, D.; Munoz, J.; Braam, S.R.; Pinkse, M.W.; Linding, R.; Heck, A.J.; Mummery, C.L.; Krijgsveld, J. Phosphorylation dynamics during early differentiation of human embryonic stem cells. Cell Stem Cell 2009, 5, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef]
- Kim, W.; Bennett, E.J.; Huttlin, E.L.; Guo, A.; Li, J.; Possemato, A.; Sowa, M.E.; Rad, R.; Rush, J.; Comb, M.J.; et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 2011, 44, 325–340. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Nielsen, M.L.; Cox, J.; Mann, M.; Choudhary, C. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol. Cell Proteom. 2011, 10, M111.013284. [Google Scholar] [CrossRef]
- Wagner, S.A.; Beli, P.; Weinert, B.T.; Scholz, C.; Kelstrup, C.D.; Young, C.; Nielsen, M.L.; Olsen, J.V.; Brakebusch, C.; Choudhary, C. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol. Cell Proteom. 2012, 11, 1578–1585. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Capra, J.A.; Pollard, K.S.; Verdin, E. SIRT1 and SIRT3 deacetylate homologous substrates: AceCS1,2 and HMGCS1,2. Aging (Albany NY) 2011, 3, 635–642. [Google Scholar] [CrossRef]
- Chang, C.C.; Hsu, W.H.; Wang, C.C.; Chou, C.H.; Kuo, M.Y.; Lin, B.R.; Chen, S.T.; Tai, S.K.; Kuo, M.L.; Yang, M.H. Connective tissue growth factor activates pluripotency genes and mesenchymal-epithelial transition in head and neck cancer cells. Cancer Res. 2013, 73, 4147–4157. [Google Scholar] [CrossRef] [PubMed]
- Yeh, T.S.; Wu, C.W.; Hsu, K.W.; Liao, W.J.; Yang, M.C.; Li, A.F.; Wang, A.M.; Kuo, M.L.; Chi, C.W. The activated Notch1 signal pathway is associated with gastric cancer progression through cyclooxygenase-2. Cancer Res. 2009, 69, 5039–5048. [Google Scholar] [CrossRef] [PubMed]
- Hsu, K.W.; Hsieh, R.H.; Wu, C.W.; Chi, C.W.; Lee, Y.H.; Kuo, M.L.; Wu, K.J.; Yeh, T.S. MBP-1 suppresses growth and metastasis of gastric cancer cells through COX-2. Mol. Biol. Cell 2009, 20, 5127–5137. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.M.; Huang, T.T.; Hsu, K.W.; Huang, K.H.; Fang, W.L.; Yang, M.H.; Lo, S.S.; Chi, C.W.; Lin, J.J.; Yeh, T.S. Yin Yang 1 is a target of microRNA-34 family and contributes to gastric carcinogenesis. Oncotarget 2014, 5, 5002–5016. [Google Scholar] [CrossRef]
- Hsu, K.W.; Hsieh, R.H.; Lee, Y.H.; Chao, C.H.; Wu, K.J.; Tseng, M.J.; Yeh, T.S. The activated Notch1 receptor cooperates with alpha-enolase and MBP-1 in modulating c-myc activity. Mol. Cell. Biol. 2008, 28, 4829–4842. [Google Scholar] [CrossRef]
- Tseng, Y.C.; Tsai, Y.H.; Tseng, M.J.; Hsu, K.W.; Yang, M.C.; Huang, K.H.; Li, A.F.; Chi, C.W.; Hsieh, R.H.; Ku, H.H.; et al. Notch2-induced COX-2 expression enhancing gastric cancer progression. Mol. Carcinog. 2012, 51, 939–951. [Google Scholar] [CrossRef]
- Hsu, K.W.; Fang, W.L.; Huang, K.H.; Huang, T.T.; Lee, H.C.; Hsieh, R.H.; Chi, C.W.; Yeh, T.S. Notch1 pathway-mediated microRNA-151-5p promotes gastric cancer progression. Oncotarget 2016, 7, 38036–38051. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, I.-H.; Huang, T.-T.; Chen, J.-L.; Chu, L.-W.; Ping, Y.-H.; Hsu, K.-W.; Huang, K.-H.; Fang, W.-L.; Lee, H.-C.; Chen, C.-F.; et al. Mevalonate Pathway Enzyme HMGCS1 Contributes to Gastric Cancer Progression. Cancers 2020, 12, 1088. https://doi.org/10.3390/cancers12051088
Wang I-H, Huang T-T, Chen J-L, Chu L-W, Ping Y-H, Hsu K-W, Huang K-H, Fang W-L, Lee H-C, Chen C-F, et al. Mevalonate Pathway Enzyme HMGCS1 Contributes to Gastric Cancer Progression. Cancers. 2020; 12(5):1088. https://doi.org/10.3390/cancers12051088
Chicago/Turabian StyleWang, I-Han, Tzu-Ting Huang, Ji-Lin Chen, Li-Wei Chu, Yueh-Hsin Ping, Kai-Wen Hsu, Kuo-Hung Huang, Wen-Liang Fang, Hsin-Chen Lee, Chian-Feng Chen, and et al. 2020. "Mevalonate Pathway Enzyme HMGCS1 Contributes to Gastric Cancer Progression" Cancers 12, no. 5: 1088. https://doi.org/10.3390/cancers12051088
APA StyleWang, I.-H., Huang, T.-T., Chen, J.-L., Chu, L.-W., Ping, Y.-H., Hsu, K.-W., Huang, K.-H., Fang, W.-L., Lee, H.-C., Chen, C.-F., Liao, C.-C., Hsieh, R.-H., & Yeh, T.-S. (2020). Mevalonate Pathway Enzyme HMGCS1 Contributes to Gastric Cancer Progression. Cancers, 12(5), 1088. https://doi.org/10.3390/cancers12051088