Application of a Biphasic Mathematical Model of Cancer Cell Drug Response for Formulating Potent and Synergistic Targeted Drug Combinations to Triple Negative Breast Cancer Cells



Abstract
1. Introduction
2. Results
2.1. Profiling of MDA-MB-231 and MDA-MB-468 Responses to Kinase Inhibitors
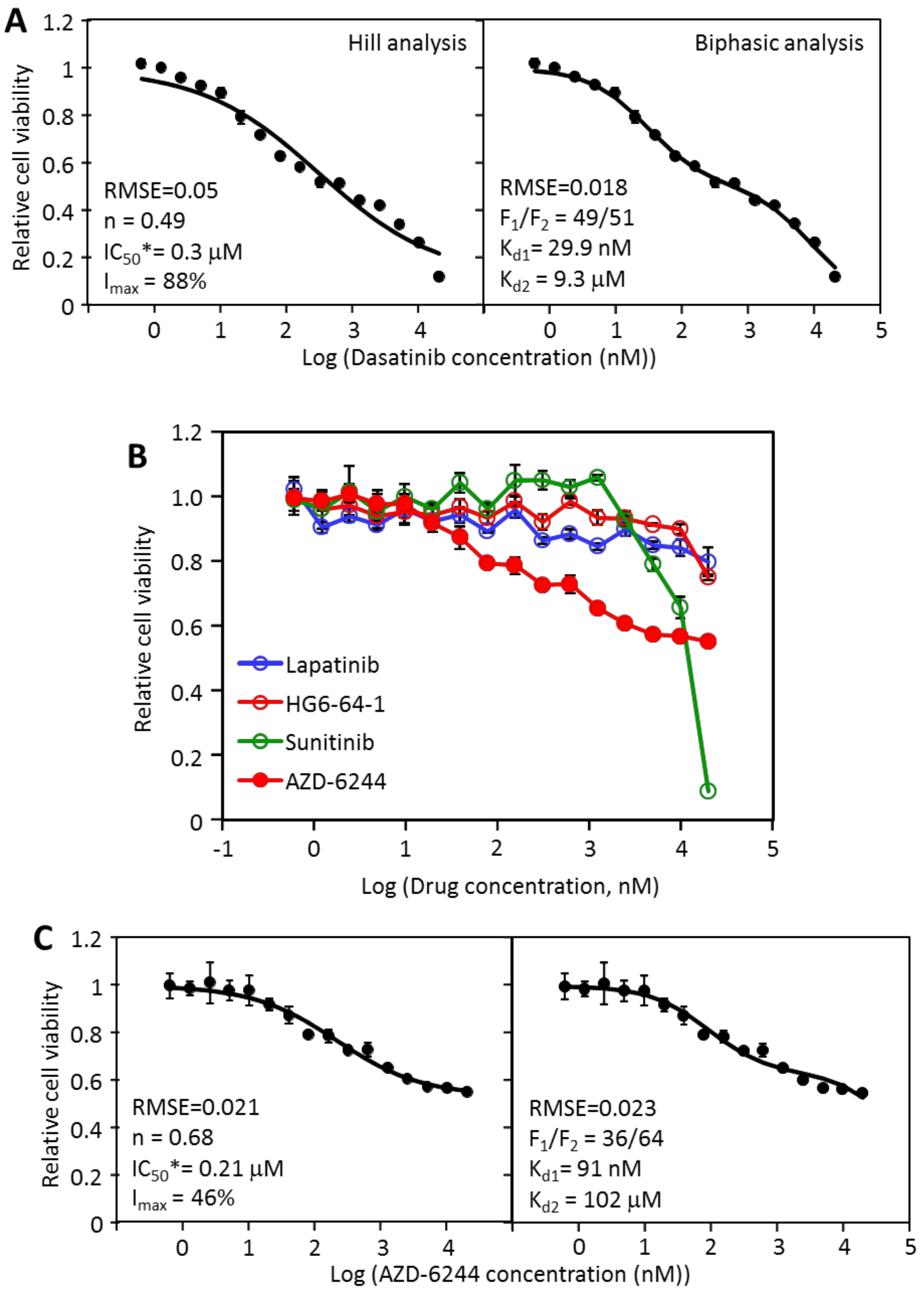
2.2. The Inhibition of MDA-MB-231 by Dasatinib Is Biphasic
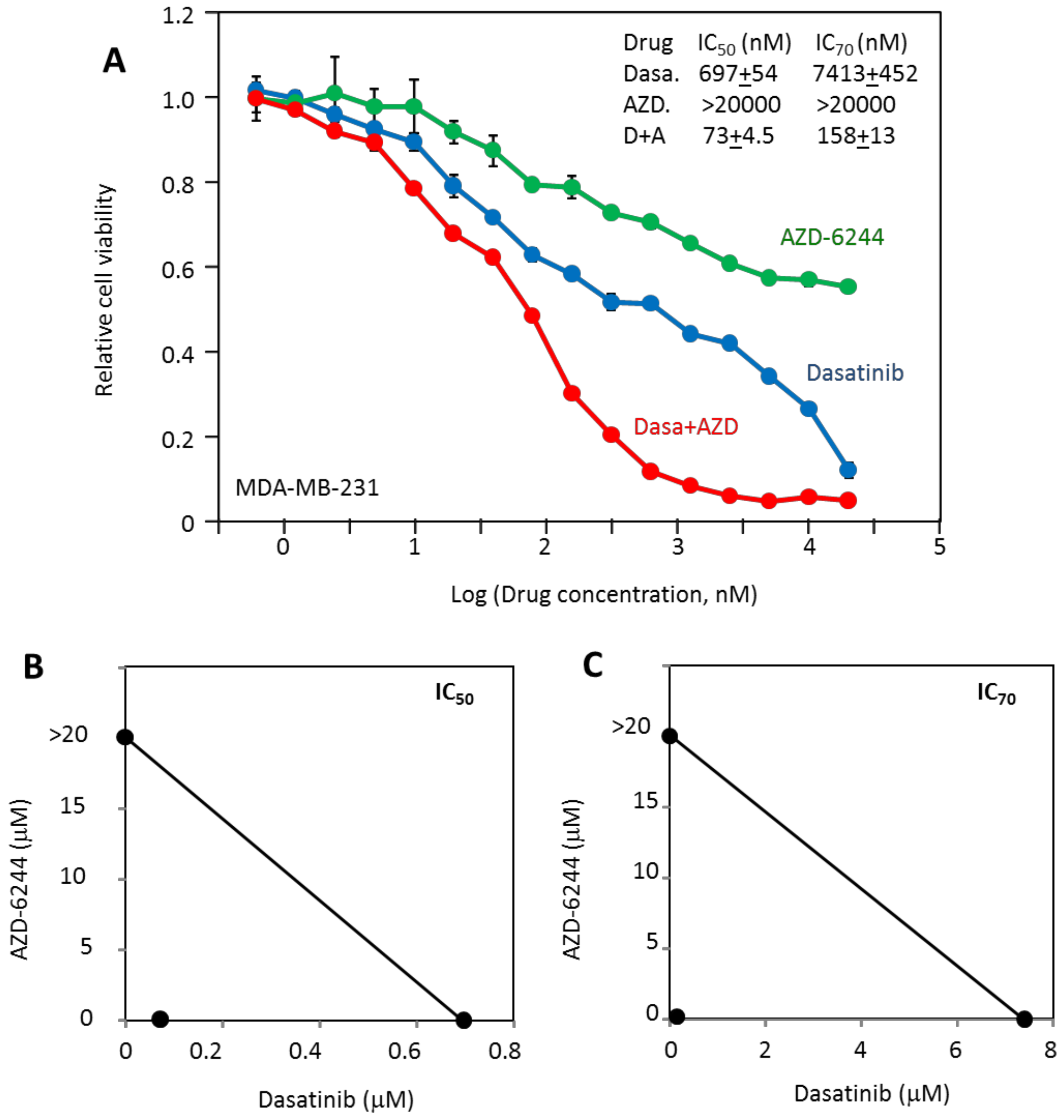
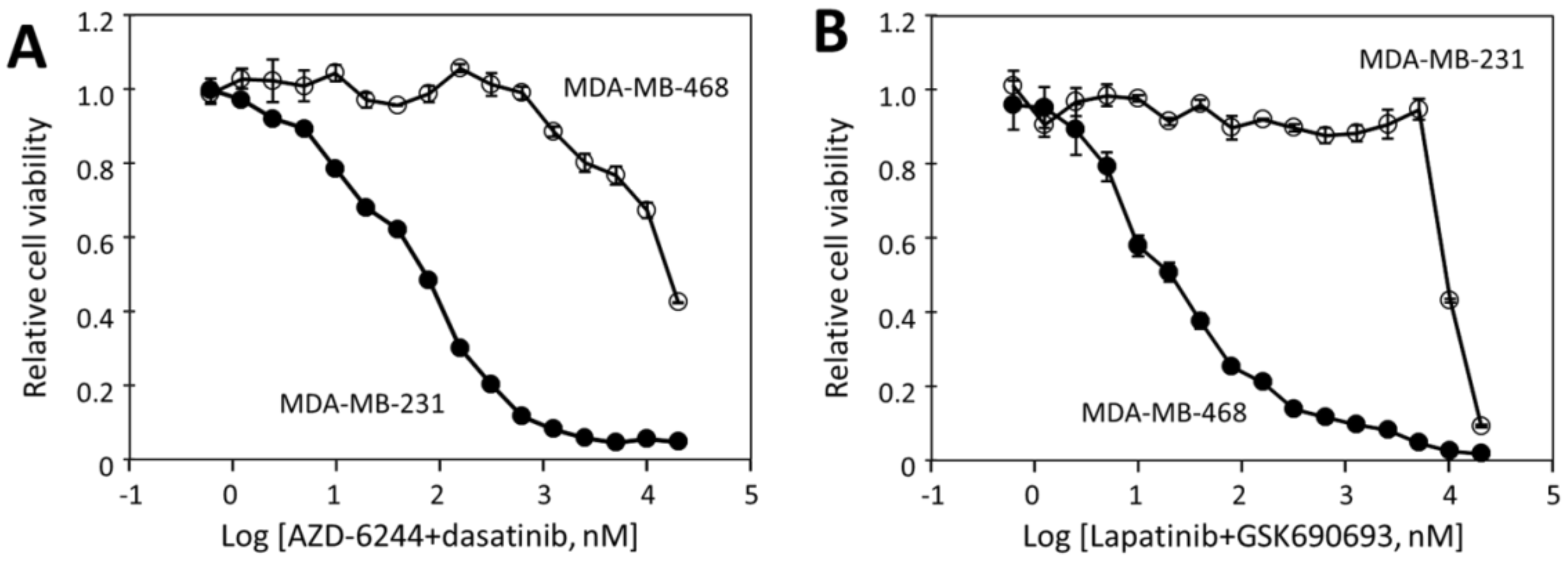
2.3. MDA-MB-231 Viability Is Potently Inhibited by the Combination of Dasatinib and AZD-6244
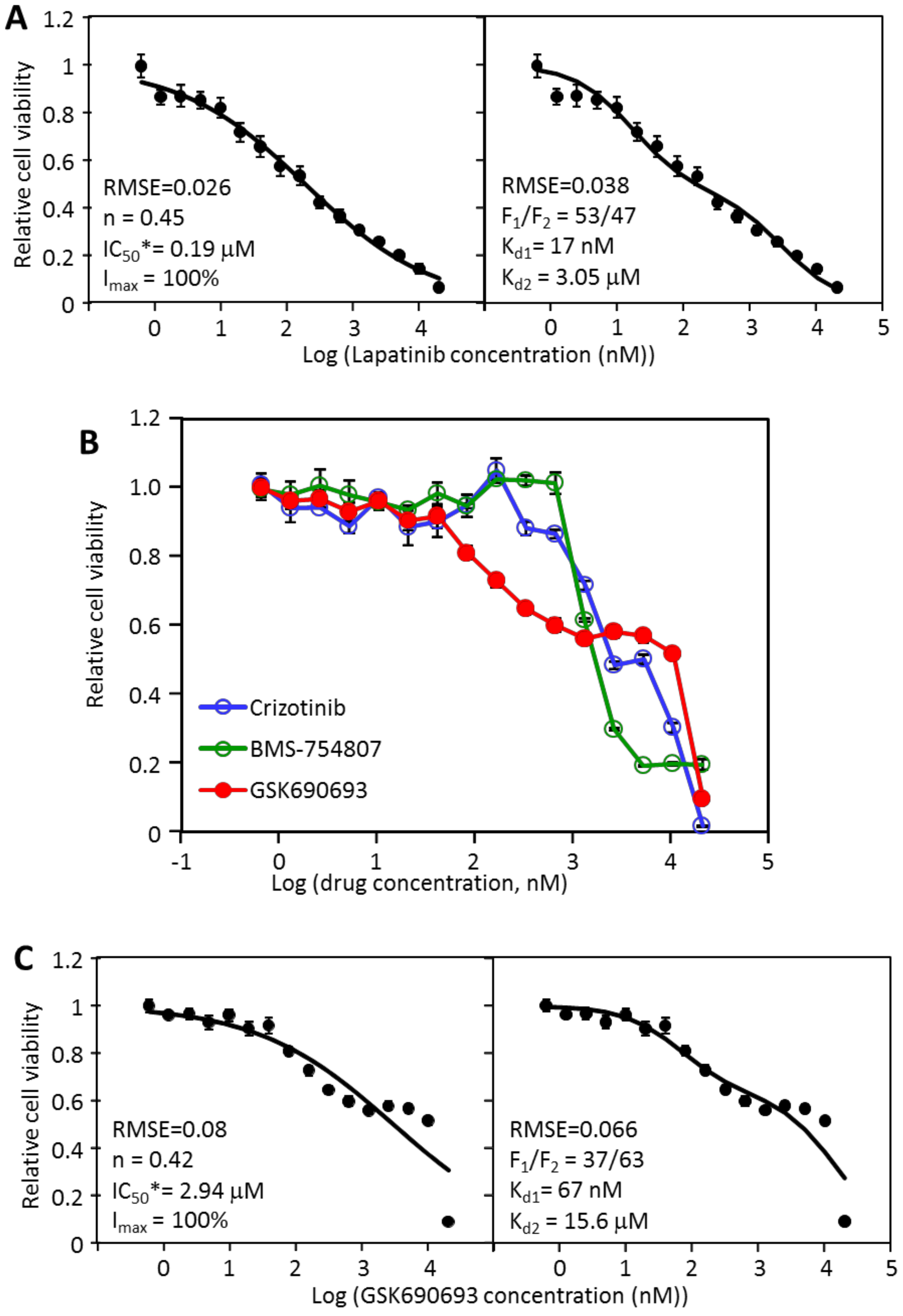
2.4. MDA-MB-468 Cells Follow Similar Inhibitory Patterns but Are Sensitive to Different Drugs
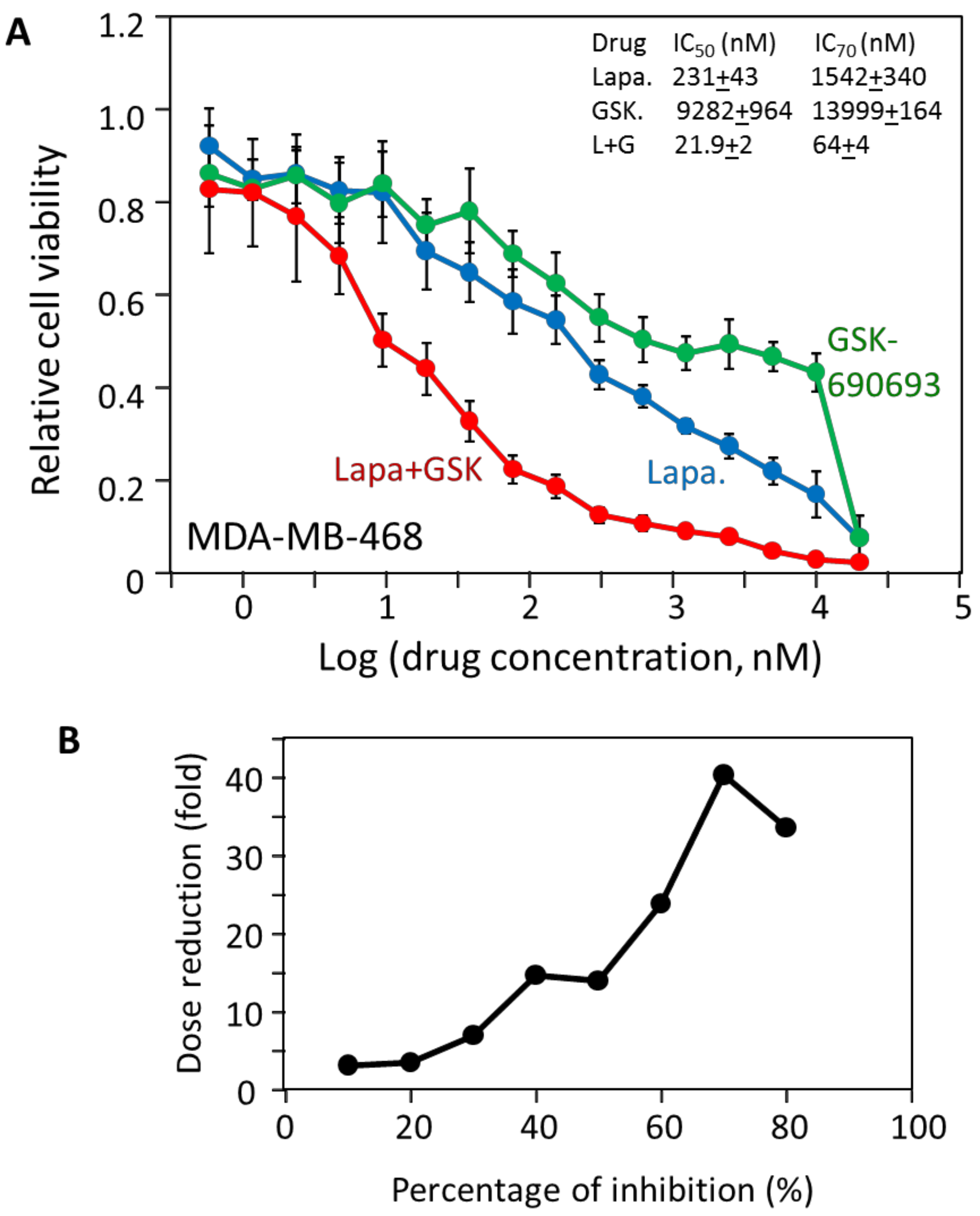
2.5. The Effectiveness of the Drug Combinations Is Cell-Specific
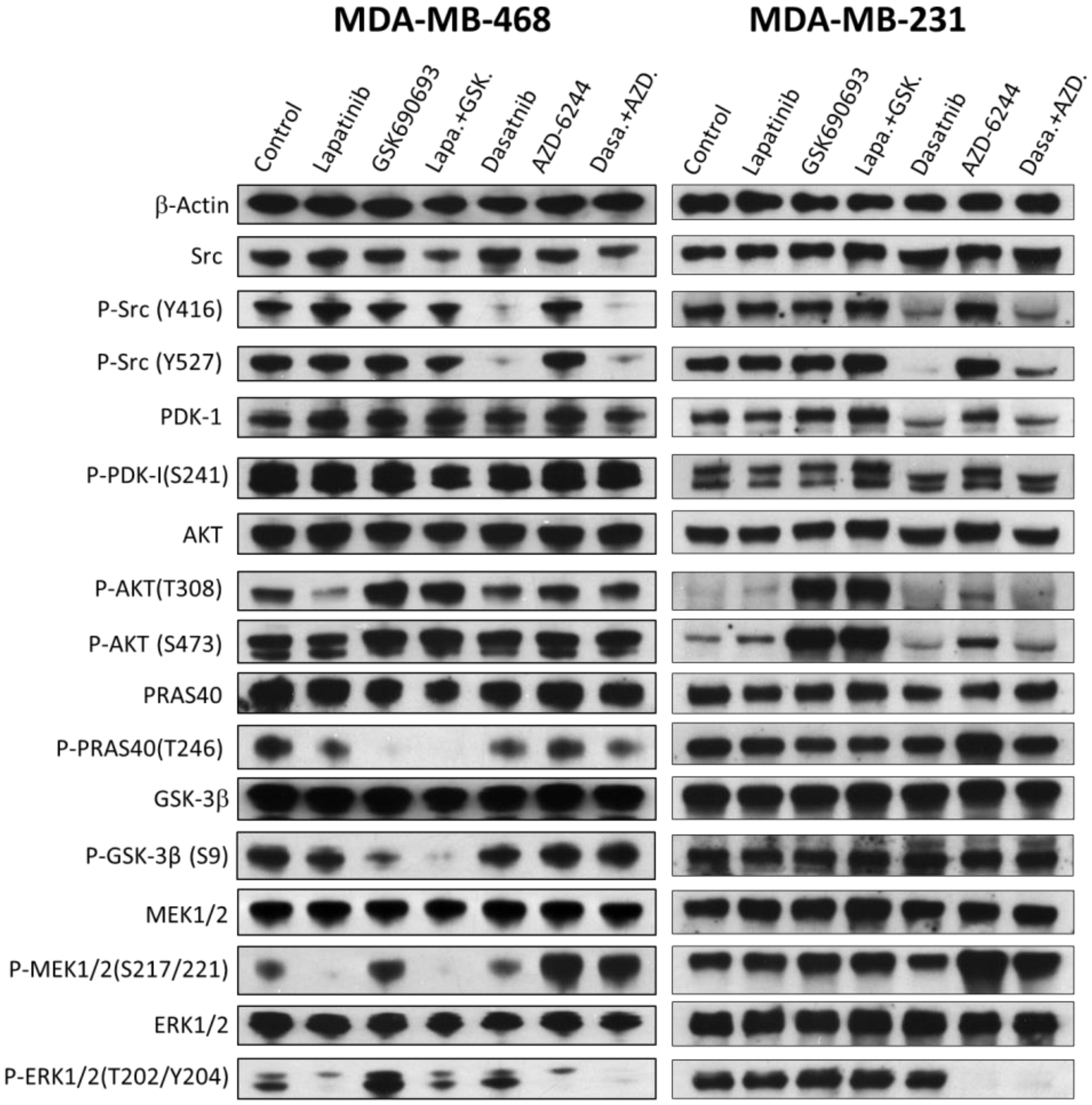
2.6. The Effects of Drugs and Their Combinations on the Signaling Pathways
2.7. The Hill and Biphasic Analyses Applied to Multi-Driver versus Mono-Driver Cancer Cells
3. Discussion
3.1. Drug Sensitivity Profiling Combined with Biphasic Analysis Can Identify Partial Drivers and Suggest Effective Drug Combinations for TNBC Cells
3.2. The Hill Analysis versus Biphasic Analysis
3.3. Heterogeneity and Cell-Specificity of TNBC Signaling
3.4. Implications of Biphasic Analysis on Targeted Therapy
4. Materials and Methods
4.1. Cell Lines, Media, and Drugs
4.2. Cell Culture and Viability Assays
4.3. Curve-Fitting by the Hill Equation and the Biphasic Equation
4.4. Drug Synergy Analysis and Combination Index Calculation
4.5. Western Blot Analysis of Drug Effects on Cell Signaling
4.6. Analyses of TNBC Cell Drug Response Data in the Genomics of Drug Sensitivity in Cancer Database
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CRC | colorectal cancer |
| DRI | dose reduction index |
| ER | estrogen receptor |
| GDSC | genomics of drug sensitivity in cancer |
| MAP kinase | mitogen-activated protein kinase |
| PR | progesterone receptor |
| RMSE | root mean square error |
| TNBC | triple negative breast cancer |
References
- American Cancer Society. How Common Is Breast Cancer? Available online: https://www.cancer.org/cancer/breast-cancer/about/how-common-is-breast-cancer.html (accessed on 8 March 2020).
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2017, 389, 1134–1150. [Google Scholar] [CrossRef]
- Burstein, H.J.; Lacchetti, C.; Griggs, J.J. Adjuvant Endocrine Therapy for Women With Hormone Receptor-Positive Breast Cancer: ASCO Clinical Practice Guideline Focused Update. J. Oncol. Pract. 2019, 15, 106–107. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Jhan, J.R.; Andrechek, E.R. Triple-negative breast cancer and the potential for targeted therapy. Pharmacogenomics 2017, 18, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Marotti, J.D.; de Abreu, F.B.; Wells, W.A.; Tsongalis, G.J. Triple-Negative Breast Cancer: Next-Generation Sequencing for Target Identification. Am. J. Pathol. 2017, 187, 2133–2138. [Google Scholar] [CrossRef]
- Network, C.G.A. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Xing, Y.; Lin, N.U.; Maurer, M.A.; Chen, H.; Mahvash, A.; Sahin, A.; Akcakanat, A.; Li, Y.; Abramson, V.; Litton, J.; et al. Phase II trial of AKT inhibitor MK-2206 in patients with advanced breast cancer who have tumors with PIK3CA or AKT mutations, and/or PTEN loss/PTEN mutation. Breast Cancer Res. 2019, 21, 78. [Google Scholar] [CrossRef]
- Goutelle, S.; Maurin, M.; Rougier, F.; Barbaut, X.; Bourguignon, L.; Ducher, M.; Maire, P. The Hill equation: A review of its capabilities in pharmacological modelling. Fundam. Clin. Pharmacol. 2008, 22, 633–648. [Google Scholar] [CrossRef]
- Hill, A. The possible effects of the aggregation of the molecules of haemoglobin on its dissociation curves. J. Physiol. 1910, 40, 4–7. [Google Scholar]
- Shen, J.; Li, L.; Yang, T.; Cohen, P.S.; Sun, G. Biphasic Mathematical Model of Cell-Drug Interaction That Separates Target-Specific and Off-Target Inhibition and Suggests Potent Targeted Drug Combinations for Multi-Driver Colorectal Cancer Cells. Cancers (Basel) 2020, 12. [Google Scholar] [CrossRef]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Lee, F.Y.; Bhalla, K.N.; Wu, J. Potent inhibition of platelet-derived growth factor-induced responses in vascular smooth muscle cells by BMS-354825 (dasatinib). Mol. Pharmacol. 2006, 69, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Schittenhelm, M.M.; Shiraga, S.; Schroeder, A.; Corbin, A.S.; Griffith, D.; Lee, F.Y.; Bokemeyer, C.; Deininger, M.W.; Druker, B.J.; Heinrich, M.C. Dasatinib (BMS-354825), a dual SRC/ABL kinase inhibitor, inhibits the kinase activity of wild-type, juxtamembrane, and activation loop mutant KIT isoforms associated with human malignancies. Cancer Res. 2006, 66, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Bailón, M.P.; Calcabrini, A.; Gómez-Domínguez, D.; Morte, B.; Martín-Forero, E.; Gómez-López, G.; Molinari, A.; Wagner, K.U.; Martín-Pérez, J. Src kinases catalytic activity regulates proliferation, migration and invasiveness of MDA-MB-231 breast cancer cells. Cell. Signal. 2012, 24, 1276–1286. [Google Scholar] [CrossRef]
- Tsai, P.C.; Chu, C.L.; Chiu, C.C.; Chang, L.S.; Lin, S.R. Inhibition of Src activation with cardiotoxin III blocks migration and invasion of MDA-MB-231 cells. Toxicon 2013, 74, 56–67. [Google Scholar] [CrossRef]
- Park, E.J.; Min, H.Y.; Chung, H.J.; Hong, J.Y.; Kang, Y.J.; Hung, T.M.; Youn, U.J.; Kim, Y.S.; Bae, K.; Kang, S.S.; et al. Down-regulation of c-Src/EGFR-mediated signaling activation is involved in the honokiol-induced cell cycle arrest and apoptosis in MDA-MB-231 human breast cancer cells. Cancer Lett. 2009, 277, 133–140. [Google Scholar] [CrossRef]
- Rusnak, D.W.; Lackey, K.; Affleck, K.; Wood, E.R.; Alligood, K.J.; Rhodes, N.; Keith, B.R.; Murray, D.M.; Knight, W.B.; Mullin, R.J.; et al. The effects of the novel, reversible epidermal growth factor receptor/ErbB-2 tyrosine kinase inhibitor, GW2016, on the growth of human normal and tumor-derived cell lines in vitro and in vivo. Mol. Cancer Ther. 2001, 1, 85–94. [Google Scholar]
- Liu, C.Y.; Hu, M.H.; Hsu, C.J.; Huang, C.T.; Wang, D.S.; Tsai, W.C.; Chen, Y.T.; Lee, C.H.; Chu, P.Y.; Hsu, C.C.; et al. Lapatinib inhibits CIP2A/PP2A/p-Akt signaling and induces apoptosis in triple negative breast cancer cells. Oncotarget 2016, 7, 9135–9149. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.Y.; Robell, K.; et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.A.; Jarboui, M.A.; Srihari, S.; Raso, C.; Bryan, K.; Dernayka, L.; Charitou, T.; Bernal-Llinares, M.; Herrera-Montavez, C.; Krstic, A.; et al. Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRAS. Nat. Commun. 2020, 11, 499. [Google Scholar] [CrossRef] [PubMed]
- Daigo, Y.; Furukawa, Y.; Kawasoe, T.; Ishiguro, H.; Fujita, M.; Sugai, S.; Nakamori, S.; Liefers, G.J.; Tollenaar, R.A.; van de Velde, C.J.; et al. Absence of genetic alteration at codon 531 of the human c-src gene in 479 advanced colorectal cancers from Japanese and Caucasian patients. Cancer Res. 1999, 59, 4222–4224. [Google Scholar]
- Irby, R.B.; Mao, W.; Coppola, D.; Kang, J.; Loubeau, J.M.; Trudeau, W.; Karl, R.; Fujita, D.J.; Jove, R.; Yeatman, T.J. Activating SRC mutation in a subset of advanced human colon cancers. Nat. Genet. 1999, 21, 187–190. [Google Scholar] [CrossRef]
- Johnson, F.M.; Gallick, G.E. SRC family nonreceptor tyrosine kinases as molecular targets for cancer therapy. Anticancer Agents Med. Chem. 2007, 7, 651–659. [Google Scholar]
- Forbes, S.A.; Beare, D.; Boutselakis, H.; Bamford, S.; Bindal, N.; Tate, J.; Cole, C.G.; Ward, S.; Dawson, E.; Ponting, L.; et al. COSMIC: Somatic cancer genetics at high-resolution. Nucleic Acids Res. 2017, 45, D777–D783. [Google Scholar] [CrossRef]
- El Guerrab, A.; Bamdad, M.; Kwiatkowski, F.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Anti-EGFR monoclonal antibodies and EGFR tyrosine kinase inhibitors as combination therapy for triple-negative breast cancer. Oncotarget 2016, 7, 73618–73637. [Google Scholar] [CrossRef]
- Weng, L.P.; Smith, W.M.; Dahia, P.L.; Ziebold, U.; Gil, E.; Lees, J.A.; Eng, C. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999, 59, 5808–5814. [Google Scholar]
- Marty, B.; Maire, V.; Gravier, E.; Rigaill, G.; Vincent-Salomon, A.; Kappler, M.; Lebigot, I.; Djelti, F.; Tourdès, A.; Gestraud, P.; et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R101. [Google Scholar] [CrossRef]
- Jang, K.; Kim, M.; Seo, H.S.; Shin, I. PTEN sensitizes MDA-MB-468 cells to inhibition of MEK/Erk signaling for the blockade of cell proliferation. Oncol. Rep. 2010, 24, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.M.; Lin, D.C.; Wei, X.; Che, Y.; Yao, Y.; Kiarash, R.; Cescon, D.W.; Fletcher, G.C.; Awrey, D.E.; Bray, M.R.; et al. Functional characterization of CFI-400945, a Polo-like kinase 4 inhibitor, as a potential anticancer agent. Cancer Cell 2014, 26, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.C.; Ho, S.C.; Tan, E.; Ng, A.W.T.; McPherson, J.R.; Goh, G.Y.L.; Teh, B.T.; Bard, F.; Rozen, S.G. Functional genomics identifies specific vulnerabilities in PTEN-deficient breast cancer. Breast Cancer Res. 2018, 20, 22. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef]
- Levy, D.S.; Kahana, J.A.; Kumar, R. AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 2009, 113, 1723–1729. [Google Scholar] [CrossRef]
- Altomare, D.A.; Zhang, L.; Deng, J.; Di Cristofano, A.; Klein-Szanto, A.J.; Kumar, R.; Testa, J.R. GSK690693 delays tumor onset and progression in genetically defined mouse models expressing activated Akt. Clin. Cancer Res. 2010, 16, 486–496. [Google Scholar] [CrossRef]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.H.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: Pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef]
- Li, Y.L.; Weng, H.C.; Hsu, J.L.; Lin, S.W.; Guh, J.H.; Hsu, L.C. The Combination of MK-2206 and WZB117 Exerts a Synergistic Cytotoxic Effect Against Breast Cancer Cells. Front. Pharmacol. 2019, 10, 1311. [Google Scholar] [CrossRef]
- Chen, X.; Cui, D.; Bi, Y.; Shu, J.; Xiong, X.; Zhao, Y. AKT inhibitor MK-2206 sensitizes breast cancer cells to MLN4924, a first-in-class NEDD8-activating enzyme (NAE) inhibitor. Cell Cycle 2018, 17, 2069–2079. [Google Scholar] [CrossRef]
- Ma, C.X.; Sanchez, C.; Gao, F.; Crowder, R.; Naughton, M.; Pluard, T.; Creekmore, A.; Guo, Z.; Hoog, J.; Lockhart, A.C.; et al. A Phase I Study of the AKT Inhibitor MK-2206 in Combination with Hormonal Therapy in Postmenopausal Women with Estrogen Receptor-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2016, 22, 2650–2658. [Google Scholar] [CrossRef]
- Lee, S.; Lin, X.; Nam, N.H.; Parang, K.; Sun, G. Determination of the substrate-docking site of protein tyrosine kinase C-terminal Src kinase. Proc. Natl. Acad. Sci. USA 2003, 100, 14707–14712. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.K.; Brown, A.; Sadeghiani, N.; Shirazi, A.N.; Bolton, J.; Tse, A.; Verkhivker, G.; Parang, K.; Sun, G. Design, Synthesis, and Evaluation of Dasatinib-Amino Acid and Dasatinib-Fatty Acid Conjugates as Protein Tyrosine Kinase Inhibitors. ChemMedChem 2017, 12, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Chiu, C.; Chiou, T.; Maeda, S.; Chiang, H.; Tzeng, C.; Sugiyama, T.; Chiang, B.N. Establishment and characterization of a human monocytoid leukemia cell line, CTV-1. GANN Jpn. J. Cancer Res. 1984, 75, 660–664. [Google Scholar]
- Drexler, H.G.; Gaedicke, G.; Maeda, S.; Chen, P.M.; Minowada, J. Monocytoid leukemia cell line CTV-1: Morphological, immunological and isoenzymatic characteristics. Tumour Biol. 1986, 6, 503–517. [Google Scholar]
- Amann, J.; Kalyankrishna, S.; Massion, P.P.; Ohm, J.E.; Girard, L.; Shigematsu, H.; Peyton, M.; Juroske, D.; Huang, Y.; Stuart Salmon, J.; et al. Aberrant epidermal growth factor receptor signaling and enhanced sensitivity to EGFR inhibitors in lung cancer. Cancer Res. 2005, 65, 226–235. [Google Scholar]
- Li, L.; Cui, Y.; Shen, J.; Dobson, H.; Sun, G. Evidence for activated Lck protein tyrosine kinase as the driver of proliferation in acute myeloid leukemia cell, CTV-1. Leuk. Res. 2019, 78, 12–20. [Google Scholar] [CrossRef]
- Palmer, A.C.; Sorger, P.K. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017, 171, 1678–1691.e1613. [Google Scholar] [CrossRef]
- O’Neil, J.; Benita, Y.; Feldman, I.; Chenard, M.; Roberts, B.; Liu, Y.; Li, J.; Kral, A.; Lejnine, S.; Loboda, A.; et al. An Unbiased Oncology Compound Screen to Identify Novel Combination Strategies. Mol. Cancer Ther. 2016, 15, 1155–1162. [Google Scholar] [CrossRef]
- Wali, V.B.; Langdon, C.G.; Held, M.A.; Platt, J.T.; Patwardhan, G.A.; Safonov, A.; Aktas, B.; Pusztai, L.; Stern, D.F.; Hatzis, C. Systematic Drug Screening Identifies Tractable Targeted Combination Therapies in Triple-Negative Breast Cancer. Cancer Res. 2017, 77, 566–578. [Google Scholar] [CrossRef]
- Menden, M.P.; Wang, D.; Mason, M.J.; Szalai, B.; Bulusu, K.C.; Guan, Y.; Yu, T.; Kang, J.; Jeon, M.; Wolfinger, R.; et al. Community assessment to advance computational prediction of cancer drug combinations in a pharmacogenomic screen. Nat. Commun. 2019, 10, 2674. [Google Scholar] [CrossRef]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive Screening Resource for the Detection of Anticancer Drug Pairs with Enhanced Therapeutic Activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.B.; Schoeberl, B.; Nielsen, U.B.; Sorger, P.K. Systems biology and combination therapy in the quest for clinical efficacy. Nat. Chem. Biol. 2006, 2, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Fallahi-Sichani, M.; Honarnejad, S.; Heiser, L.M.; Gray, J.W.; Sorger, P.K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 2013, 9, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Gaule, P.; Mukherjee, N.; Corkery, B.; Eustace, A.J.; Gately, K.; Roche, S.; O’Connor, R.; O’Byrne, K.J.; Walsh, N.; Duffy, M.J.; et al. Dasatinib Treatment Increases Sensitivity to c-Met Inhibition in Triple-Negative Breast Cancer Cells. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Qian, X.L.; Zhang, J.; Li, P.Z.; Lang, R.G.; Li, W.D.; Sun, H.; Liu, F.F.; Guo, X.J.; Gu, F.; Fu, L. Dasatinib inhibits c-src phosphorylation and prevents the proliferation of Triple-Negative Breast Cancer (TNBC) cells which overexpress Syndecan-Binding Protein (SDCBP). PLoS ONE 2017, 12, e0171169. [Google Scholar] [CrossRef]
- Pichot, C.S.; Hartig, S.M.; Xia, L.; Arvanitis, C.; Monisvais, D.; Lee, F.Y.; Frost, J.A.; Corey, S.J. Dasatinib synergizes with doxorubicin to block growth, migration, and invasion of breast cancer cells. Br. J. Cancer 2009, 101, 38–47. [Google Scholar] [CrossRef]
- Nautiyal, J.; Majumder, P.; Patel, B.B.; Lee, F.Y.; Majumdar, A.P. Src inhibitor dasatinib inhibits growth of breast cancer cells by modulating EGFR signaling. Cancer Lett. 2009, 283, 143–151. [Google Scholar] [CrossRef]
- Finn, R.S.; Dering, J.; Ginther, C.; Wilson, C.A.; Glaspy, P.; Tchekmedyian, N.; Slamon, D.J. Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res. Treat 2007, 105, 319–326. [Google Scholar] [CrossRef]
- Zhou, Y.; Lin, S.; Tseng, K.F.; Han, K.; Wang, Y.; Gan, Z.H.; Min, D.L.; Hu, H.Y. Selumetinib suppresses cell proliferation, migration and trigger apoptosis, G1 arrest in triple-negative breast cancer cells. BMC Cancer 2016, 16, 818. [Google Scholar] [CrossRef]
- Maiello, M.R.; D’Alessio, A.; Bevilacqua, S.; Gallo, M.; Normanno, N.; De Luca, A. EGFR and MEK Blockade in Triple Negative Breast Cancer Cells. J. Cell. Biochem. 2015, 116, 2778–2785. [Google Scholar] [CrossRef]
- Zhou, Y.; Hu, H.Y.; Meng, W.; Jiang, L.; Zhang, X.; Sha, J.J.; Lu, Z.; Yao, Y. MEK inhibitor effective against proliferation in breast cancer cell. Tumour Biol. 2014, 35, 9269–9279. [Google Scholar] [CrossRef]
- Hegde, P.S.; Rusnak, D.; Bertiaux, M.; Alligood, K.; Strum, J.; Gagnon, R.; Gilmer, T.M. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol. Cancer Ther. 2007, 6, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Showalter, L.E.; Oechsle, C.; Ghimirey, N.; Steele, C.; Czerniecki, B.J.; Koski, G.K. Th1 cytokines sensitize HER-expressing breast cancer cells to lapatinib. PLoS ONE 2019, 14, e0210209. [Google Scholar] [CrossRef]
- Phyu, S.M.; Tseng, C.C.; Fleming, I.N.; Smith, T.A. Probing the PI3K/Akt/mTor pathway using. Sci. Rep. 2016, 6, 36544. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Bengala, C.; Ibrahim, N.; Roché, H.; Sparano, J.; Strauss, L.C.; Fairchild, J.; Sy, O.; Goldstein, L.J. Dasatinib as a single agent in triple-negative breast cancer: Results of an open-label phase 2 study. Clin. Cancer Res. 2011, 17, 6905–6913. [Google Scholar] [CrossRef]
- Finn, R.S.; Press, M.F.; Dering, J.; Arbushites, M.; Koehler, M.; Oliva, C.; Williams, L.S.; Di Leo, A. Estrogen receptor, progesterone receptor, human epidermal growth factor receptor 2 (HER2), and epidermal growth factor receptor expression and benefit from lapatinib in a randomized trial of paclitaxel with lapatinib or placebo as first-line treatment in HER2-negative or unknown metastatic breast cancer. J. Clin. Oncol. 2009, 27, 3908–3915. [Google Scholar] [CrossRef]
- Chou, T.C. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef]
- Shen, J.; Li, L.; Yang, T.; Cheng, N.; Sun, G. Drug Sensitivity Screening and Targeted Pathway Analysis Reveal a Multi-Driver Proliferative Mechanism and Suggest a Strategy of Combination Targeted Therapy for Colorectal Cancer Cells. Molecules 2019, 24. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target 1 | IC50 (μM) 2 | |
|---|---|---|---|
| MDA-MB-231 | MDA-MB-468 | ||
| Dasatinib | Src (0.2), Abl (0.05), PDGFRs (1) | 0.578 + 0.05 | 19.7 + 0.30 |
| AZD-6244 | Mek (99) | >20 | >20 |
| Lapatinib | EGFR (2.4) ErbB2 (7) | >20 | 0.30 + 0.06 |
| GSK690693 | Akt (2–3) | 16.7 + 0.40 | 9.28 + 0.96 |
| BX-912 | PDK1 (26) | 14.6 + 0.63 | 1.62 + 0.037 |
| AZD6482 | PI 3-Kβ (10) | >20 | >20 |
| HG6-64-1 | BRAF (90) | >20 | >20 |
| Bosutinib | Src (1), Abl (0.12), etc | 10.3 + 0.21 | >20 |
| BMS-754807 | IR (1.7), IGF-1R (1.8), Met (5.6) | 7.47 + 0.15 | 1.59 + 0.021 |
| Crizotinib | Alk (3.3), Met (2.1) | 5.63 + 0.41 | 2.39 + 0.11 |
| Erlotinib | EGFR (0.7) | >20 | 15.9 + 0.63 |
| Linsitinib | IR (75), IGF-1R (35) | >20 | >20 |
| Linifanib | PDGFRs (0.6–10), VEGFRs (7–8) | >20 | 6.29 + 0.46 |
| Sorafenib | PDGFRs (13), VEGFRs (31) | 9.20 + 0.25 | 7.12 + 0.14 |
| Sunitinib | PDGFRs (0.1–2), VEGFRs (1–2) | 12.1 + 0.33 | 15.5 + 0.35 |
| Pazopanib | PDGFRs (2-8), VEGFRs (14) | >20 | >20 |
| Gefitinib | EGFR (1) | >20 | 19.1 + 0.86 |
| BGJ398 | FGFRs (0.9–1.4) | >20 | 13.3 + 0.27 |
| Cell | Drug | Hill Analysis | Biphasic Analysis | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RMSE | Imax (%) | IC50* (nM) | n | RMSE | F1/F2 | Kd1 (nM) | Kd2 (μM) | ||
| HT-29 | AZD-6244 | 0.027 | 73 | 248 | 0.52 | 0.029 | 55/45 | 92 | 54.1 |
| BMS-754807 | 0.023 | 100 | 585 | 0.34 | 0.047 | 44/56 | 12 | 8.4 | |
| Dasatinib | 0.032 | 67 | 147 | 0.62 | 0.026 | 51/49 | 59 | 33.3 | |
| HG6-64-1 | 0.024 | 51 | 16 | 0.84 | 0.026 | 50/50 | 14 | >100 | |
| SK-CO-1 | AZD-6244 | 0.025 | 83 | 543 | 0.69 | 0.025 | 55/45 | 169 | 13.6 |
| BMS-754807 | 0.034 | 68 | 200 | 0.5 | 0.048 | 44/56 | 32 | 29.3 | |
| NCI-H747 | AZD-6244 | 0.019 | 75 | 71 | 0.62 | 0.029 | 61/39 | 34 | 25.6 |
| BMS-754807 | 0.028 | 75 | 231 | 0.6 | 0.031 | 54/46 | 75 | 22.4 | |
| MDA-MB-231 | AZD-6244 | 0.021 | 46 | 210 | 0.68 | 0.023 | 36/64 | 91 | >100 |
| Dasatinib | 0.05 | 88 | 300 | 0.49 | 0.018 | 49/51 | 30 | 9.3 | |
| MDA-MB-468 | GSK690693 | 0.08 | 100 | 2940 | 0.42 | 0.06 | 37/63 | 67 | 15.6 |
| Lapatinib | 0.026 | 100 | 190 | 0.45 | 0.038 | 53/47 | 17 | 3.1 | |
| HCC-827 | Gefitinib | 0.027 | 89 | 10 | 1.73 | 0.056 | Aug-92 | 11 | >100 |
| Erlotinib | 0.029 | 90 | 14 | 1.87 | 0.062 | Jul-93 | 16 | >100 | |
| Dasatinib | 0.049 | 93 | 149 | 1.88 | 0.075 | Feb-98 | 157 | >100 | |
| CTV-1 | Bosutinib | 0.033 | 96 | 51 | 2.71 | 0.1 | 100/0 | 56 | None |
| Dasatinib | 0.043 | 97 | 9.1 | 2.72 | 0.11 | 100/0 | 9.1 | None | |
| WH-4-023 | 0.043 | 98 | 712 | 2.49 | 0.096 | 100/0 | 716 | None | |
| Drug | Target | Hill Analysis | Biphasic Analysis | ||||||
|---|---|---|---|---|---|---|---|---|---|
| RMSE | Imax (%) | IC50 (nM) | n | RMSE | F1/F2 | Kd1 (nM) | Kd2 (μM) | ||
| AZ628 | BRAF | 0.011 | 91 | 27 | 1.81 | 0.055 | 95/5 | 26 | 3469 |
| PLX4720 | BRAF | 0.035 | 90 | 378 | 1.41 | 0.052 | 98/2 | 472 | 32448 |
| SB590885 | BRAF | 0.081 | 75 | 353 | 1.47 | 0.088 | 86/14 | 524 | 60 |
| RDEA119 | Mek | 0.030 | 86 | 37 | 1.62 | 0.055 | 90/10 | 37 | 75644 |
| CI-1040 | Mek | 0.024 | 88 | 116 | 1.63 | 0.054 | 93/7 | 131 | 50525 |
| PD-0325901 | Mek | 0.044 | 81 | 2.4 | 2.30 | 0.094 | 87/13 | 2.6 | 2169 |
| VX-11e | Erk | 0.029 | 98 | 80 | 1.36 | 0.049 | 100/0 | 80 | None |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, J.; Li, L.; Howlett, N.G.; Cohen, P.S.; Sun, G. Application of a Biphasic Mathematical Model of Cancer Cell Drug Response for Formulating Potent and Synergistic Targeted Drug Combinations to Triple Negative Breast Cancer Cells. Cancers 2020, 12, 1087. https://doi.org/10.3390/cancers12051087
Shen J, Li L, Howlett NG, Cohen PS, Sun G. Application of a Biphasic Mathematical Model of Cancer Cell Drug Response for Formulating Potent and Synergistic Targeted Drug Combinations to Triple Negative Breast Cancer Cells. Cancers. 2020; 12(5):1087. https://doi.org/10.3390/cancers12051087
Chicago/Turabian StyleShen, Jinyan, Li Li, Niall G. Howlett, Paul S. Cohen, and Gongqin Sun. 2020. "Application of a Biphasic Mathematical Model of Cancer Cell Drug Response for Formulating Potent and Synergistic Targeted Drug Combinations to Triple Negative Breast Cancer Cells" Cancers 12, no. 5: 1087. https://doi.org/10.3390/cancers12051087
APA StyleShen, J., Li, L., Howlett, N. G., Cohen, P. S., & Sun, G. (2020). Application of a Biphasic Mathematical Model of Cancer Cell Drug Response for Formulating Potent and Synergistic Targeted Drug Combinations to Triple Negative Breast Cancer Cells. Cancers, 12(5), 1087. https://doi.org/10.3390/cancers12051087