Iron Causes Lipid Oxidation and Inhibits Proteasome Function in Multiple Myeloma Cells: A Proof of Concept for Novel Combination Therapies

 ,
,  ,
,
Abstract

1. Introduction
2. Results
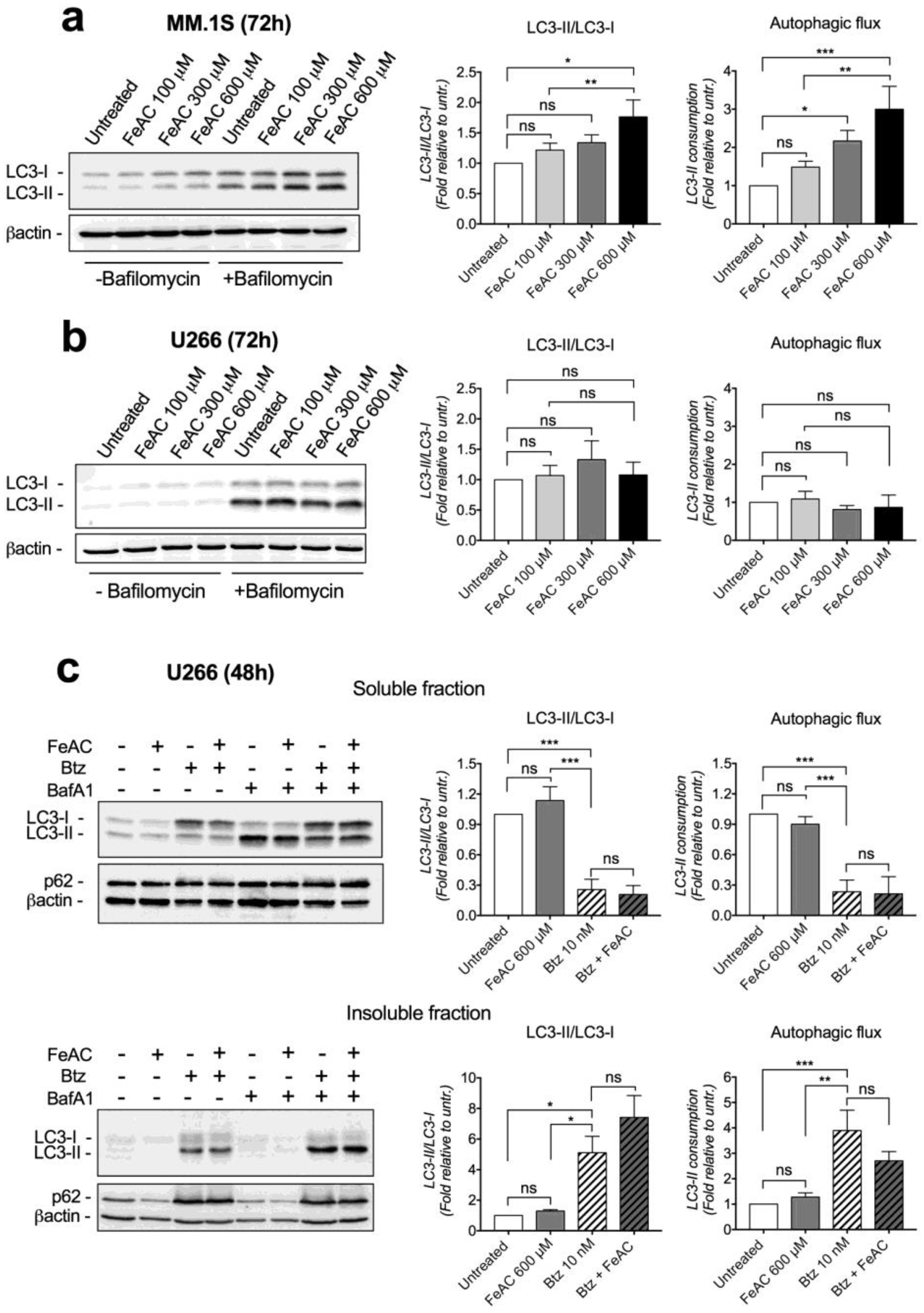
2.1. Iron Causes Lipid Damage and Impairs Proteasome Functionality in MM Cell Lines
2.2. Iron Increases Carfilzomib Efficacy in MM Cells
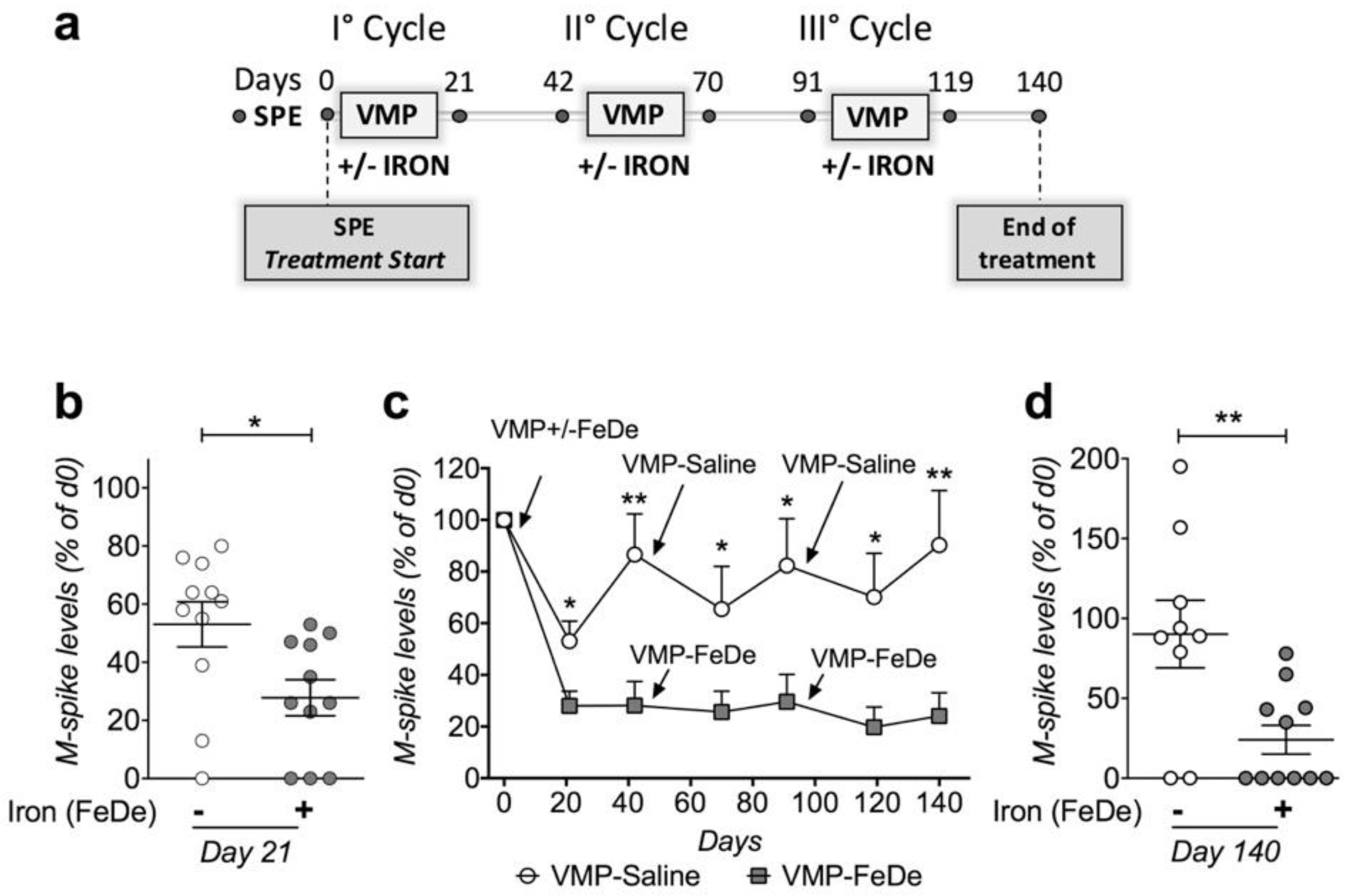
2.3. Iron Increases the Efficacy of Bortezomib-Melphalan-Prednisone Regimen in Vk*MYC Mice
3. Discussion
3.1. Molecular Mechanisms of Iron Toxicity in MM Cells
3.2. Iron Is a Potential Candidate for MM Combination Therapy
4. Materials and Methods
4.1. Cell Culture
4.2. Lipid Peroxidation
4.3. 26S peptidase Activity Measurements
4.4. Mouse Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gu, Z.; Wang, H.; Xia, J.; Yang, Y.; Jin, Z.; Xu, H.; Shi, J.; De Domenico, I.; Tricot, G.; Zhan, F. Decreased ferroportin promotes myeloma cell growth and osteoclast differentiation. Cancer Res. 2015, 75, 2211–2221. [Google Scholar] [CrossRef] [PubMed]
- Tesfay, L.; Clausen, K.A.; Kim, J.W.; Hegde, P.; Wang, X.; Miller, L.D.; Deng, Z.; Blanchette, N.; Arvedson, T.; Miranti, C.K.; et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015, 75, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Whitnall, M.; Howard, J.; Ponka, P.; Richardson, D.R. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc. Natl. Acad. Sci. USA 2006, 103, 14901–14906. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, Z.; Kalinowski, D.S.; Lovejoy, D.B.; Yu, Y.; Suryo Rahmanto, Y.; Sharpe, P.C.; Bernhardt, P.V.; Richardson, D.R. The Medicinal Chemistry of Novel Iron Chelators for the Treatment of Cancer. Curr. Top. Med. Chem. 2011, 11, 483–499. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to Tango: Regulation of Mammalian Iron Metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Breuer, W.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron. Int. J. Biochem. Cell Biol. 2008, 40, 350–354. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; Dimopoulos, M.A.; Palumbo, A.; Blade, J.; Merlini, G.; Mateos, M.V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Bordini, J.; Galvan, S.; Ponzoni, M.; Bertilaccio, M.T.S.; Chesi, M.; Bergsagel, P.L.; Camaschella, C.; Campanella, A. Induction of iron excess restricts malignant plasma cells expansion and potentiates bortezomib effect in models of multiple myeloma. Leukemia 2017, 31, 967–970. [Google Scholar] [CrossRef]
- Campanella, A.; Santambrogio, P.; Fontana, F.; Frenquelli, M.; Cenci, S.; Marcatti, M.; Sitia, R.; Tonon, G.; Camaschella, C. Iron increases the susceptibility of multiple myeloma cells to bortezomib. Haematologica 2013, 98, 971–979. [Google Scholar] [CrossRef]
- Benbrook, D.M.; Long, A. Integration of autophagy, proteasomal degradation, unfolded protein response and apoptosis. Exp. Oncol. 2012, 34, 286–297. [Google Scholar]
- Suh, D.H.; Kim, M.-K.; Kim, H.S.; Chung, H.H.; Song, Y.S. Unfolded protein response to autophagy as a promising druggable target for anticancer therapy. Ann. N. Y. Acad. Sci. 2012, 1271, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin–proteasome system. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Meister, S.; Schubert, U.; Neubert, K.; Herrmann, K.; Burger, R.; Gramatzki, M.; Hahn, S.; Schreiber, S.; Wilhelm, S.; Herrmann, M.; et al. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007, 67, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Oliva, L.; Cascio, P.; Pengo, N.; Fontana, F.; Cerruti, F.; Orsi, A.; Pasqualetto, E.; Mezghrani, A.; Calbi, V.; et al. The proteasome load versus capacity balance determines apoptotic sensitivity of multiple myeloma cells to proteasome inhibition. Blood 2009, 113, 3040–3049. [Google Scholar] [CrossRef] [PubMed]
- Pengo, N.; Scolari, M.; Oliva, L.; Milan, E.; Mainoldi, F.; Raimondi, A.; Fagioli, C.; Merlini, A.; Mariani, E.; Pasqualetto, E.; et al. Plasma cells require autophagy for sustainable immunoglobulin production. Nat. Immunol. 2013, 14, 298–305. [Google Scholar] [CrossRef]
- Kawaguchi, T.; Miyazawa, K.; Moriya, S.; Ohtomo, T.; Che, X.F.; Naito, M.; Itoh, M.; Tomoda, A. Combined treatment with bortezomib plus bafilomycin A1enhances the cytocidal effect and induces endoplasmic reticulum stress in U266 myeloma cells: Crosstalk among proteasome, autophagy-lysosome and ER stress. Int. J. Oncol. 2011, 38, 643–654. [Google Scholar]
- Chesi, M.; Matthews, G.M.; Garbitt, V.M.; Palmer, S.E.; Shortt, J.; Lefebure, M.; Stewart, A.K.; Johnstone, R.W.; Bergsagel, P.L. Drug response in a genetically engineered mouse model of multiple myeloma is predictive of clinical efficacy. Blood 2012, 120, 376–385. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Cenci, S.; Sitia, R. Managing and exploiting stress in the antibody factory. FEBS Lett. 2007, 581, 3652–3657. [Google Scholar] [CrossRef]
- Shimizu, Y.; Hendershot, L.M. Oxidative Folding: Cellular Strategies for Dealing with the Resultant Equimolar Production of Reactive Oxygen Species. Antioxid. Redox Signal. 2009, 11, 2317–2331. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [PubMed]
- Bordini, J.; Bertilaccio, M.T.S.; Ponzoni, M.; Fermo, I.; Chesi, M.; Bergsagel, P.L.; Camaschella, C.; Campanella, A. Erythroblast apoptosis and microenvironmental iron restriction trigger anemia in the VK*MYC model of multiple myeloma. Haematologica 2015, 100, 534–841. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Levi, S. Cytosolic and mitochondrial ferritins in the regulation of cellular iron homeostasis and oxidative damage. Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 783–792. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Sriramaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwel, B.R. Synthetic Lethal Screening Identifies Compounds Activating IronDependent, Nonapoptotic Cell Death in Oncogenic-RASHarboring Cancer Cells. Cell 2008, 15, 234–245. [Google Scholar]
- Bridges, R.J.; Natale, N.R.; Patel, S.A. System xc-cystine/glutamate antiporter: An update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 2012, 165, 20–34. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated necrosis: Disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef]
- Wu, D.; Chen, L. Ferroptosis: A novel cell death form will be a promising therapy target for diseases. Acta Biochim. Biophys. Sin. 2015, 47, 857–859. [Google Scholar] [CrossRef]
- Yu, Y.; Xie, Y.; Cao, L.; Yang, L.; Yang, M.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2015, 2, 1–7. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef]
- Rückrich, T.; Kraus, M.; Gogel, J.; Beck, A.; Ovaa, H.; Verdoes, M.; Overkleeft, H.S.; Kalbacher, H.; Driesse, C. Characterization of the ubiquitin—proteasome system in bortezomib-adapted cells. Leukemia 2009, 23, 1098–1105. [Google Scholar] [CrossRef]
- Kumar, S.K.; Berdeja, J.G.; Niesvizky, R.; Lonial, S.; Laubach, J.P.; Hamadani, M.; Stewart, A.K.; Hari, P.; Roy, V.; Vescio, R.; et al. Safety and tolerability of ixazomib, an oral proteasome inhibitor, in combination with lenalidomide and dexamethasone in patients with previously untreated multiple myeloma: An open-label phase 1/2 study. Lancet Oncol. 2014, 15, 1503–1512. [Google Scholar] [CrossRef]
- Camaschella, C. Iron-Deficiency Anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, T.; Tschechne, B.; Harlin, O.; Klement, B.; Franzem, M.; Wamhoff, J.; Tesch, H.; Rohrberg, R.; Marschner, N. Clinical experience with ferric carboxymaltose in the treatment of cancer- and chemotherapy-associated anaemia. Ann. Oncol. 2013, 24, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Favrat, B.; Balck, K.; Breymann, C.; Hedenus, M.; Keller, T.; Mezzacasa, A.; Gasche, C. Evaluation of a single dose of ferric carboxymaltose in fatigued, iron-deficient women—PREFER a randomized, placebo-controlled study. PLoS ONE 2014, 9, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Toledano, A.; Luporsi, E.; Morere, J.F.; Scotté, F.; Laribi, K.; Barrière, J.; Huot-Marchand, P.; Duvillié, L.; Concas, V.H.; Bugat, R. Clinical use of ferric carboxymaltose in patients with solid tumours or haematological malignancies in France. Support. Care Cancer 2016, 24, 67–75. [Google Scholar] [CrossRef]
- Raule, M.; Cerruti, F.; Benaroudj, N.; Migotti, R.; Kikuchi, J.; Bachi, A.; Navon, A.; Dittmar, G.; Cascio, P. PA28αβ Reduces Size and Increases Hydrophilicity of 20S Immunoproteasome Peptide Products. Chem. Biol. 2014, 21, 470–480. [Google Scholar] [CrossRef]
- Cascio, P.; Goldberg, A.L. Preparation of Hybrid (19S-20S-PA28) Proteasome Complexes and Analysis of Peptides Generated during Protein Degradation. Methods Enzymol. 2005, 398, 336–352. [Google Scholar]
- Cerruti, F.; Martano, M.; Petterino, C.; Bollo, E.; Morello, E.; Bruno, R.; Buracco, P.; Cascio, P. Enhanced Expression of Interferon-γ-Induced Antigen-Processing Machinery Components in a Spontaneously Occurring Cancer. Neoplasia 2007, 9, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Raule, M.; Cerruti, F.; Cascio, P. Enhanced rate of degradation of basic proteins by 26S immunoproteasomes. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 1942–1947. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peripheral Blood Markers | Treatment Start | After Two Cycles | End of Treatments | References Values | |||
|---|---|---|---|---|---|---|---|
| VMP−Saline | VMP−FeDe | VMP−Saline | VMP−FeDe | VMP−Saline | VMP−FeDe | ||
| Albumin (g/dl) | 3.1 ± 0.35 | 3.1 ± 0.46 | 3.1 ± 0.26 | 3.3 ± 0.26 | 3.0 ± 0.42 | 2.7 ± 0.36 | 2.7–3.6 |
| Cholinesterase (U/L) | 4490 ± 1001 | 5343 ± 1623 | 5449 ± 305 | 5956 ± 1495 | 4939 ± 297 | 4998 ± 1376 | 1400–6300 |
| Alanine aminotransferase (U/L) | 51 ± 14 | 41 ± 13 | 45 ± 11 | 46 ± 14 | 31 ± 6 | 55 ± 9 | <70 |
| Creatinine (mg/dL) | − | − | − | − | 0.31 ± 0.03 | 0.29 ± 0.02 | 0.31–0.40 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bordini, J.; Morisi, F.; Cerruti, F.; Cascio, P.; Camaschella, C.; Ghia, P.; Campanella, A. Iron Causes Lipid Oxidation and Inhibits Proteasome Function in Multiple Myeloma Cells: A Proof of Concept for Novel Combination Therapies. Cancers 2020, 12, 970. https://doi.org/10.3390/cancers12040970
Bordini J, Morisi F, Cerruti F, Cascio P, Camaschella C, Ghia P, Campanella A. Iron Causes Lipid Oxidation and Inhibits Proteasome Function in Multiple Myeloma Cells: A Proof of Concept for Novel Combination Therapies. Cancers. 2020; 12(4):970. https://doi.org/10.3390/cancers12040970
Chicago/Turabian StyleBordini, Jessica, Federica Morisi, Fulvia Cerruti, Paolo Cascio, Clara Camaschella, Paolo Ghia, and Alessandro Campanella. 2020. "Iron Causes Lipid Oxidation and Inhibits Proteasome Function in Multiple Myeloma Cells: A Proof of Concept for Novel Combination Therapies" Cancers 12, no. 4: 970. https://doi.org/10.3390/cancers12040970
APA StyleBordini, J., Morisi, F., Cerruti, F., Cascio, P., Camaschella, C., Ghia, P., & Campanella, A. (2020). Iron Causes Lipid Oxidation and Inhibits Proteasome Function in Multiple Myeloma Cells: A Proof of Concept for Novel Combination Therapies. Cancers, 12(4), 970. https://doi.org/10.3390/cancers12040970