The Role of Circulating Tumor Cells in the Metastatic Cascade: Biology, Technical Challenges, and Clinical Relevance

 ,
, 
Abstract
1. Introduction
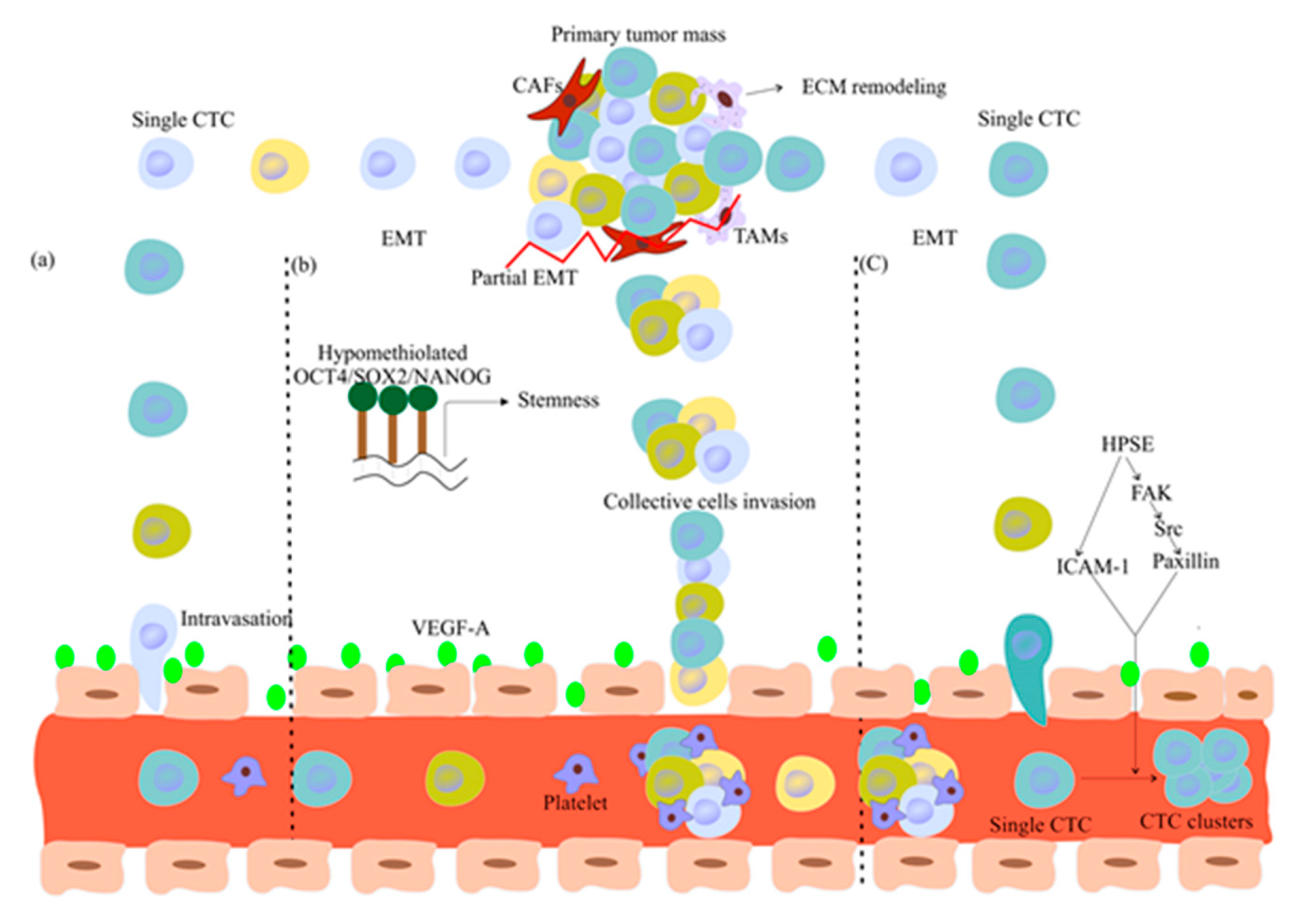
2. CTC Biology and Tumor Metastases
2.1. Do CTCs Have Cancer Stem Cell Features?
2.2. Are Single CTCs or CTC Clusters Involved in Metastasis Formation?
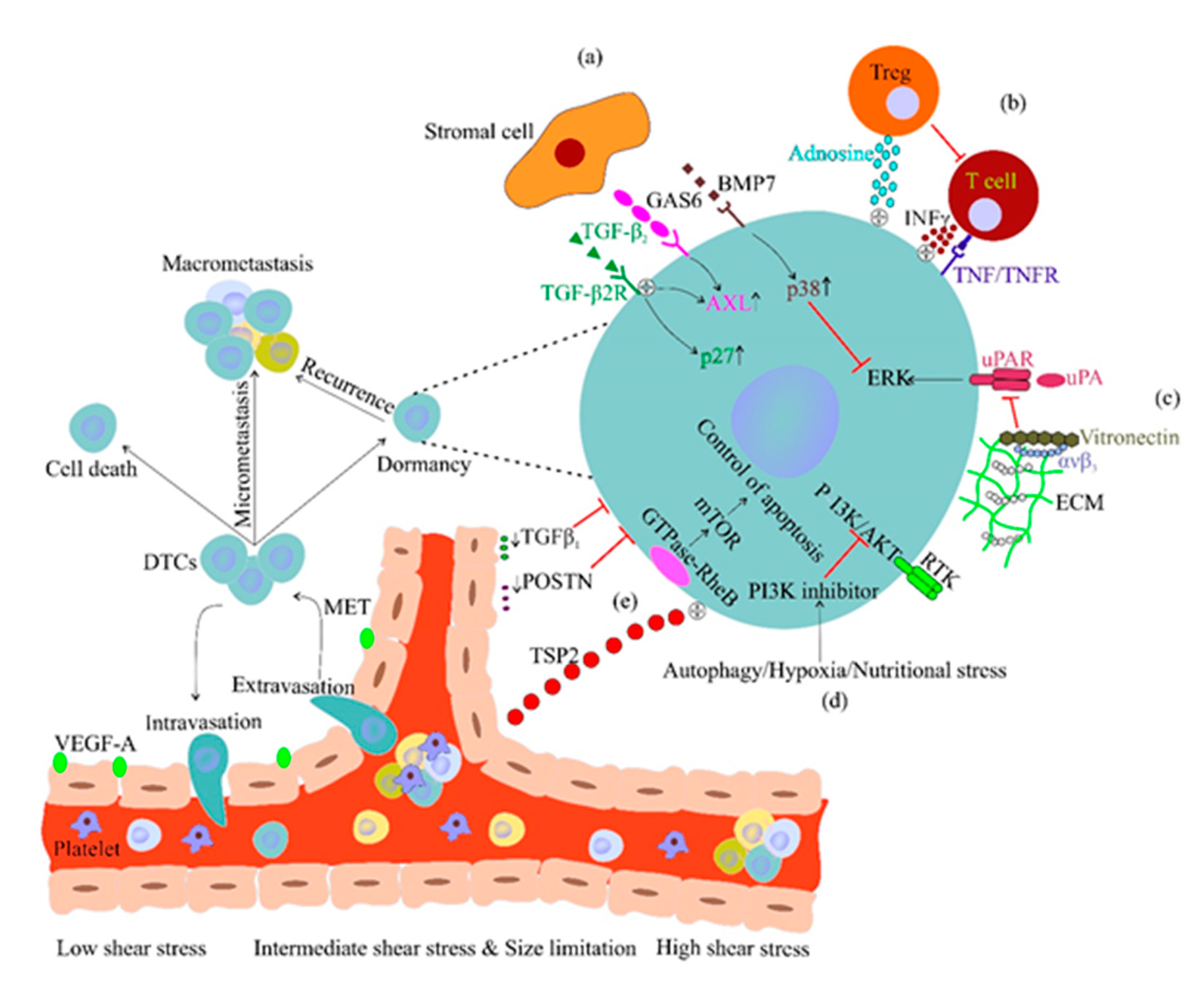
2.3. CTC Dissemination and Dormancy
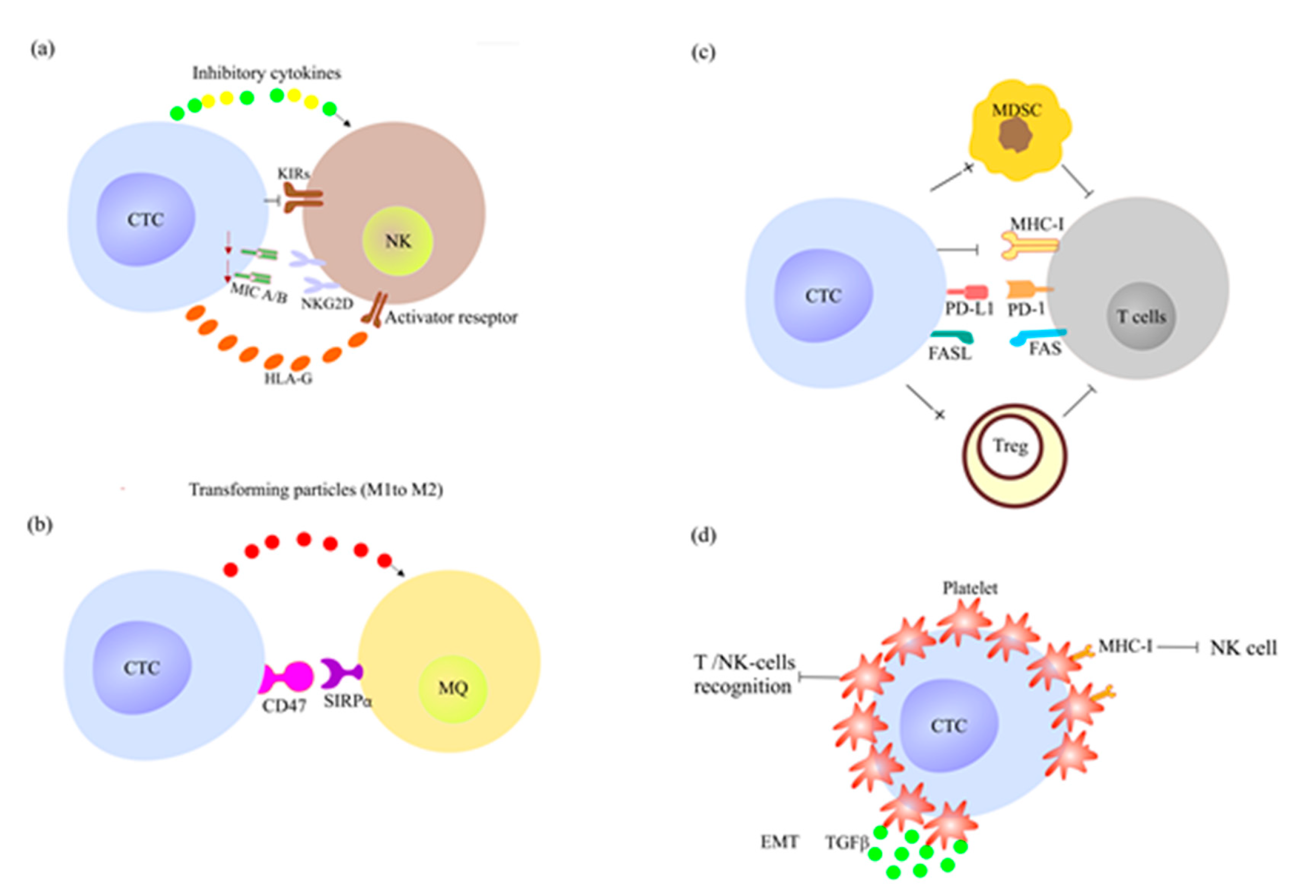
2.4. How CTCs Escape the Immune System Surveillance and Therapy?
2.4.1. CTCs Escape the Innate Immune System Response
2.4.2. CTCs Escape the Adoptive Immune System Response
2.4.3. CTCs Induce Resistance to Chemotherapy
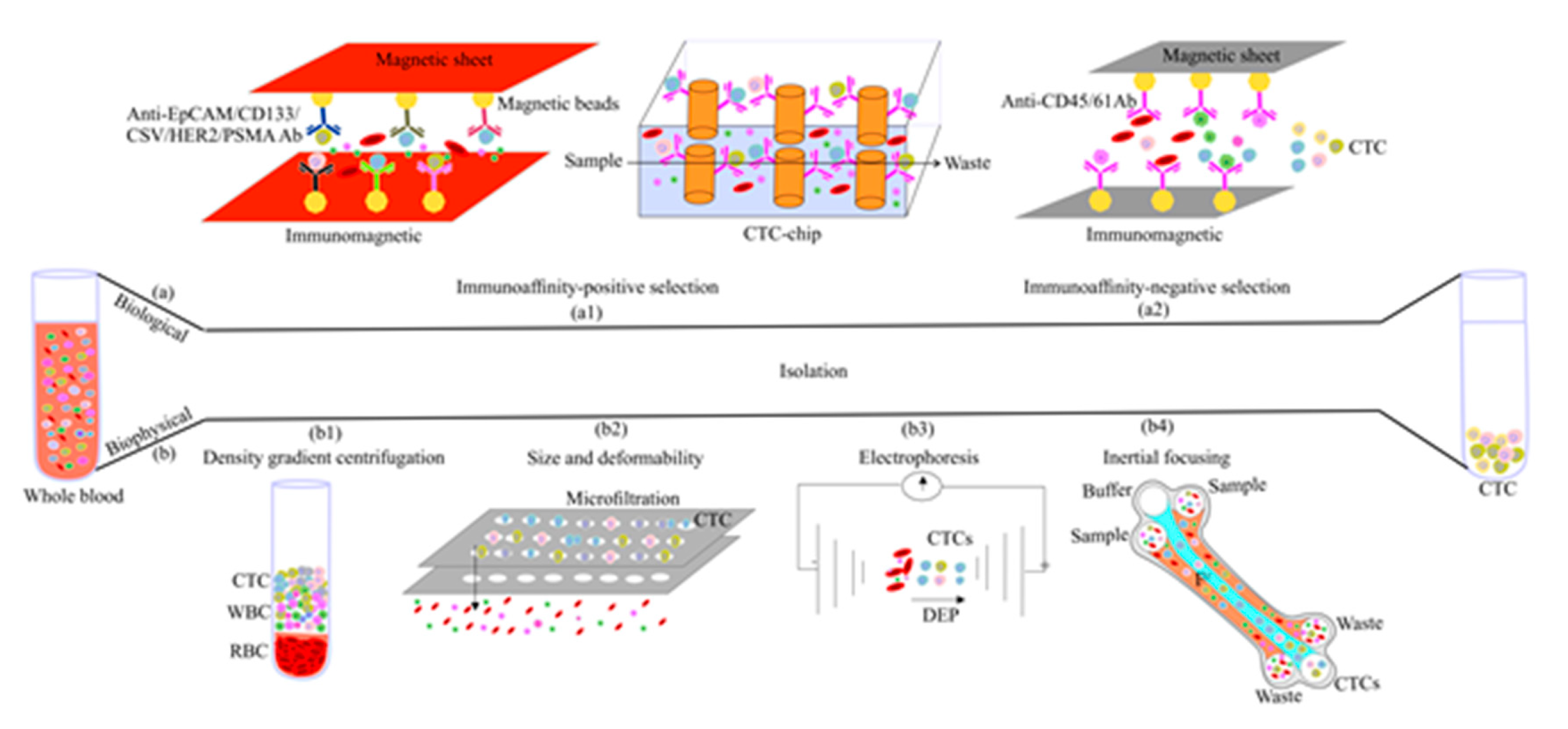
3. CTC Enrichment Strategies
3.1. Biological Feature-Based CTC Enrichment
3.1.1. Positive Enrichment
3.1.2. Negative Enrichment
3.2. Physical Feature-Based CTC Enrichment
3.2.1. Density-Based Enrichment
3.2.2. Size-Based Enrichment
3.2.3. Dielectrophoresis
3.2.4. Inertial Focusing
3.3. Combined Methods
3.4. Challenge with DTC Isolation
4. CTC Clinical Relevance
4.1. How CTCs Improve Metastasis Prediction and Diagnosis?
4.2. Can CTC Profiling Guide the Therapeutic Strategy?
4.2.1. CTC Proteomic Analysis
4.2.2. CTC Genomic Analysis
4.3. Which One Is Better, CTC or ctDNA?
4.4. CTC Targeting for Metastasis Therapy
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yu, M.; Bardia, A.; Wittner, B.S.; Stott, S.L.; Smas, M.E.; Ting, D.T.; Isakoff, S.J.; Ciciliano, J.C.; Wells, M.N.; Shah, A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 2013, 339, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Micalizzi, D.S.; Maheswaran, S.; Haber, D.A. A conduit to metastasis: Circulating tumor cell biology. Genes Dev. 2017, 31, 1827–1840. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.; Cao, T.; Nagrath, S.; King, M.R. Circulating Tumor Cells: Diagnostic and Therapeutic Applications. Annu. Rev. Biomed. Eng. 2018, 20, 329–352. [Google Scholar] [CrossRef] [PubMed]
- Ashworth, T. A case of cancer in which cells similar to those in the tumours were seen in the blood after death. Aust. Med. J. 1869, 14, 146. [Google Scholar]
- Pantel, K.; Speicher, M. The biology of circulating tumor cells. Oncogene 2016, 35, 1216. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—Latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef]
- Dianat-Moghadam, H.; Heidarifard, M.; Jahanban-Esfahlan, R.; Panahi, Y.; Hamishehkar, H.; Pouremamali, F.; Rahbarghazi, R.; Nouri, M. Cancer stem cells-emanated therapy resistance: Implications for liposomal drug delivery systems. J. Control. Release 2018, 288, 62–83. [Google Scholar] [CrossRef]
- Kasimir-Bauer, S.; Hoffmann, O.; Wallwiener, D.; Kimmig, R.; Fehm, T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012, 14, R15. [Google Scholar] [CrossRef]
- Schölch, S.; García, S.A.; Iwata, N.; Niemietz, T.; Betzler, A.M.; Nanduri, L.K.; Bork, U.; Kahlert, C.; Thepkaysone, M.-L.; Swiersy, A. Circulating tumor cells exhibit stem cell characteristics in an orthotopic mouse model of colorectal cancer. Oncotarget 2016, 7, 27232. [Google Scholar] [CrossRef] [PubMed]
- Masuda, T.; Hayashi, N.; Iguchi, T.; Ito, S.; Eguchi, H.; Mimori, K. Clinical and biological significance of circulating tumor cells in cancer. Mol. Oncol. 2016, 10, 408–417. [Google Scholar] [CrossRef]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the roles of CD44/CD24 and ALDH1 as cancer stem cell markers in tumorigenesis and metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, Y.; Xu, J.; Zhang, A.; Wang, X.; Tang, R.; Zhang, X.; Yin, H.; Liu, M.; Wang, D.D.; et al. Quantified postsurgical small cell size CTCs and EpCAM+ circulating tumor stem cells with cytogenetic abnormalities in hepatocellular carcinoma patients determine cancer relapse. Cancer Lett. 2018, 412, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Cheng, J.; Feng, X.; He, S.; Wang, Y.; Huang, Q. The viable circulating tumor cells with cancer stem cells feature, where is the way out? J. Exp. Clin. Cancer Res. 2018, 37, 38. [Google Scholar] [CrossRef] [PubMed]
- Bonnomet, A.; Brysse, A.; Tachsidis, A.; Waltham, M.; Thompson, E.W.; Polette, M.; Gilles, C. Epithelial-to-mesenchymal transitions and circulating tumor cells. J. Mammary Gland Biol. Neoplasia 2010, 15, 261–273. [Google Scholar] [CrossRef]
- Adorno-Cruz, V.; Kibria, G.; Liu, X.; Doherty, M.; Junk, D.J.; Guan, D.; Hubert, C.; Venere, M.; Mulkearns-Hubert, E.; Sinyuk, M.; et al. Cancer stem cells: Targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015, 75, 924–929. [Google Scholar] [CrossRef]
- Lianidou, E.; Hoon, D. Circulating tumor cells and circulating tumor DNA. In Principles and Applications of Molecular Diagnostics; Elsevier: Amsterdam, The Netherlands, 2018; pp. 235–281. [Google Scholar]
- E Melo, F.D.S.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H. A distinct role for Lgr5+ stem cells in primary and metastatic colon cancer. Nature 2017, 543, 676. [Google Scholar] [CrossRef]
- Kowalik, A.; Kowalewska, M.; Góźdź, S. Current approaches for avoiding the limitations of circulating tumor cells detection methods—Implications for diagnosis and treatment of patients with solid tumors. Transl. Res. 2017, 185, 58–84.e15. [Google Scholar] [CrossRef]
- Gkountela, S.; Aceto, N. Stem-like features of cancer cells on their way to metastasis. Biol. Direct 2016, 11, 33. [Google Scholar] [CrossRef]
- Gkountela, S.; Castro-Giner, F.; Szczerba, B.M.; Vetter, M.; Landin, J.; Scherrer, R.; Krol, I.; Scheidmann, M.C.; Beisel, C.; Stirnimann, C.U.; et al. Circulating Tumor Cell Clustering Shapes DNA Methylation to Enable Metastasis Seeding. Cell 2019, 176, 98–112.e114. [Google Scholar] [CrossRef]
- Hou, J.-M.; Krebs, M.G.; Lancashire, L.; Sloane, R.; Backen, A.; Swain, R.K.; Priest, L.; Greystoke, A.; Zhou, C.; Morris, K. Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 2012, 30, 525–532. [Google Scholar] [CrossRef]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J. Cancer cell invasion and EMT marker expression: A three-dimensional study of the human cancer—Host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Mader, S.; Pantel, K. Epithelial-mesenchymal plasticity in circulating tumor cells. J. Mol. Med. 2017, 95, 133–142. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Teddy, J.M.; Kulesa, P.M. In vivo evidence for short-and long-range cell communication in cranial neural crest cells. Development 2004, 131, 6141–6151. [Google Scholar] [CrossRef]
- Chapnick, D.A.; Liu, X. Leader cell positioning drives wound-directed collective migration in TGFβ-stimulated epithelial sheets. Mol. Biol. Cell 2014, 25, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Twomey, J.D.; Zhang, B. Circulating Tumor Cells Develop Resistance to TRAIL-Induced Apoptosis Through Autophagic Removal of Death Receptor 5: Evidence from an In Vitro Model. Cancers 2019, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Li, S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell. Mol. Life Sci. 2011, 68, 3033–3046. [Google Scholar] [CrossRef] [PubMed]
- Lecharpentier, A.; Vielh, P.; Perez-Moreno, P.; Planchard, D.; Soria, J.C.; Farace, F. Detection of circulating tumour cells with a hybrid (epithelial/mesenchymal) phenotype in patients with metastatic non-small cell lung cancer. Br. J. Cancer 2011, 105, 1338–1341. [Google Scholar] [CrossRef] [PubMed]
- Satelli, A.; Mitra, A.; Brownlee, Z.; Xia, X.; Bellister, S.; Overman, M.J.; Kopetz, S.; Ellis, L.M.; Meng, Q.H.; Li, S. Epithelial–Mesenchymal Transitioned Circulating Tumor Cells Capture for Detecting Tumor Progression. Clin. Cancer Res. 2015, 21, 899. [Google Scholar] [CrossRef]
- Sarioglu, A.F.; Aceto, N.; Kojic, N.; Donaldson, M.C.; Zeinali, M.; Hamza, B.; Engstrom, A.; Zhu, H.; Sundaresan, T.K.; Miyamoto, D.T. A microfluidic device for label-free, physical capture of circulating tumor cell clusters. Nat. Methods 2015, 12, 685. [Google Scholar] [CrossRef]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Mahauad-Fernandez, W.D.; Naushad, W.; Panzner, T.D.; Bashir, A.; Lal, G.; Okeoma, C.M. BST-2 promotes survival in circulation and pulmonary metastatic seeding of breast cancer cells. Sci. Rep. 2018, 8, 17608. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.J.; Gabrielson, E.; Werb, Z.; Ewald, A.J. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell 2013, 155, 1639–1651. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.-R.; Sun, D.-N.; Yang, H.; Yan, J.; Zhang, X.; Zheng, X.-L.; Fu, X.-H.; Geng, M.-Y.; Huang, X.; Ding, J. CTC clusters induced by heparanase enhance breast cancer metastasis. Acta Pharmacol. Sin. 2018, 39, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Aceto, N.; Bardia, A.; Miyamoto, D.T.; Donaldson, M.C.; Wittner, B.S.; Spencer, J.A.; Yu, M.; Pely, A.; Engstrom, A.; Zhu, H. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 2014, 158, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Maddipati, R.; Stanger, B.Z. Pancreatic cancer metastases harbor evidence of polyclonality. Cancer Discov. 2015, 5, 1086–1097. [Google Scholar] [CrossRef]
- Umer, M.; Vaidyanathan, R.; Nguyen, N.-T.; Shiddiky, M.J. Circulating tumor microemboli: Progress in molecular understanding and enrichment technologies. Biotechnol. Adv. 2018, 36, 1367–1389. [Google Scholar] [CrossRef]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef]
- Cheung, K.J.; Ewald, A.J. A collective route to metastasis: Seeding by tumor cell clusters. Science 2016, 352, 167–169. [Google Scholar] [CrossRef]
- Wang, C.; Mu, Z.; Chervoneva, I.; Austin, L.; Ye, Z.; Rossi, G.; Palazzo, J.P.; Sun, C.; Abu-Khalaf, M.; Myers, R.E.; et al. Longitudinally collected CTCs and CTC-clusters and clinical outcomes of metastatic breast cancer. Breast Cancer Res. Treat. 2017, 161, 83–94. [Google Scholar] [CrossRef]
- Jansson, S.; Bendahl, P.-O.; Larsson, A.-M.; Aaltonen, K.E.; Rydén, L. Prognostic impact of circulating tumor cell apoptosis and clusters in serial blood samples from patients with metastatic breast cancer in a prospective observational cohort. BMC Cancer 2016, 16, 433. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-C.; Chang, Y.-T.; Chen, J.-Y.; Jeng, Y.-M.; Yang, C.-Y.; Tien, Y.-W.; Yang, S.-H.; Chen, H.-L.; Liang, T.-Y.; Wang, C.-F. Clinical significance of circulating tumor microemboli as a prognostic marker in patients with pancreatic ductal adenocarcinoma. Clin. Chem. 2016, 62, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.; Högnäs, G.; Annala, M. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353. [Google Scholar] [CrossRef] [PubMed]
- Achrol, A.S.; Rennert, R.C.; Anders, C.; Soffietti, R.; Ahluwalia, M.S.; Nayak, L.; Peters, S.; Arvold, N.D.; Harsh, G.R.; Steeg, P.S. Brain metastases. Nat. Rev. Dis. Primers 2019, 5, 5. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.-L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329. [Google Scholar] [CrossRef]
- Noh, Y.-W.; Kong, S.-H.; Choi, D.-Y.; Park, H.S.; Yang, H.-K.; Lee, H.-J.; Kim, H.C.; Kang, K.W.; Sung, M.-H.; Lim, Y.T. Near-infrared emitting polymer nanogels for efficient sentinel lymph node mapping. ACS Nano 2012, 6, 7820–7831. [Google Scholar] [CrossRef]
- Olmeda, D.; Cerezo-Wallis, D.; Riveiro-Falkenbach, E.; Pennacchi, P.C.; Contreras-Alcalde, M.; Ibarz, N.; Cifdaloz, M.; Catena, X.; Calvo, T.G.; Cañón, E. Whole-body imaging of lymphovascular niches identifies pre-metastatic roles of midkine. Nature 2017, 546, 676. [Google Scholar] [CrossRef]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Gao, H.; Chakraborty, G.; Lee-Lim, A.P.; Mo, Q.; Decker, M.; Vonica, A.; Shen, R.; Brogi, E.; Brivanlou, A.H.; Giancotti, F.G. The BMP inhibitor Coco reactivates breast cancer cells at lung metastatic sites. Cell 2012, 150, 764–779. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Valiente, M.; Obenauf, A.C.; Jin, X.; Chen, Q.; Zhang, X.H.-F.; Lee, D.J.; Chaft, J.E.; Kris, M.G.; Huse, J.T.; Brogi, E. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell 2014, 156, 1002–1016. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.W.; Amin, R.; Deasy, S.; Ha, N.-H.; Wakefield, L. Genetic insights into the morass of metastatic heterogeneity. Nat. Rev. Cancer 2018, 18, 211. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J. GAS6/AXL axis regulates prostate cancer invasion, proliferation, and survival in the bone marrow niche. Neoplasia 2010, 12, 116. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Okuda, H.; Xing, F.; Pandey, P.R.; Watabe, M.; Hirota, S.; Pai, S.K.; Liu, W.; Fukuda, K.; Chambers, C. Bone morphogenetic protein 7 in dormancy and metastasis of prostate cancer stem-like cells in bone. J. Exp. Med. 2011, 208, 2641–2655. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150high bone marrow Tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell 2018, 22, 445–453.e445. [Google Scholar] [CrossRef] [PubMed]
- Müller-Hermelink, N.; Braumüller, H.; Pichler, B.; Wieder, T.; Mailhammer, R.; Schaak, K.; Ghoreschi, K.; Yazdi, A.; Haubner, R.; Sander, C.A. TNFR1 signaling and IFN-γ signaling determine whether T cells induce tumor dormancy or promote multistage carcinogenesis. Cancer Cell 2008, 13, 507–518. [Google Scholar] [CrossRef]
- Putz, E.; Witter, K.; Offner, S.; Stosiek, P.; Zippelius, A.; Johnson, J.; Zahn, R.; Riethmüller, G.; Pantel, K. Phenotypic characteristics of cell lines derived from disseminated cancer cells in bone marrow of patients with solid epithelial tumors: Establishment of working models for human micrometastases. Cancer Res. 1999, 59, 241–248. [Google Scholar]
- Aguirre-Ghiso, J.A.; Liu, D.; Mignatti, A.; Kovalski, K.; Ossowski, L. Urokinase receptor and fibronectin regulate the ERKMAPK to p38MAPK activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol. Biol. Cell 2001, 12, 863–879. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Pan, T.-C.; Ruth, J.; Feng, Y.; Zhou, A.; Pant, D.; Grimley, J.S.; Wandless, T.J.; DeMichele, A.; Chodosh, L.A. Par-4 downregulation promotes breast cancer recurrence by preventing multinucleation following targeted therapy. Cancer Cell 2013, 24, 30–44. [Google Scholar] [CrossRef]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269. [Google Scholar] [CrossRef]
- Schewe, D.M.; Aguirre-Ghiso, J.A. ATF6α-Rheb-mTOR signaling promotes survival of dormant tumor cells in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 10519–10524. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Khong, H.T.; Restifo, N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002, 3, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.F.; Mannam, V.K.; Craft, B.S.; Puneky, L.V.; Sheehan, N.T.; Lewis, R.E.; Cruse, J.M. Comparative analysis of innate immune system function in metastatic breast, colorectal, and prostate cancer patients with circulating tumor cells. Exp. Mol. Pathol. 2014, 96, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Schlimok, G.; Kutter, D.; Schaller, G.; Genz, T.; Wiebecke, B.; Backmann, R.; Funke, I.; Riethmüller, G. Frequent down-regulation of major histocompatibility class I antigen expression on individual micrometastatic carcinoma cells. Cancer Res. 1991, 51, 4712–4715. [Google Scholar] [PubMed]
- Wang, B.; Wang, Q.; Wang, Z.; Jiang, J.; Yu, S.C.; Ping, Y.F.; Yang, J.; Xu, S.L.; Ye, X.Z.; Xu, C.; et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014, 74, 5746–5757. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Hamilton, T.K.; Li, X.; Cotechini, T.; Miles, E.A.; Siemens, D.R.; Graham, C.H. Hypoxia induces escape from innate immunity in cancer cells via increased expression of ADAM10: Role of nitric oxide. Cancer Res. 2011, 71, 7433–7441. [Google Scholar] [CrossRef]
- König, L.; Kasimir-Bauer, S.; Hoffmann, O.; Bittner, A.-K.; Wagner, B.; Manvailer, L.F.S.; Schramm, S.; Bankfalvi, A.; Giebel, B.; Kimmig, R. The prognostic impact of soluble and vesicular HLA-G and its relationship to circulating tumor cells in neoadjuvant treated breast cancer patients. Hum. Immunol. 2016, 77, 791–799. [Google Scholar] [CrossRef]
- Lin, A.; Yan, W.-H. Human leukocyte antigen-G (HLA-G) expression in cancers: Roles in immune evasion, metastasis and target for therapy. Mol. Med. 2015, 21, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef]
- Steinert, G.; Scholch, S.; Niemietz, T.; Iwata, N.; Garcia, S.A.; Behrens, B.; Voigt, A.; Kloor, M.; Benner, A.; Bork, U.; et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 2014, 74, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Huh, S.J.; Liang, S.; Sharma, A.; Dong, C.; Robertson, G.P. Transiently entrapped circulating tumor cells interact with neutrophils to facilitate lung metastasis development. Cancer Res. 2010, 70, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Hanley, W.D.; Napier, S.L.; Burdick, M.M.; Schnaar, R.L.; Sackstein, R.; Konstantopoulos, K. Variant isoforms of CD44 are P-and L-selectin ligands on colon carcinoma cells. FASEB J. 2006, 20, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef]
- Patel, S.P.; Kurzrock, R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef]
- Mazel, M.; Jacot, W.; Pantel, K.; Bartkowiak, K.; Topart, D.; Cayrefourcq, L.; Rossille, D.; Maudelonde, T.; Fest, T.; Alix-Panabières, C. Frequent expression of PD-L1 on circulating breast cancer cells. Mol. Oncol. 2015, 9, 1773–1782. [Google Scholar] [CrossRef]
- Ock, C.-Y.; Kim, S.; Keam, B.; Kim, M.; Kim, T.M.; Kim, J.-H.; Jeon, Y.K.; Lee, J.-S.; Kwon, S.K.; Hah, J.H. PD-L1 expression is associated with epithelial-mesenchymal transition in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 15901. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Shin, J.H.; Longmire, M.; Wang, H.; Kohrt, H.E.; Chang, H.Y.; Sunwoo, J.B. CD44+ Cells in Head and Neck Squamous Cell Carcinoma Suppress T-Cell-Mediated Immunity by Selective Constitutive and Inducible Expression of PD-L1. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3571–3581. [Google Scholar] [CrossRef]
- Zheng, H.X.; Cai, Y.D.; Wang, Y.D.; Cui, X.B.; Xie, T.T.; Li, W.J.; Peng, L.; Zhang, Y.; Wang, Z.Q.; Wang, J.; et al. Fas signaling promotes motility and metastasis through epithelial–mesenchymal transition in gastrointestinal cancer. Oncogene 2013, 32, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Gruber, I.; El Yousfi, S.; Dürr-Störzer, S.; Wallwiener, D.; Solomayer, E.; Fehm, T. Down-regulation of CD28, TCR-zeta (ζ) and up-regulation of FAS in peripheral cytotoxic T-cells of primary breast cancer patients. Anticancer Res. 2008, 28, 779–784. [Google Scholar] [PubMed]
- Gruber, I.; Landenberger, N.; Staebler, A.; Hahn, M.; Wallwiener, D.; Fehm, T. Relationship between circulating tumor cells and peripheral T-cells in patients with primary breast cancer. Anticancer Res. 2013, 33, 2233–2238. [Google Scholar]
- Jiang, H.; Gebhardt, C.; Umansky, L.; Beckhove, P.; Schulze, T.J.; Utikal, J.; Umansky, V. Elevated chronic inflammatory factors and myeloid-derived suppressor cells indicate poor prognosis in advanced melanoma patients. Int. J. Cancer 2015, 136, 2352–2360. [Google Scholar] [CrossRef]
- Smith, H.A.; Kang, Y. The metastasis-promoting roles of tumor-associated immune cells. J. Mol. Med. 2013, 91, 411–429. [Google Scholar] [CrossRef]
- Tseng, J.-Y.; Yang, C.-Y.; Liang, S.-C.; Liu, R.-S.; Yang, S.-H.; Lin, J.-K.; Chen, Y.-M.; Wu, Y.-C.; Jiang, J.-K.; Lin, C.-H. Interleukin-17A modulates circulating tumor cells in tumor draining vein of colorectal cancers and affects metastases. Clin. Cancer Res. 2014, 20, 2885–2897. [Google Scholar] [CrossRef]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Männel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.-G.; Kopp, H.-G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [PubMed]
- Klameth, L.; Rath, B.; Hochmaier, M.; Moser, D.; Redl, M.; Mungenast, F.; Gelles, K.; Ulsperger, E.; Zeillinger, R.; Hamilton, G. Small cell lung cancer: Model of circulating tumor cell tumorospheres in chemoresistance. Sci. Rep. 2017, 7, 5337. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, C.; Muniz, M.C.; Thomas, D.G.; Griffith, K.A.; Kidwell, K.M.; Tokudome, N.; Brown, M.E.; Aung, K.; Miller, M.C.; Blossom, D.L. Development of circulating tumor cell-endocrine therapy index in patients with hormone receptor—Positive breast cancer. Clin. Cancer Res. 2015, 21, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351. [Google Scholar] [CrossRef]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697. [Google Scholar] [CrossRef]
- Mego, M.; Mani, S.A.; Lee, B.N.; Li, C.; Evans, K.W.; Cohen, E.N.; Gao, H.; Jackson, S.A.; Giordano, A.; Hortobagyi, G.N. Expression of epithelial–mesenchymal transition-inducing transcription factors in primary breast cancer: The effect of neoadjuvant therapy. Int. J. Cancer 2012, 130, 808–816. [Google Scholar] [CrossRef]
- Yokobori, T.; Iinuma, H.; Shimamura, T.; Imoto, S.; Sugimachi, K.; Ishii, H.; Iwatsuki, M.; Ota, D.; Ohkuma, M.; Iwaya, T. Plastin3 is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res. 2013, 73, 2059–2069. [Google Scholar] [CrossRef]
- Satelli, A.; Brownlee, Z.; Mitra, A.; Meng, Q.H.; Li, S. Circulating Tumor Cell Enumeration with a Combination of Epithelial Cell Adhesion Molecule–and Cell-Surface Vimentin–Based Methods for Monitoring Breast Cancer Therapeutic Response. Clin. Chem. 2015, 61, 259–266. [Google Scholar] [CrossRef]
- Rana, K.; Liesveld, J.L.; King, M.R. Delivery of apoptotic signal to rolling cancer cells: A novel biomimetic technique using immobilized TRAIL and E-selectin. Biotechnol. Bioeng. 2009, 102, 1692–1702. [Google Scholar] [CrossRef]
- Allard, W.J.; Matera, J.; Miller, M.C.; Repollet, M.; Connelly, M.C.; Rao, C.; Tibbe, A.G.; Uhr, J.W.; Terstappen, L.W. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2004, 10, 6897–6904. [Google Scholar] [CrossRef]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235. [Google Scholar] [CrossRef] [PubMed]
- Praharaj, P.P.; Bhutia, S.K.; Nagrath, S.; Bitting, R.L.; Deep, G. Circulating tumor cell-derived organoids: Current challenges and promises in medical research and precision medicine. Biochim. Biophys. Acta (BBA) Rev. Cancer 2018, 1869, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.M.; Ramani, V.C.; Jeffrey, S.S. Circulating tumor cell technologies. Mol. Oncol. 2016, 10, 374–394. [Google Scholar] [CrossRef] [PubMed]
- Earhart, C.M.; Hughes, C.E.; Gaster, R.S.; Ooi, C.C.; Wilson, R.J.; Zhou, L.Y.; Humke, E.W.; Xu, L.; Wong, D.J.; Willingham, S.B. Isolation and mutational analysis of circulating tumor cells from lung cancer patients with magnetic sifters and biochips. Lab Chip 2014, 14, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Rostami, P.; Kashaninejad, N.; Moshksayan, K.; Saidi, M.S.; Firoozabadi, B.; Nguyen, N.-T. Novel approaches in cancer management with circulating tumor cell clusters. J. Sci. Adv. Mater. Devices 2019, 4, 1–18. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Challenges in circulating tumour cell research. Nat. Rev. Cancer 2014, 14, 623. [Google Scholar] [CrossRef]
- Mitra, A.; Satelli, A.; Xia, X.; Cutrera, J.; Mishra, L.; Li, S. Cell-surface Vimentin: A mislocalized protein for isolating csVimentin+CD133− novel stem-like hepatocellular carcinoma cells expressing EMT markers. Int. J. Cancer 2015, 137, 491–496. [Google Scholar] [CrossRef]
- Noh, H.; Yan, J.; Hong, S.; Kong, L.-Y.; Gabrusiewicz, K.; Xia, X.; Heimberger, A.B.; Li, S. Discovery of cell surface vimentin targeting mAb for direct disruption of GBM tumor initiating cells. Oncotarget 2016, 7, 72021. [Google Scholar] [CrossRef]
- Witek, M.A.; Aufforth, R.D.; Wang, H.; Kamande, J.W.; Jackson, J.M.; Pullagurla, S.R.; Hupert, M.L.; Usary, J.; Wysham, W.Z.; Hilliard, D.; et al. Discrete microfluidics for the isolation of circulating tumor cell subpopulations targeting fibroblast activation protein alpha and epithelial cell adhesion molecule. NPJ Precis. Oncol. 2017, 1, 24. [Google Scholar] [CrossRef]
- Skirecki, T.; Hoser, G.; Kawiak, J.; Dziedzic, D.; Domagała-Kulawik, J. Flow Cytometric Analysis of CD133- and EpCAM-Positive Cells in the Peripheral Blood of Patients with Lung Cancer. Arch. Immunol. Ther. Exp. 2014, 62, 67–75. [Google Scholar] [CrossRef]
- Varillas, J.I.; Zhang, J.; Chen, K.; Barnes, I.I.; Liu, C.; George, T.J.; Fan, Z.H. Microfluidic isolation of circulating tumor cells and cancer stem-like cells from patients with pancreatic ductal adenocarcinoma. Theranostics 2019, 9, 1417. [Google Scholar] [CrossRef] [PubMed]
- Danila, D.C.; Samoila, A.; Patel, C.; Schreiber, N.; Herkal, A.; Anand, A.; Bastos, D.; Heller, G.; Fleisher, M.; Scher, H.I. Clinical validity of detecting circulating tumor cells by AdnaTest assay compared to direct detection of tumor mRNA in stabilized whole blood, as a biomarker predicting overall survival for metastatic castration-resistant prostate cancer patients. Cancer J. (Sudbury, Mass.) 2016, 22, 315. [Google Scholar] [CrossRef] [PubMed]
- Talasaz, A.H.; Powell, A.A.; Huber, D.E.; Berbee, J.G.; Roh, K.-H.; Yu, W.; Xiao, W.; Davis, M.M.; Pease, R.F.; Mindrinos, M.N. Isolating highly enriched populations of circulating epithelial cells and other rare cells from blood using a magnetic sweeper device. Proc. Natl. Acad. Sci. USA 2009, 106, 3970–3975. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.C.; Niederacher, D.; Topp, S.A.; Honisch, E.; Schumacher, S.; Schmitz, N.; Föhrding, L.Z.; Vay, C.; Hoffmann, I.; Kasprowicz, N.S. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc. Natl. Acad. Sci. USA 2013, 110, 16580–16585. [Google Scholar] [CrossRef]
- Saucedo-Zeni, N.; Mewes, S.; Niestroj, R.; Gasiorowski, L.; Murawa, D.; Nowaczyk, P.; Tomasi, T.; Weber, E.; Dworacki, G.; Morgenthaler, N.G. A novel method for the in vivo isolation of circulating tumor cells from peripheral blood of cancer patients using a functionalized and structured medical wire. Int. J. Oncol. 2012, 41, 1241–1250. [Google Scholar]
- Vermesh, O.; Aalipour, A.; Ge, T.J.; Saenz, Y.; Guo, Y.; Alam, I.S.; Park, S.-M.; Adelson, C.N.; Mitsutake, Y.; Vilches-Moure, J.; et al. An intravascular magnetic wire for the high-throughput retrieval of circulating tumour cells in vivo. Nat. Biomed. Eng. 2018, 2, 696–705. [Google Scholar] [CrossRef]
- Kim, T.H.; Wang, Y.; Oliver, C.R.; Thamm, D.H.; Cooling, L.; Paoletti, C.; Smith, K.J.; Nagrath, S.; Hayes, D.F. A temporary indwelling intravascular aphaeretic system for in vivo enrichment of circulating tumor cells. Nat. Commun. 2019, 10, 1478. [Google Scholar] [CrossRef]
- Pantel, K.; Denève, E.; Nocca, D.; Coffy, A.; Vendrell, J.-P.; Maudelonde, T.; Riethdorf, S.; Alix-Panabières, C. Circulating Epithelial Cells in Patients with Benign Colon Diseases. Clin. Chem. 2012, 58, 936–940. [Google Scholar] [CrossRef]
- Rosenberg, R.; Gertler, R.; Friederichs, J.; Fuehrer, K.; Dahm, M.; Phelps, R.; Thorban, S.; Nekarda, H.; Siewert, J. Comparison of two density gradient centrifugation systems for the enrichment of disseminated tumor cells in blood. Cytom. J. Int. Soc. Anal. Cytol. 2002, 49, 150–158. [Google Scholar] [CrossRef]
- Munz, M.; Baeuerle, P.A.; Gires, O. The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res. 2009, 69, 5627–5629. [Google Scholar] [CrossRef]
- Low, W.S.; Abas, W.; Bakar, W.A. Benchtop technologies for circulating tumor cells separation based on biophysical properties. BioMed Res. Int. 2015, 2015, 239362. [Google Scholar] [CrossRef]
- El-Heliebi, A.; Kroneis, T.; Zöhrer, E.; Haybaeck, J.; Fischereder, K.; Kampel-Kettner, K.; Zigeuner, R.; Pock, H.; Riedl, R.; Stauber, R. Are morphological criteria sufficient for the identification of circulating tumor cells in renal cancer? J. Transl. Med. 2013, 11, 214. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, N.; Dulmage, K.; Pletcher, C.H.; Wang, L.; DeMuth, W.; Sen, M.; Balli, D.; Yee, S.S.; Sa, S.; Tong, F. An integrated flow cytometry-based platform for isolation and molecular characterization of circulating tumor single cells and clusters. Sci. Rep. 2018, 8, 5035. [Google Scholar] [CrossRef] [PubMed]
- Au, S.H.; Edd, J.; Stoddard, A.E.; Wong, K.H.; Fachin, F.; Maheswaran, S.; Haber, D.A.; Stott, S.L.; Kapur, R.; Toner, M. Microfluidic isolation of circulating tumor cell clusters by size and asymmetry. Sci. Rep. 2017, 7, 2433. [Google Scholar] [CrossRef] [PubMed]
- Edd, J.F.; Mishra, A.; Dubash, T.D.; Herrera, S.; Mohammad, R.; Williams, E.K.; Hong, X.; Mutlu, B.R.; Walsh, J.R.; Machado de Carvalho, F.; et al. Microfluidic concentration and separation of circulating tumor cell clusters from large blood volumes. Lab Chip 2020, 20, 558–567. [Google Scholar] [CrossRef]
- Peeters, D.; De Laere, B.; Van den Eynden, G.; Van Laere, S.; Rothe, F.; Ignatiadis, M.; Sieuwerts, A.; Lambrechts, D.; Rutten, A.; Van Dam, P. Semiautomated isolation and molecular characterisation of single or highly purified tumour cells from CellSearch enriched blood samples using dielectrophoretic cell sorting. Br. J. Cancer 2013, 108, 1358. [Google Scholar] [CrossRef]
- Zhou, J.; Kulasinghe, A.; Bogseth, A.; O’Byrne, K.; Punyadeera, C.; Papautsky, I. Isolation of circulating tumor cells in non-small-cell-lung-cancer patients using a multi-flow microfluidic channel. Microsyst. Nanoeng. 2019, 5, 8. [Google Scholar] [CrossRef]
- Chen, H.; Cao, B.; Sun, B.; Cao, Y.; Yang, K.; Lin, Y.-S. Highly-sensitive capture of circulating tumor cells using micro-ellipse filters. Sci. Rep. 2017, 7, 610. [Google Scholar] [CrossRef]
- Zeinali, M.; Lee, M.; Nadhan, A.; Mathur, A.; Hedman, C.; Lin, E.; Harouaka, R.; Wicha, M.S.; Zhao, L.; Palanisamy, N. High-Throughput Label-Free Isolation of Heterogeneous Circulating Tumor Cells and CTC Clusters from Non-Small-Cell Lung Cancer Patients. Cancers 2020, 12, 127. [Google Scholar] [CrossRef]
- Hou, H.W.; Warkiani, M.E.; Khoo, B.L.; Li, Z.R.; Soo, R.A.; Tan, D.S.-W.; Lim, W.-T.; Han, J.; Bhagat, A.A.S.; Lim, C.T. Isolation and retrieval of circulating tumor cells using centrifugal forces. Sci. Rep. 2013, 3, 1259. [Google Scholar] [CrossRef]
- Sollier, E.; Go, D.E.; Che, J.; Gossett, D.R.; O’Byrne, S.; Weaver, W.M.; Kummer, N.; Rettig, M.; Goldman, J.; Nickols, N. Size-selective collection of circulating tumor cells using Vortex technology. Lab Chip 2014, 14, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Gwak, H.; Kim, J.; Kashefi-Kheyrabadi, L.; Kwak, B.; Hyun, K.-A.; Jung, H.-I. Progress in circulating tumor cell research using microfluidic devices. Micromachines 2018, 9, 353. [Google Scholar] [CrossRef] [PubMed]
- Kalinich, M.; Bhan, I.; Kwan, T.T.; Miyamoto, D.T.; Javaid, S.; LiCausi, J.A.; Milner, J.D.; Hong, X.; Goyal, L.; Sil, S. An RNA-based signature enables high specificity detection of circulating tumor cells in hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 1123–1128. [Google Scholar] [CrossRef]
- Cortés-Hernández, L.E.; Eslami-S, Z.; Alix-Panabières, C. Circulating tumor cell as the functional aspect of liquid biopsy to understand the metastatic cascade in solid cancer. Mol. Asp. Med. 2019, 100816. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Kanatani, S.; Tomer, R.; Sahlgren, C.; Kronqvist, P.; Kaczynska, D.; Louhivuori, L.; Kis, L.; Lindh, C.; Mitura, P. Whole-tissue biopsy phenotyping of three-dimensional tumours reveals patterns of cancer heterogeneity. Nat. Biomed. Eng. 2017, 1, 796–806. [Google Scholar] [CrossRef]
- Agerbæk, M.Ø.; Bang-Christensen, S.R.; Yang, M.-H.; Clausen, T.M.; Pereira, M.A.; Sharma, S.; Ditlev, S.B.; Nielsen, M.A.; Choudhary, S.; Gustavsson, T. The VAR2CSA malaria protein efficiently retrieves circulating tumor cells in an EpCAM-independent manner. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef]
- Liu, Z.; Fusi, A.; Klopocki, E.; Schmittel, A.; Tinhofer, I.; Nonnenmacher, A.; Keilholz, U. Negative enrichment by immunomagnetic nanobeads for unbiased characterization of circulating tumor cells from peripheral blood of cancer patients. J. Transl. Med. 2011, 9, 70. [Google Scholar] [CrossRef]
- Wu, Y.; Deighan, C.J.; Miller, B.L.; Balasubramanian, P.; Lustberg, M.B.; Zborowski, M.; Chalmers, J.J. Isolation and analysis of rare cells in the blood of cancer patients using a negative depletion methodology. Methods 2013, 64, 169–182. [Google Scholar] [CrossRef]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Chen, H. A Triplet Parallelizing Spiral Microfluidic Chip for Continuous Separation of Tumor Cells. Sci. Rep. 2018, 8, 4042. [Google Scholar] [CrossRef]
- Fabbri, F.; Carloni, S.; Zoli, W.; Ulivi, P.; Gallerani, G.; Fici, P.; Chiadini, E.; Passardi, A.; Frassineti, G.L.; Ragazzini, A.; et al. Detection and recovery of circulating colon cancer cells using a dielectrophoresis-based device: KRAS mutation status in pure CTCs. Cancer Lett. 2013, 335, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, P.; Kinders, R.J.; Kummar, S.; Gupta, V.; Hasegawa, D.; Menachery, A.; Lawrence, S.M.; Wang, L.; Ferry-Galow, K.; Davis, D. Antibody-independent capture of circulating tumor cells of non-epithelial origin with the ApoStream® system. PLoS ONE 2017, 12, e0175414. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Cheng, B.; Fu, L. Clinical applications of circulating tumor cells in pharmacotherapy: Challenges and perspectives. Mol. Pharmacol. 2017, 92, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.; Kienle, P.; Lacroix, J.; Willeke, F.; Benner, A.; Lehnert, T.; Herfarth, C.; von Knebel Doeberitz, M. Dissemination of tumor cells in patients undergoing surgery for colorectal cancer. Clin. Cancer Res. 1998, 4, 343–348. [Google Scholar]
- Zharov, V.P.; Galanzha, E.I.; Shashkov, E.V.; Kim, J.-W.; Khlebtsov, N.G.; Tuchin, V.V. Photoacoustic flow cytometry: Principle and application for real-time detection of circulating single nanoparticles, pathogens, and contrast dyes in vivo. J. Biomed. Opt. 2007, 12, 051503. [Google Scholar] [CrossRef]
- Kirby, B.J.; Jodari, M.; Loftus, M.S.; Gakhar, G.; Pratt, E.D.; Chanel-Vos, C.; Gleghorn, J.P.; Santana, S.M.; Liu, H.; Smith, J.P. Functional characterization of circulating tumor cells with a prostate-cancer-specific microfluidic device. PLoS ONE 2012, 7, e35976. [Google Scholar] [CrossRef]
- Karabacak, N.M.; Spuhler, P.S.; Fachin, F.; Lim, E.J.; Pai, V.; Ozkumur, E.; Martel, J.M.; Kojic, N.; Smith, K.; Chen, P.-I.; et al. Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protoc. 2014, 9, 694–710. [Google Scholar] [CrossRef]
- Alix-Panabières, C. EPISPOT assay: Detection of viable DTCs/CTCs in solid tumor patients. In Minimal Residual Disease and Circulating Tumor Cells in Breast Cancer; Springer: Berlin/Heidelberg, Germany, 2012; pp. 69–76. [Google Scholar]
- Armstrong, A.J.; Marengo, M.S.; Oltean, S.; Kemeny, G.; Bitting, R.L.; Turnbull, J.D.; Herold, C.I.; Marcom, P.K.; George, D.J.; Garcia-Blanco, M.A. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol. Cancer Res. 2011, 9, 997–1007. [Google Scholar] [CrossRef]
- Vona, G.; Sabile, A.; Louha, M.; Sitruk, V.; Romana, S.; Schütze, K.; Capron, F.; Franco, D.; Pazzagli, M.; Vekemans, M. Isolation by size of epithelial tumor cells: A new method for the immunomorphological and molecular characterization of circulating tumor cells. Am. J. Pathol. 2000, 156, 57–63. [Google Scholar] [CrossRef]
- Sharma, S.; Zhuang, R.; Long, M.; Pavlovic, M.; Kang, Y.; Ilyas, A.; Asghar, W. Circulating tumor cell isolation, culture, and downstream molecular analysis. Biotechnol. Adv. 2018, 36, 1063–1078. [Google Scholar] [CrossRef]
- Kaifi, J.T.; Kunkel, M.; Das, A.; Harouaka, R.A.; Dicker, D.T.; Li, G.; Zhu, J.; Clawson, G.A.; Yang, Z.; Reed, M.F. Circulating tumor cell isolation during resection of colorectal cancer lung and liver metastases: A prospective trial with different detection techniques. Cancer Biol. Ther. 2015, 16, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.C.; Robinson, P.S.; Wagner, C.; O’Shannessy, D.J. The Parsortix™ cell separation system—A versatile liquid biopsy platform. Cytom. Part A 2018, 93, 1234–1239. [Google Scholar] [CrossRef] [PubMed]
- Castro, J.; Sanchez, L.; Nuñez, M.T.; Lu, M.; Castro, T.; Sharifi, H.R.; Ericsson, C. Screening circulating tumor cells as a noninvasive cancer test in 3388 individuals from high-risk groups (ICELLATE2). Dis. Mark. 2018, 2018, 4653109. [Google Scholar] [CrossRef]
- Joosse, S.A.; Beyer, B.; Gasch, C.; Nastały, P.; Kuske, A.; Isbarn, H.; Horst, L.J.; Hille, C.; Gorges, T.M.; Cayrefourcq, L.; et al. Tumor-Associated Release of Prostatic Cells into the Blood after Transrectal Ultrasound-Guided Biopsy in Patients with Histologically Confirmed Prostate Cancer. Clin. Chem. 2019. [Google Scholar] [CrossRef]
- Oklu, R.; Sheth, R.; Albadawi, H.; Bhan, I.; Sarioglu, A.F.; Choz, M.; Zeinali, M.; Deshpande, V.; Maheswaran, S.; Haber, D.A. Relationship between hepatocellular carcinoma circulating tumor cells and tumor volume. Cancer Converg. 2018, 2, 1–9. [Google Scholar] [CrossRef]
- Stott, S.L.; Lee, R.J.; Nagrath, S.; Yu, M.; Miyamoto, D.T.; Ulkus, L.; Inserra, E.J.; Ulman, M.; Springer, S.; Nakamura, Z. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci. Transl. Med. 2010, 2, 25ra23. [Google Scholar] [CrossRef]
- Rack, B.; Schindlbeck, C.; Jückstock, J.; Andergassen, U.; Hepp, P.; Zwingers, T.; Friedl, T.W.; Lorenz, R.; Tesch, H.; Fasching, P.A. Circulating tumor cells predict survival in early average-to-high risk breast cancer patients. J. Natl. Cancer Inst. 2014, 106, dju066. [Google Scholar] [CrossRef]
- Murlidhar, V.; Reddy, R.M.; Fouladdel, S.; Zhao, L.; Ishikawa, M.K.; Grabauskiene, S.; Zhang, Z.; Lin, J.; Chang, A.C.; Carrott, P. Poor prognosis indicated by venous circulating tumor cell clusters in early-stage lung cancers. Cancer Res. 2017, 77, 5194–5206. [Google Scholar] [CrossRef]
- He, W.; Hou, M.; Zhang, H.; Zeng, C.; He, S.; Chen, X.; Xu, M.; Sun, C.; Jiang, W.; Wang, H. Clinical significance of circulating tumor cells in predicting disease progression and chemotherapy resistance in patients with gestational choriocarcinoma. Int. J. Cancer 2019, 144, 1421–1431. [Google Scholar] [CrossRef]
- Tanaka, F.; Yoneda, K.; Kondo, N.; Hashimoto, M.; Takuwa, T.; Matsumoto, S.; Okumura, Y.; Rahman, S.; Tsubota, N.; Tsujimura, T. Circulating tumor cell as a diagnostic marker in primary lung cancer. Clin. Cancer Res. 2009, 15, 6980–6986. [Google Scholar] [CrossRef]
- Nastały, P.; Ruf, C.; Becker, P.; Bednarz-Knoll, N.; Stoupiec, M.; Kavsur, R.; Isbarn, H.; Matthies, C.; Wagner, W.; Höppner, D. Circulating tumor cells in patients with testicular germ cell tumors. Clin. Cancer Res. 2014, 20, 3830–3841. [Google Scholar] [CrossRef] [PubMed]
- Hartkopf, A.D.; Wallwiener, M.; Hahn, M.; Fehm, T.N.; Brucker, S.Y.; Taran, F.-A. Simultaneous detection of disseminated and circulating tumor cells in primary breast cancer patients. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2016, 48, 115. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.-C.; Peeters, D.J.; Fehm, T.; Nolé, F.; Gisbert-Criado, R.; Mavroudis, D.; Grisanti, S.; Generali, D.; Garcia-Saenz, J.A.; Stebbing, J.; et al. Clinical validity of circulating tumour cells in patients with metastatic breast cancer: A pooled analysis of individual patient data. Lancet Oncol. 2014, 15, 406–414. [Google Scholar] [CrossRef]
- Balakrishnan, A.; Koppaka, D.; Anand, A.; Deb, B.; Grenci, G.; Viasnoff, V.; Thompson, E.W.; Gowda, H.; Bhat, R.; Rangarajan, A.; et al. Circulating Tumor Cell cluster phenotype allows monitoring response to treatment and predicts survival. Sci. Rep. 2019, 9, 7933. [Google Scholar] [CrossRef] [PubMed]
- Sawabata, N.; Susaki, Y.; Nakamura, T.; Kawaguchi, T.; Yasukawa, M.; Taniguchi, S. Cluster circulating tumor cells in surgical cases of lung cancer. Gen. Thorac. Cardiovasc. Surg. 2020, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Schwarzenbach, H.; Pantel, K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- Babayan, A.; Hannemann, J.; Spötter, J.; Müller, V.; Pantel, K.; Joosse, S.A. Heterogeneity of estrogen receptor expression in circulating tumor cells from metastatic breast cancer patients. PLoS ONE 2013, 8, e75038. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef]
- Hille, C.; Gorges, T.M.; Riethdorf, S.; Mazel, M.; Steuber, T.; Von Amsberg, G.; König, F.; Peine, S.; Alix-Panabières, C.; Pantel, K. Detection of Androgen Receptor Variant 7 (ARV7) mRNA Levels in EpCAM-Enriched CTC Fractions for Monitoring Response to Androgen Targeting Therapies in Prostate Cancer. Cells 2019, 8, 1067. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef]
- Dhar, M.; Wong, J.; Che, J.; Matsumoto, M.; Grogan, T.; Elashoff, D.; Garon, E.B.; Goldman, J.W.; Christen, E.S.; Di Carlo, D. Evaluation of PD-L1 expression on vortex-isolated circulating tumor cells in metastatic lung cancer. Sci. Rep. 2018, 8, 2592. [Google Scholar] [CrossRef]
- Wang, Y.; Kim, T.H.; Fouladdel, S.; Zhang, Z.; Soni, P.; Qin, A.; Zhao, L.; Azizi, E.; Lawrence, T.S.; Ramnath, N.; et al. PD-L1 Expression in Circulating Tumor Cells Increases during Radio(chemo)therapy and Indicates Poor Prognosis in Non-small Cell Lung Cancer. Sci. Rep. 2019, 9, 566. [Google Scholar] [CrossRef]
- Satelli, A.; Brownlee, Z.; Noh, H.; Meng, Q.H.; Kopetz, S.; Overman, M.; Li, S. Abstract 1596: Detection of PD-L1 in cell surface vimentin positive circulating tumor cells is associated with poor survival in cancer patients. Cancer Res. 2015, 75, 1596. [Google Scholar] [CrossRef]
- Todaro, M.; Gaggianesi, M.; Catalano, V.; Benfante, A.; Iovino, F.; Biffoni, M.; Apuzzo, T.; Sperduti, I.; Volpe, S.; Cocorullo, G. CD44v6 is a marker of constitutive and reprogrammed cancer stem cells driving colon cancer metastasis. Cell Stem Cell 2014, 14, 342–356. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.-F.; Bachmann, M.H.; Shimono, Y. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef]
- Pestrin, M.; Salvianti, F.; Galardi, F.; De Luca, F.; Turner, N.; Malorni, L.; Pazzagli, M.; Di Leo, A.; Pinzani, P. Heterogeneity of PIK3CA mutational status at the single cell level in circulating tumor cells from metastatic breast cancer patients. Mol. Oncol. 2015, 9, 749–757. [Google Scholar] [CrossRef]
- Maheswaran, S.; Sequist, L.V.; Nagrath, S.; Ulkus, L.; Brannigan, B.; Collura, C.V.; Inserra, E.; Diederichs, S.; Iafrate, A.J.; Bell, D.W. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 2008, 359, 366–377. [Google Scholar] [CrossRef]
- Wu, C.; Wu, P.; Zhao, H.; Liu, W.; Li, W. Clinical Applications of and Challenges in Single-Cell Analysis of Circulating Tumor Cells. DNA Cell Biol. 2018, 37, 78–89. [Google Scholar] [CrossRef]
- Sakaizawa, K.; Goto, Y.; Kiniwa, Y.; Uchiyama, A.; Harada, K.; Shimada, S.; Saida, T.; Ferrone, S.; Takata, M.; Uhara, H.; et al. Mutation analysis of BRAF and KIT in circulating melanoma cells at the single cell level. Br. J. Cancer 2012, 106, 939. [Google Scholar] [CrossRef]
- Lianidou, E.S.; Markou, A. Circulating tumor cells in breast cancer: Detection systems, molecular characterization, and future challenges. Clin. Chem. 2011, 57, 1242–1255. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Aieta, M.; Facchinetti, A.; De Faveri, S.; Manicone, M.; Tartarone, A.; Possidente, L.; Lerose, R.; Mambella, G.; Calderone, G.; Zamarchi, R.; et al. Monitoring and Characterization of Circulating Tumor Cells (CTCs) in a Patient With EML4-ALK–Positive Non–Small Cell Lung Cancer (NSCLC). Clin. Lung Cancer 2016, 17, e173–e177. [Google Scholar] [CrossRef]
- Eslami-S, Z.; Cortés-Hernández, L.E.; Cayrefourcq, L.; Alix-Panabières, C. The Different Facets of Liquid Biopsy: A Kaleidoscopic View. Cold Spring Harb. Perspect. Med. 2019, a037333. [Google Scholar] [CrossRef]
- Calabuig-Farinas, S.; Jantus-Lewintre, E.; Herreros-Pomares, A.; Camps, C. Circulating tumor cells versus circulating tumor DNA in lung cancer—Which one will win? Transl. Lung Cancer Res. 2016, 5, 466. [Google Scholar] [CrossRef]
- Razavi, P.; Li, B.T.; Brown, D.N.; Jung, B.; Hubbell, E.; Shen, R.; Abida, W.; Juluru, K.; De Bruijn, I.; Hou, C.; et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019, 25, 1928–1937. [Google Scholar] [CrossRef]
- Guibert, N.; Pradines, A.; Farella, M.; Casanova, A.; Gouin, S.; Keller, L.; Favre, G.; Mazieres, J. Monitoring KRAS mutations in circulating DNA and tumor cells using digital droplet PCR during treatment of KRAS-mutated lung adenocarcinoma. Lung Cancer 2016, 100, 1–4. [Google Scholar] [CrossRef]
- Xu, R.; Zhong, G.; Huang, T.; He, W.; Kong, C.; Zhang, X.; Wang, Y.; Liu, M.; Xu, M.; Chen, S. Sequencing of circulating tumor DNA for dynamic monitoring of gene mutations in advanced non-small cell lung cancer. Oncol. Lett. 2018, 15, 3726–3734. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, H.; Zhan, Y.; Long, M.; Liu, S.; Lu, J.; Zang, H.; Fan, S. Detection and application of circulating tumor cell and circulating tumor DNA in the non-small cell lung cancer. Am. J. Cancer Res. 2018, 8, 2377. [Google Scholar]
- Rossi, G.; Mu, Z.; Rademaker, A.W.; Austin, L.K.; Strickland, K.S.; Costa, R.L.B.; Nagy, R.J.; Zagonel, V.; Taxter, T.J.; Behdad, A.; et al. Cell-Free DNA and Circulating Tumor Cells: Comprehensive Liquid Biopsy Analysis in Advanced Breast Cancer. Clin. Cancer Res. 2018, 24, 560. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, J.; Liu, S.; Wang, J.; Han, X.; Qin, H.; Lang, J.; Cheng, K.; Li, Y.; Qi, Y. Inhibition of platelet function using liposomal nanoparticles blocks tumor metastasis. Theranostics 2017, 7, 1062. [Google Scholar] [CrossRef]
- Li, J.; Sharkey, C.C.; Wun, B.; Liesveld, J.L.; King, M.R. Genetic engineering of platelets to neutralize circulating tumor cells. J. Control. Release 2016, 228, 38–47. [Google Scholar] [CrossRef]
- Phipps, L.E.; Hino, S.; Muschel, R.J. Targeting cell spreading: A method of sensitizing metastatic tumor cells to TRAIL-induced apoptosis. Mol. Cancer Res. 2011, 9, 249–258. [Google Scholar] [CrossRef]
- Mitchell, M.J.; King, M.R. Fluid shear stress sensitizes cancer cells to receptor-mediated apoptosis via trimeric death receptors. New J. Phys. 2013, 15, 015008. [Google Scholar] [CrossRef]
- Wayne, E.C.; Chandrasekaran, S.; Mitchell, M.J.; Chan, M.F.; Lee, R.E.; Schaffer, C.B.; King, M.R. TRAIL-coated leukocytes that prevent the bloodborne metastasis of prostate cancer. J. Control. Release 2016, 223, 215–223. [Google Scholar] [CrossRef]
- Van Trappen, P.O.; Pepper, M.S. Lymphatic dissemination of tumour cells and the formation of micrometastases. Lancet Oncol. 2002, 3, 44–52. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Chan, M.F.; Li, J.; King, M.R. Super natural killer cells that target metastases in the tumor draining lymph nodes. Biomaterials 2016, 77, 66–76. [Google Scholar] [CrossRef]
- Kauder, S.E.; Kuo, T.C.; Harrabi, O.; Chen, A.; Sangalang, E.; Doyle, L.; Rocha, S.S.; Bollini, S.; Han, B.; Sim, J. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS ONE 2018, 13, e0201832. [Google Scholar] [CrossRef]
- Ingram, J.R.; Blomberg, O.S.; Sockolosky, J.T.; Ali, L.; Schmidt, F.I.; Pishesha, N.; Espinosa, C.; Dougan, S.K.; Garcia, K.C.; Ploegh, H.L. Localized CD47 blockade enhances immunotherapy for murine melanoma. Proc. Natl. Acad. Sci. USA 2017, 114, 10184–10189. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Veillette, A.; Chen, J. SIRPα–CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018, 39, 173–184. [Google Scholar] [CrossRef]
- Chowdhury, S.; Castro, S.; Coker, C.; Hinchliffe, T.E.; Arpaia, N.; Danino, T. Programmable bacteria induce durable tumor regression and systemic antitumor immunity. Nat. Med. 2019, 25, 1057–1063. [Google Scholar] [CrossRef]
- Hosseini, H.; Obradović, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552. [Google Scholar] [CrossRef]
- Agudo, J.; Park, E.S.; Rose, S.A.; Alibo, E.; Sweeney, R.; Dhainaut, M.; Kobayashi, K.S.; Sachidanandam, R.; Baccarini, A.; Merad, M.; et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity 2018, 48, 271–285.e275. [Google Scholar] [CrossRef]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Höfer, T.; Riddell, S.R.; Busch, D.H. Serial transfer of single-cell-derived immunocompetence reveals stemness of CD8+ central memory T cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef]
- Berger, C.; Jensen, M.C.; Lansdorp, P.M.; Gough, M.; Elliott, C.; Riddell, S.R. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J. Clin. Investig. 2008, 118, 294–305. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M. CD19 CAR–T cells of defined CD4+: CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A.; Bragado, P.; Sosa, M.S. Metastasis Awakening: Targeting dormant cancer. Nat. Med. 2013, 19, 276. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kittler, O.; Ragg, T.; Daskalakis, A.; Granzow, M.; Ahr, A.; Blankenstein, T.J.; Kaufmann, M.; Diebold, J.; Arnholdt, H.; Müller, P. From latent disseminated cells to overt metastasis: Genetic analysis of systemic breast cancer progression. Proc. Natl. Acad. Sci. USA 2003, 100, 7737–7742. [Google Scholar] [CrossRef] [PubMed]
- Demicheli, R.; Miceli, R.; Moliterni, A.; Zambetti, M.; Hrushesky, W.; Retsky, M.; Valagussa, P.; Bonadonna, G. Breast cancer recurrence dynamics following adjuvant CMF is consistent with tumor dormancy and mastectomy-driven acceleration of the metastatic process. Ann. Oncol. 2005, 16, 1449–1457. [Google Scholar] [CrossRef] [PubMed]
- Almog, N.; Henke, V.; Flores, L.; Hlatky, L.; Kung, A.L.; Wright, R.D.; Berger, R.; Hutchinson, L.; Naumov, G.N.; Bender, E. Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis. FASEB J. 2006, 20, 947–949. [Google Scholar] [CrossRef]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Alencar, V.H.M.; Badran, A.; Bonfill, X. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302. [Google Scholar] [CrossRef]
- Buscail, E.; Alix-Panabières, C.; Quincy, P.; Cauvin, T.; Chauvet, A.; Degrandi, O.; Caumont, C.; Verdon, S.; Lamrissi, I.; Moranvillier, I.; et al. High Clinical Value of Liquid Biopsy to Detect Circulating Tumor Cells and Tumor Exosomes in Pancreatic Ductal Adenocarcinoma Patients Eligible for Up-Front Surgery. Cancers 2019, 11, 1656. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enrichment Method | Selection Criteria | Detection Method | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|
| (a) Biological-based methods for CTC enrichment | |||||
| Immunoaffinity-positive enrichment | |||||
| CellSearch® | EpCAM | Flow cytometry | ● FDA-approved method, ● Clinically relevant automated system, ● Quantitative, ● Easy, ● Highly robust and reproducible | ● Do not detect EpCAM-negative CTCs, ● Expensive and subjective image evaluation, ● Cell preservative limits RNA analysis | [16,18,103,135] |
| CellCollector® | EpCAM | Immunocytochemical staining | ● Can isolate rare CTCs in early cancer stages, ● Provide more CTCs, ● No need of blood sampling, ● Detect CTCs in vivo | ● Cannot isolate EMT-CTCs, ● CTCs cannot be released from the wire | [115,116] |
| MagWIRE system | EpCAM | qPCR | ● Large-scale CTC capture in vivo, ● Very fast processing, ● Completely self-contained, ● Flexible | ●Require additional biocompatibility testing, ● Capture EpCAM-positive CTCs only, ● Possible systemic exposure to excess iron | [116,117] |
| rVAR2-based CTC isolation Ligand-receptor affinity | Oncofetal chondroitin sulfate (ofCS) | Flowcytometry/ddPCR/Western blotting/Four-color immunofluorescence staining | ● Not dependent on the expression of a single marker, ● Low contamination of PBMCs, ● CTC enumeration correlates with disease stage | ● Need to be followed by CTC detection methods, ● Need redesign for each tumor type, ● Not commercially available | [137] |
| Immunoaffinity-negative enrichment | |||||
| EasySep™ | CD45 depletion | Flow cytometry | ● Simple, ● Easy-to-use batch separation, ● Do not bias the sample according to selection markers | ● False positive results due to CD45– endothelial cells, ● Do not achieve the same high purity levels | [138] |
| Quadrupole Magnetic Separator (QMS) | CD45 depletion | IF staining | ● High-throughput magnetic cell sorter, ● Continuous flow | ● RBC lysis required | [139] |
| (b) Physical methods for CTC enrichment | |||||
| Size and deformability | |||||
| ISET technology | Size/ Deformability | IF staining | ● Easy and fast processing time, ● Sensitivity threshold of 1 CTC/mL of blood, ● Label-independent isolation, ● Isolation of intact CTCs, ● Isolation of EpCAM-negative CTCs, ● CTCs can be further analyzed by multiplexed imaging and genetic analysis | ● Low specificity, ● Retention only of CTCs larger than the leukocyte range, ● Bigger leukocytes may be captured, ● Blood cells need to be fixed, ● False-positive results, ● Low recovery and purity, ● Need the pathologists’ expertise for CTC detection | [3,16,18,140] |
| Spiral- Slits Chip | Size and deformability | RT-PCR | ● Avoid clogging, ● Continuous separation, ● Minimal contamination, ● Detection with optical spectroscopy, ● Fast processing | ● False-positive results, ● Need optimizing the structure geometry, ● Low sensitivity | [141] |
| Cluster-Chip | Strength of cell-cell junctions | RNA sequencing | ● Label-free isolation ● Potential study of tumor-immune system interactions ● Chemistry-free approach | ● Lack of biological characterization and clinical significance, ● Not commercially available, ● Shear stress is needed to release the majority of clusters from micropillars | [31] |
| Nanotube-CTC-chip | Preferential adherence or phenotype | IF staining | ● Antigen/Size-independent CTC capture, ● Better capture sensitivity from droplets, ● No cell loss, ● Surface architecture lends itself to easier CTC downstream analysis, ● CTC isolation with high purity and 100% sensitivity, ● Can capture CTCs with different phenotypes | ● Need development for surface architecture, ● Not commercially available, ● Cannot isolate EMT-CTCs, ● Long set-up times | [141] |
| Dielectric properties | |||||
| DEPArray™ | Electrical signature | ● DEP cages allow the recovery and manipulation of viable single cells | ● Requires pre-enrichment | [142] | |
| ApoStream® | Conductivity Morphology and Membrane surface area | IF staining | ● Label-independent isolation, ● Continuous flow, ● Captures viable cells | ● Cells need to be pre-purified because whole blood has high electrical conductivity | [18,143,144] |
| Density | |||||
| OncoQuick® | Density and filtration | - | ● Porous membrane prevent mixing, ● Simple, ● Inexpensive | ● Relative low yield and enrichment | [120] |
| Ficoll-Paque® | Density | RT-PCR | ● Inexpensive, ● Easy-to-use | ● Significant CTC loss | [145] |
| Inertial Focusing | |||||
| Labyrinth device | Size | IF staining | Can capture viable cells, label free | Prior RBC depletion required | [130] |
| Multi-flow microfluidic device | Size and inertial forces | IF staining | Predictable and tunable cutoff size, isolation of CTC clusters, one-step isolation | Relative low flow rate, | [128] |
| Micro-ellipse filters | Size, deformability and inertial forces | IF staining | Robust, large sample processing, on-chip culture | RBC lysis required | [129] |
| ClearCell® FX | Size and inertial forces | Flow cytometry | ● Can captures viable cells, ● Easy to manufacture, ● Can process a 7.5 mL sample in 8 min, ● Exerts minimal stress on captured cells | ● RBC lysis required | [131] |
| Vortex | Size | IF staining | ● No RBC lysis required, ● Can capture viable cells, ● Easy to manufacture, ● Detect clusters | ● Low sensitivity and reproducibility | [132] |
| Photoacoustic flow cytometry | |||||
| Ultrasound and a pulsed near-infrared laser | - | Flow cytometry | ● Can count CTCs in blood vessels up to 3 mm deep, ● Label-free | ● Only used for CTC count, ● No molecular analysis | [146] |
| (c) Combined methods | |||||
| GEDI chip | Size and immunoaffinity | cDNA sequencing and immunostaining | ● Relative high yield and enrichment | ● Requires surface chemistry modifications, ● Requires high-resolution imaging method | [147] |
| CTC-iChip (Microfluidic immunomagnetic-based) | Size inertial focusing Negative enrichment | Mass cytometry | ● Allows the sequential separation of different blood components through micropillar array, ● Hydrodynamic size-based sorting/magnetophoresis, ● Simplicity, ● Can sort rare CTCs | ● Samples not suitable for DNA sequencing, ● Expensive, ● Long set-up times, ● Difficult to implement in clinical settings, ● Multiple manually interconnected chips | [148] |
| RosetteSep system | Negative enrichment EPISPOT (protein secretion) | IF staining | ● Captures and detects viable CTCs at the single-cell level, ● Does not need whole-genome or transcriptome amplification, ● Limited number of markers | ● Proteins must be actively secreted ● Unbiased enrichment independent of CTC/ DTC phenotype | [140,142,149] |
| RosetteSep system EPIDROP | Secreted proteins Density centrifugation Immunoaffinity | IF staining | ● Simultaneous proteomic and secretomic analysis of single viable CTCs, ● Can test different drugs in a single patient, ● More reliable and sensitive than EPISPOT, ● Automatic detection of positive events using the appropriate software | ● Prototype development still in progress | [6] |
| Device | Ref. | Enrichment/Detection Method | Condition | Status | Primary Endpoints | Trail |
|---|---|---|---|---|---|---|
| GILUPI CellCollector® | [116] | Immunoaffinity (anti-EpCAM Ab) | Locally advanced breast cancer | Completed | PFS, OS | NCT03732339 |
| EMT-marker based ferrofluid device | [150] | N-cadherin or O-cadherin based analysis | Metastatic breast and prostate cancers | Completed | Clinical stage, Screening | NCT02025413 |
| ISET® technology | [151] | Immunocytochemistry (PD-L1 expression analysis) FISH analysis of ALK | Lung cancer Lung Neoplasms | Recruiting Active | Not provided Validation of ALK analysis | NCT02827344 NCT02372448 |
| Culture system | [152] | Affinity-based WBC deletion | Early detection of cancer | Recruiting | Early diagnosis and screening | NCT03843450 |
| Flexible Micro Spring Array (FMSA) | [153] | Filtration (or size-exclusion of viable CTCs) | Stage IV colorectal cancer | Completed | PFS, OS, response to therapy | NCT01722903 |
| Ficoll enrichment | [145] | Density/ PCR | Pancreatic ductal adenocarcinoma | Completed | PFS, OS, response to therapy | NCT02150746 |
| CellSearch® | [154] | Immunoaffinity (anti-EpCAM Ab) | Prostate cancer Metastatic breast cancer | Recruiting Completed | EMT markers, PFS and OS CTC-Endocrine Therapy Index | NCT04021394 NCT01701050 |
| CTC-Chip | [102] | Size or Immunoaffinity | Prostate cancer (Prostatectomy) | Recruiting | Examine chromosome translocation | NCT01961713 |
| Parsortix™ | [155] | Cellular size and deformability | Ovarian neoplasms | Completed | Estimate risk of cancer | NCT02785731 |
| IsoPic™ microfluidic system | [156] | Flow rate, surface interactions, plasticity, and elasticity | Unknown primary cancer | Recruiting | Prediction of molecularly targeted therapies | NCT04025970 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dianat-Moghadam, H.; Azizi, M.; Eslami-S, Z.; Cortés-Hernández, L.E.; Heidarifard, M.; Nouri, M.; Alix-Panabières, C. The Role of Circulating Tumor Cells in the Metastatic Cascade: Biology, Technical Challenges, and Clinical Relevance. Cancers 2020, 12, 867. https://doi.org/10.3390/cancers12040867
Dianat-Moghadam H, Azizi M, Eslami-S Z, Cortés-Hernández LE, Heidarifard M, Nouri M, Alix-Panabières C. The Role of Circulating Tumor Cells in the Metastatic Cascade: Biology, Technical Challenges, and Clinical Relevance. Cancers. 2020; 12(4):867. https://doi.org/10.3390/cancers12040867
Chicago/Turabian StyleDianat-Moghadam, Hassan, Mehdi Azizi, Zahra Eslami-S, Luis Enrique Cortés-Hernández, Maryam Heidarifard, Mohammad Nouri, and Catherine Alix-Panabières. 2020. "The Role of Circulating Tumor Cells in the Metastatic Cascade: Biology, Technical Challenges, and Clinical Relevance" Cancers 12, no. 4: 867. https://doi.org/10.3390/cancers12040867
APA StyleDianat-Moghadam, H., Azizi, M., Eslami-S, Z., Cortés-Hernández, L. E., Heidarifard, M., Nouri, M., & Alix-Panabières, C. (2020). The Role of Circulating Tumor Cells in the Metastatic Cascade: Biology, Technical Challenges, and Clinical Relevance. Cancers, 12(4), 867. https://doi.org/10.3390/cancers12040867