The Inhibitory Mechanisms of Tumor PD-L1 Expression by Natural Bioactive Gallic Acid in Non-Small-Cell Lung Cancer (NSCLC) Cells


 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. GA Downregulates the PD-L1 Expression in NSCLC Cells
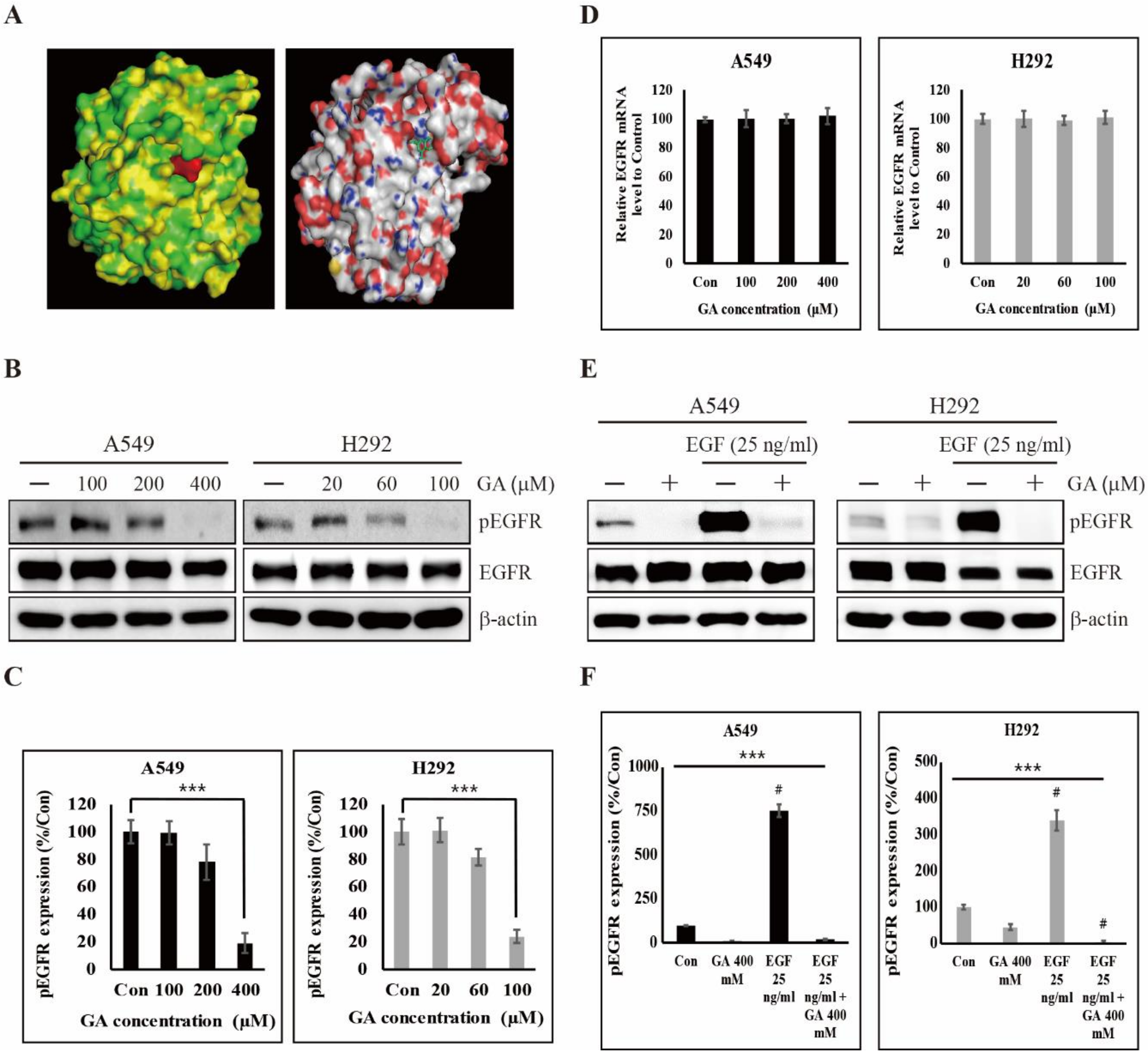
2.2. GA Binds to EGFR and Then Inhibits its Phosphorylation
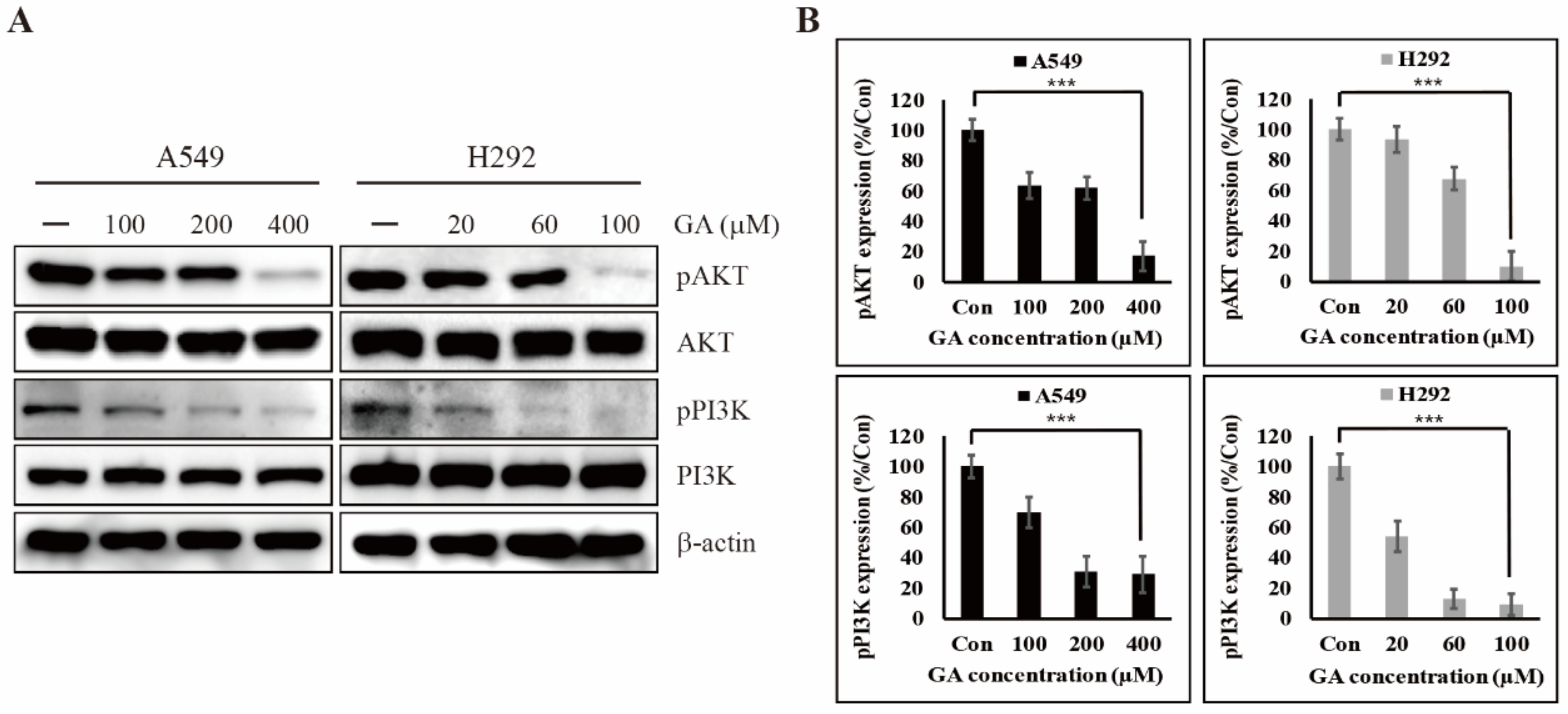
2.3. GA Reduces the Phosphorylation of PI3K/AKT That is One of the Downstream Targets of EGFR Signaling
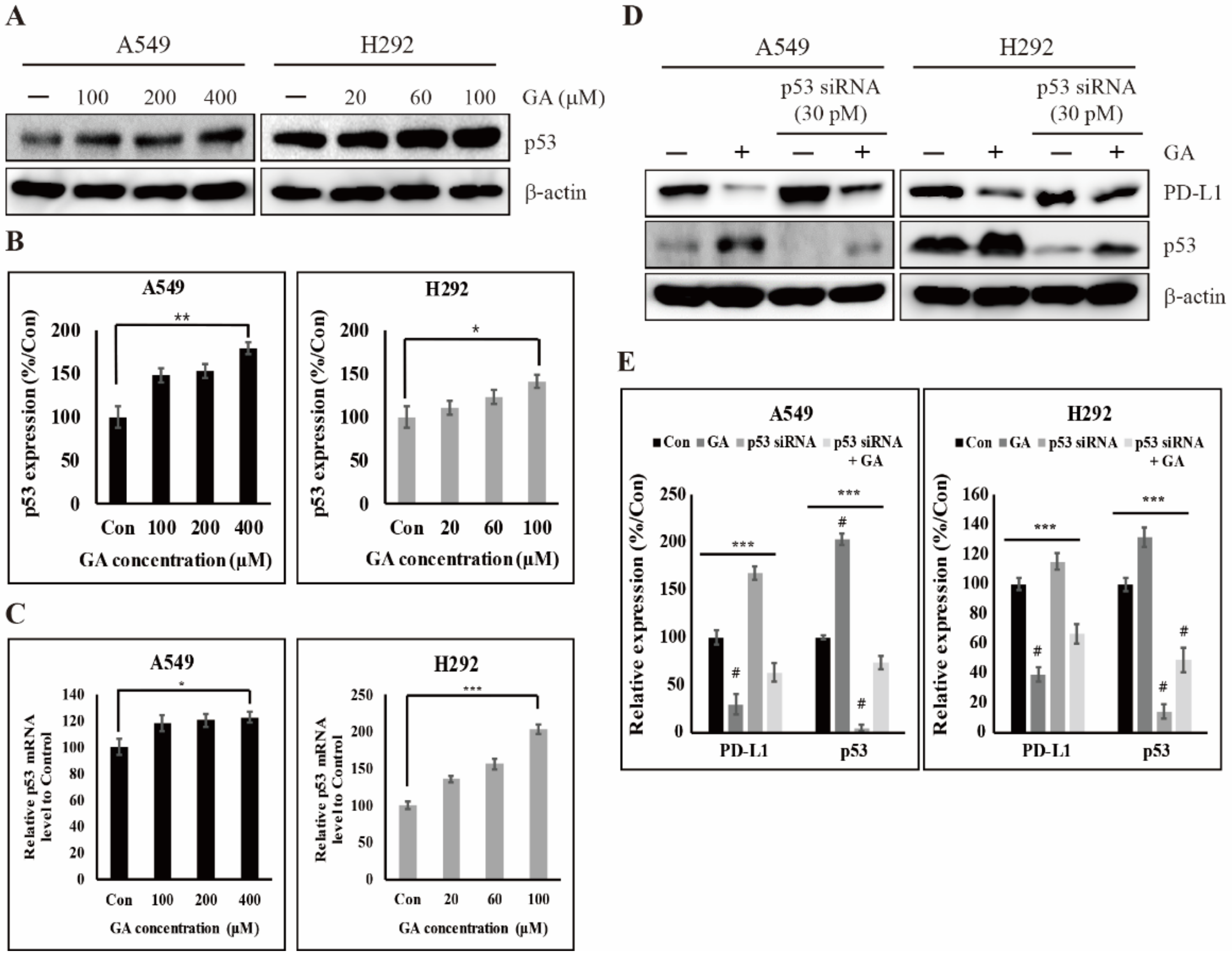
2.4. GA Activates the Expression of Tumor Suppressor Factor p53 for Inhibiting the Expression of PD-L1
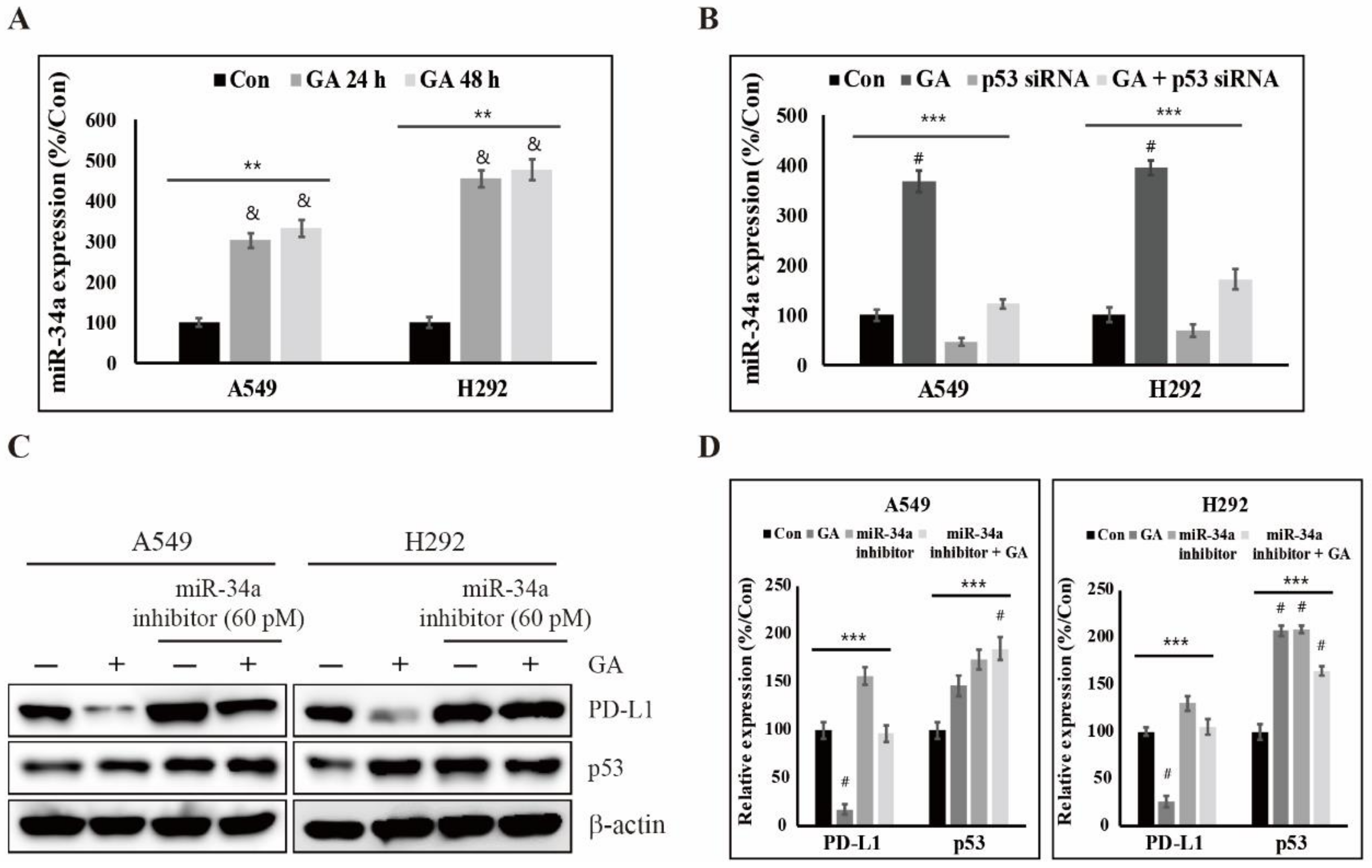
2.5. GA Upregulates p53-Dependent MiR-34a for Inhibiting the Expression of PD-L1
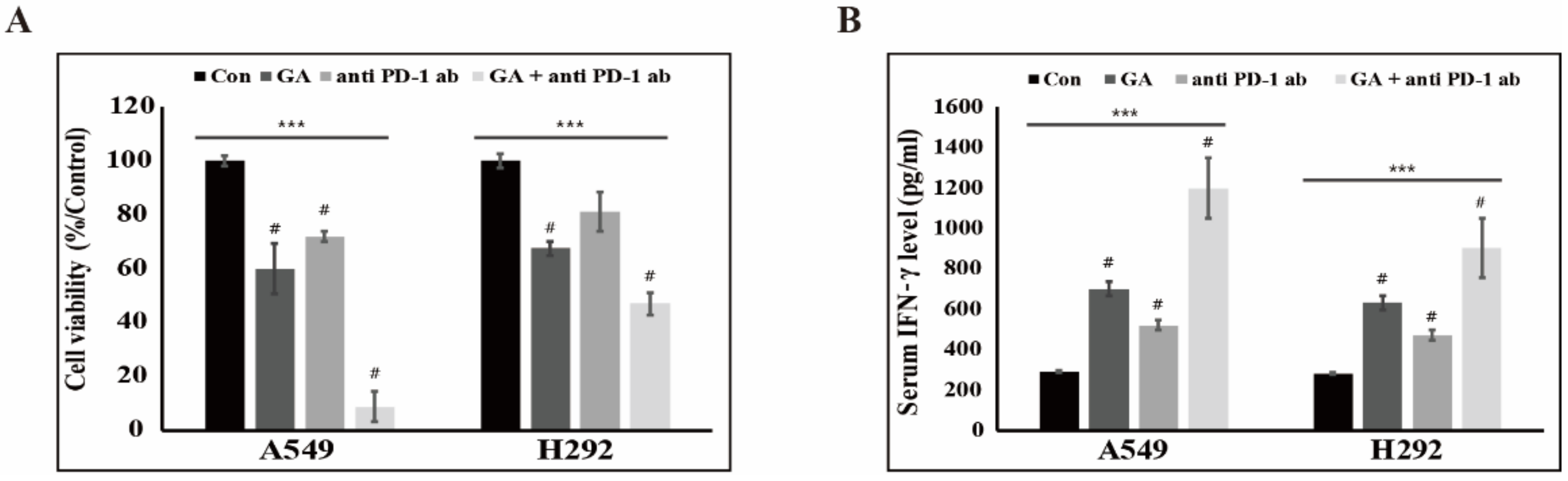
2.6. The Downregulation of PD-L1 Expression by GA Induces the Combination Effect with PD-1 Blockade
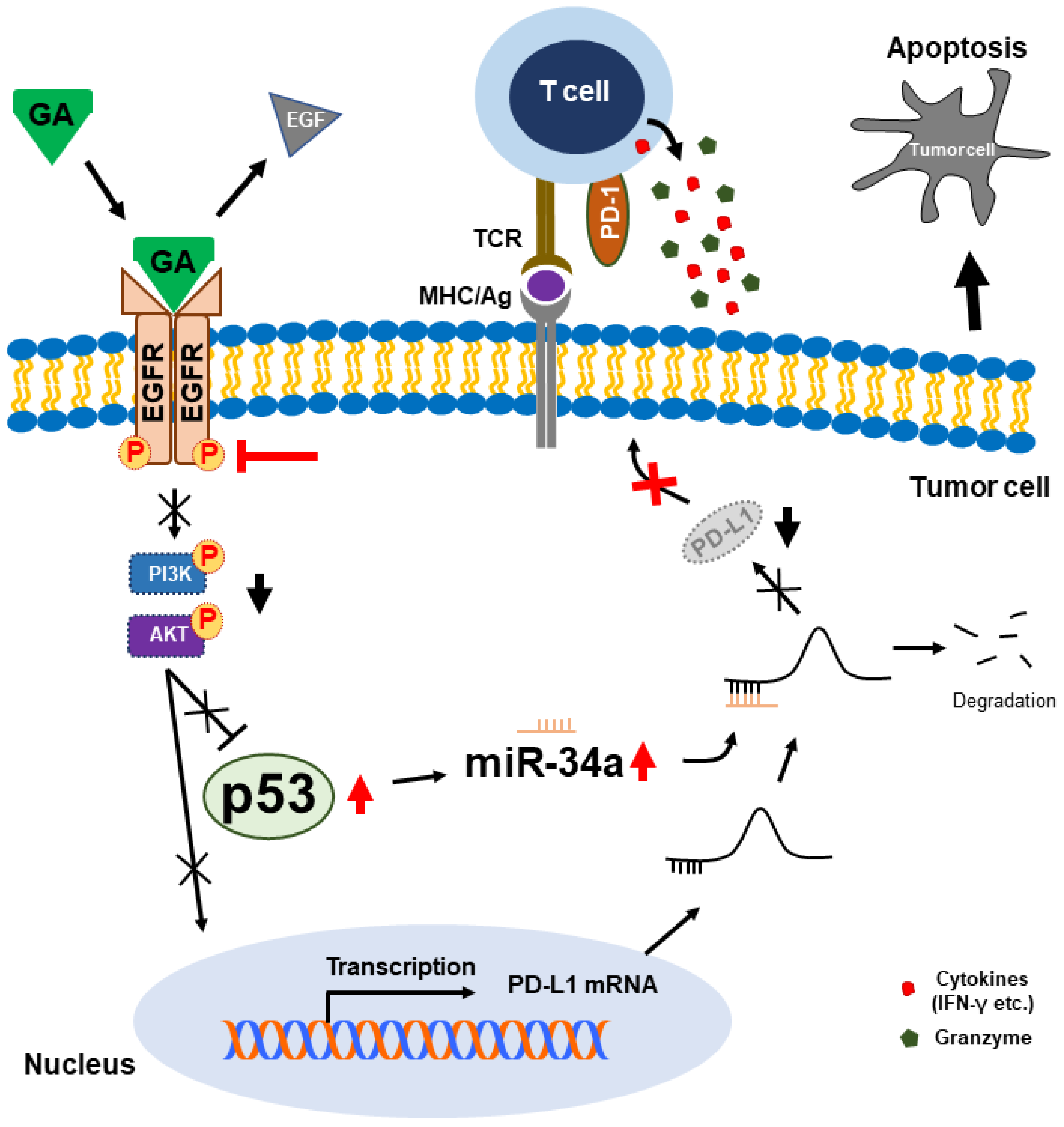
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Immunoblotting
4.3. Real Time Quantitative PCR (qPCR)
4.4. Transfections of siRNA and miRNA
4.5. NSCLC-Cell and PBMC Co-Culture Experiments
4.6. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reck, M.; Heigener, D.F.; Mok, T.; Soria, J.C.; Rabe, K.F. Management of non-small-cell lung cancer: Recent developments. Lancet 2013, 382, 709–719. [Google Scholar] [CrossRef]
- Bender, E. Epidemiology: The dominant malignancy. Nature 2014, 513, S2–S3. [Google Scholar] [CrossRef] [PubMed]
- Inamura, K. Lung Cancer: Understanding its molecular pathology and the 2015 WHO classification. Front. Oncol. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, E.; Travis, W.D.; Colby, T.V.; Corrin, B.; Shimosato, Y. The new World Health Organization classification of lung tumours. Eur. Respir. J. 2001, 18, 1059–1068. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Akerley, W.; Borghaei, H.; Chang, A.C.; Cheney, R.T.; Chirieac, L.R.; D’Amico, T.A.; Demmy, T.L.; Govindan, R.; Grannis, F.W.; et al. Non-Small Cell Lung Cancer, Version 2.2013 Featured Updates to the NCCN Guidelines. J. Natl. Compr. Cancer Netw. 2013, 11, 645–653. [Google Scholar] [CrossRef]
- Di, S.Y.; Fan, C.X.; Yang, Y.; Jiang, S.; Liang, M.M.; Wu, G.L.; Wang, B.D.; Xin, Z.L.; Hu, W.; Zhu, Y.F.; et al. Activation of endoplasmic reticulum stress is involved in the activity of icariin against human lung adenocarcinoma cells. Apoptosis 2015, 20, 1229–1241. [Google Scholar] [CrossRef]
- Ma, Z.Q.; Yang, Y.; Fan, C.X.; Han, J.; Wang, D.J.; Di, S.Y.; Hu, W.; Liu, D.; Li, X.F.; Reiter, R.J.; et al. Melatonin as a potential anticarcinogen for non-small-cell lung cancer. Oncotarget 2016, 7, 46768–46784. [Google Scholar] [CrossRef]
- Aung, T.N.; Qu, Z.P.; Kortschak, R.D.; Adelson, D.L. Understanding the Effectiveness of Natural Compound Mixtures in Cancer through Their Molecular Mode of Action. Int. J. Mol. Sci. 2017, 18, 656. [Google Scholar] [CrossRef]
- Shahrzad, S.; Aoyagi, K.; Winter, A.; Koyama, A.; Bitsch, I. Pharmacokinetics of gallic acid and its relative bioavailability from tea in healthy humans. J. Nutr. 2001, 131, 1207–1210. [Google Scholar] [CrossRef]
- Abdelwahed, A.; Bouhlel, I.; Skandrani, I.; Valenti, K.; Kadri, M.; Guiraud, P.; Steiman, R.; Mariotte, A.M.; Ghedira, K.; Laporte, F.; et al. Study of antimutagenic and antioxidant activities of gallic acid and 1,2,3,4,6-pentagalloylglucose from Pistacia lentiscus. Confirmation by microarray expression profiling. Chem. Biol. Interact. 2007, 165, 1–13. [Google Scholar] [CrossRef]
- Velderrain-Rodriguez, G.R.; Torres-Moreno, H.; Villegas-Ochoa, M.A.; Ayala-Zavala, J.F.; Robles-Zepeda, R.E.; Wall-Medrano, A.; Gonzalez-Aguilar, G.A. Gallic Acid Content and an Antioxidant Mechanism Are Responsible for the Antiproliferative Activity of ‘Ataulfo’ Mango Peel on LS180 Cells. Molecules 2018, 23, 695. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Han, Y.W.; Lee, S.T.; Jeong, H.J.; Kim, S.H.; Kim, I.H.; Lee, S.O.; Kim, D.G.; Kim, S.Z.; Park, W.H. A superoxide anion generator, pyrogallol, inhibits the growth of HeLa cells via cell cycle arrest and apoptosis. Mol. Carcinog. 2008, 47, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, E.; Succi, M.; Tipaldi, L.; Pannella, G.; Maiuro, L.; Sturchio, M.; Coppola, R.; Tremonte, P. Antimicrobial activity of gallic acid against food-related Pseudomonas strains and its use as biocontrol tool to improve the shelf life of fresh black truffles. Int. J. Food Microbiol. 2018, 266, 183–189. [Google Scholar] [CrossRef]
- Lee, J.H.; Oh, M.; Seok, J.H.; Kim, S.; Lee, D.B.; Bae, G.; Bae, H.I.; Bae, S.Y.; Hong, Y.M.; Kwon, S.O.; et al. Antiviral Effects of Black Raspberry (Rubus coreanus) Seed and Its Gallic Acid against Influenza Virus Infection. Viruses-Basel 2016, 8, 157. [Google Scholar] [CrossRef] [PubMed]
- Dludla, P.V.; Nkambule, B.B.; Jack, B.; Mkandla, Z.; Mutize, T.; Silvestri, S.; Orlando, P.; Tiano, L.; Louw, J.; Mazibuko-Mbeje, S.E. Inflammation and Oxidative Stress in an Obese State and the Protective Effects of Gallic Acid. Nutrients 2019, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- You, B.R.; Moon, H.J.; Han, Y.H.; Park, W.H. Gallic acid inhibits the growth of HeLa cervical cancer cells via apoptosis and/or necrosis. Food Chem. Toxicol. 2010, 48, 1334–1340. [Google Scholar] [CrossRef]
- Subramanian, A.P.; Jaganathan, S.K.; Mandal, M.; Supriyanto, E.; Muhamad, I.I. Gallic acid induced apoptotic events in HCT-15 colon cancer cells. World J. Gastroentero. 2016, 22, 3952–3961. [Google Scholar] [CrossRef]
- Tang, H.M.; Cheung, P.C.K. Gallic Acid Triggers Iron-Dependent Cell Death with Apoptotic, Ferroptotic, and Necroptotic Features. Toxins 2019, 11, 492. [Google Scholar] [CrossRef]
- Phan, A.N.; Hua, T.N.; Kim, M.K.; Vo, V.T.; Choi, J.W.; Kim, H.W.; Rho, J.K.; Kim, K.W.; Jeong, Y. Gallic acid inhibition of Src-Stat3 signaling overcomes acquired resistance to EGF receptor tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget 2016, 7, 54702–54713. [Google Scholar] [CrossRef]
- Liao, C.C.; Chen, S.C.; Huang, H.P.; Wang, C.J. Gallic acid inhibits bladder cancer cell proliferation and migration via regulating fatty acid synthase (FAS). J. Food Drug Anal. 2018, 26, 620–627. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Muhammad, N.; Steele, R.; Peng, G.; Ray, R.B. Immunomodulatory role of bitter melon extract in inhibition of head and neck squamous cell carcinoma growth. Oncotarget 2016, 7, 33202–33209. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Rajamanickam, S.; Deep, G.; Singh, M.; Agarwal, R.; Agarwal, C. Chemopreventive effects of oral gallic acid feeding on tumor growth and progression in TRAMP mice. Mol. Cancer Ther. 2008, 7, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, L.; Baumgaertner, P.; Devevre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8(+) T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Pauken, K.E.; Wherry, E.J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015, 36, 265–276. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl Med. 2016, 8, 328rv4–328rv4. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Swaika, A.; Hammond, W.A.; Joseph, R.W. Current state of anti-PD-L1 and anti-PD-1 agents in cancer therapy. Mol. Immunol. 2015, 67, 4–17. [Google Scholar] [CrossRef]
- Escors, D.; Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Garcia-Granda, M.J.; Vera, R.; Kochan, G. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct. Target. Ther. 2018, 3, 26. [Google Scholar] [CrossRef]
- Pascual, M.; Mena-Varas, M.; Robles, E.F.; Garcia-Barchino, M.J.; Panizo, C.; Hervas-Stubbs, S.; Alignani, D.; Sagardoy, A.; Martinez-Ferrandis, J.I.; Bunting, K.L.; et al. PD-1/PD-L1 immune checkpoint and p53 loss facilitate tumor progression in activated B-cell diffuse large B-cell lymphomas. Blood 2019, 133, 2401–2412. [Google Scholar] [CrossRef]
- Cha, Y.J.; Kim, H.R.; Lee, C.Y.; Cho, B.C.; Shim, H.S. Clinicopathological and prognostic significance of programmed cell death ligand-1 expression in lung adenocarcinoma and its relationship with p53 status. Lung Cancer 2016, 97, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Thiem, A.; Hesbacher, S.; Kneitz, H.; di Primio, T.; Heppt, M.V.; Hermanns, H.M.; Goebeler, M.; Meierjohann, S.; Houben, R.; Schrama, D. IFN-gamma-induced PD-L1 expression in melanoma depends on p53 expression. J. Exp. Clin. Cancer Res. 2019, 38, 397. [Google Scholar] [CrossRef] [PubMed]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, X.; Sun, R.; Tang, C.; Tzankov, A.; Zhang, J.; Manyam, G.C.; Xiao, M.; Miao, Y.; Jabbar, K.; et al. Clinical Significance of PTEN Deletion, Mutation, and Loss of PTEN Expression in De Novo Diffuse Large B-Cell Lymphoma. Neoplasia 2018, 20, 574–593. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Fillmore, C.M.; Koyama, S.; Wu, H.; Zhao, Y.; Chen, Z.; Herter-Sprie, G.S.; Akbay, E.A.; Tchaicha, J.H.; Altabef, A.; et al. Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer cell 2014, 25, 590–604. [Google Scholar] [CrossRef]
- Buchakjian, M.R.; Merritt, N.M.; Moose, D.L.; Dupuy, A.J.; Tanas, M.R.; Henry, M.D. A Trp53fl/flPtenfl/fl mouse model of undifferentiated pleomorphic sarcoma mediated by adeno-Cre injection and in vivo bioluminescence imaging. PLoS ONE 2017, 12, e0183469. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Tian, K.; Chen, X.; Zhang, R.; Mu, X.; Wu, Y.; Wang, D.; Wang, S.; Liu, F.; et al. Apigenin suppresses PD-L1 expression in melanoma and host dendritic cells to elicit synergistic therapeutic effects. J. Exp. Clin. Cancer Res. 2018, 37, 261. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Bell, D.W.; Lynch, T.J.; Haserlat, S.M.; Harris, P.L.; Okimoto, R.A.; Brannigan, B.W.; Sgroi, D.C.; Muir, B.; Riemenschneider, M.J.; Iacona, R.B.; et al. Epidermal growth factor receptor mutations and gene amplification in non-small-cell lung cancer: Molecular analysis of the IDEAL/INTACT gefitinib trials. J. Clin. Oncol. 2005, 23, 8081–8092. [Google Scholar] [CrossRef]
- Erlichman, C.; Hidalgo, M.; Boni, J.P.; Martins, P.; Quinn, S.E.; Zacharchuk, C.; Amorusi, P.; Adjei, A.A.; Rowinsky, E.K. Phase I study of EKB-569, an irreversible inhibitor of the epidermal growth factor receptor, in patients with advanced solid tumors. J. Clin. Oncol. 2006, 24, 2252–2260. [Google Scholar] [CrossRef]
- Sp, N.; Kang, D.Y.; Joung, Y.H.; Park, J.H.; Kim, W.S.; Lee, H.K.; Song, K.D.; Park, Y.M.; Yang, Y.M. Nobiletin Inhibits Angiogenesis by Regulating Src/FAK/STAT3-Mediated Signaling through PXN in ER+ Breast Cancer Cells. Int. J. Mol. Sci. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.Y.; Sp, N.; Kim, D.H.; Joung, Y.H.; Lee, H.G.; Park, Y.M.; Yang, Y.M. Salidroside inhibits migration, invasion and angiogenesis of MDAMB 231 TNBC cells by regulating EGFR/Jak2/STAT3 signaling via MMP2. Int. J. Oncol. 2018, 53, 877–885. [Google Scholar] [PubMed]
- Chen, J.; Jiang, C.C.; Jin, L.; Zhang, X.D. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann. Oncol. 2016, 27, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, S.; Ogura, K.; Yokoyama, S.; Saiki, I.; Hayakawa, Y. AKT-STAT3 Pathway as a Downstream Target of EGFR Signaling to Regulate PD-L1 Expression on NSCLC cells. J. Cancer 2016, 7, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Ogawara, Y.; Kishishita, S.; Obata, T.; Isazawa, Y.; Suzuki, T.; Tanaka, K.; Masuyama, N.; Gotoh, Y. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J. Biol. Chem. 2002, 277, 21843–21850. [Google Scholar] [CrossRef] [PubMed]
- Gong, A.Y.; Zhou, R.; Hu, G.; Li, X.; Splinter, P.L.; O’Hara, S.P.; LaRusso, N.F.; Soukup, G.A.; Dong, H.; Chen, X.M. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J. Immunol. 2009, 182, 1325–1333. [Google Scholar] [CrossRef]
- Guo, W.; Tan, W.; Liu, S.; Huang, X.; Lin, J.; Liang, R.; Su, L.; Su, Q.; Wang, C. MiR-570 inhibited the cell proliferation and invasion through directly targeting B7-H1 in hepatocellular carcinoma. Tumour Biol. 2015, 36, 9049–9057. [Google Scholar] [CrossRef]
- Lowe, S.W.; Bodis, S.; Mcclatchey, A.; Remington, L.; Ruley, H.E.; Fisher, D.E.; Housman, D.E.; Jacks, T. P53 Status and the Efficacy of Cancer-Therapy in-Vivo. Science 1994, 266, 807–810. [Google Scholar] [CrossRef]
- Santoro, A.; Vlachou, T.; Luzi, L.; Melloni, G.; Mazzarella, L.; D’Elia, E.; Aobuli, X.; Pasi, C.E.; Reavie, L.; Bonetti, P.; et al. p53 Loss in Breast Cancer Leads to Myc Activation, Increased Cell Plasticity, and Expression of a Mitotic Signature with Prognostic Value. Cell Rep. 2019, 26, 624. [Google Scholar] [CrossRef]
- Chiarugi, V.; Ruggiero, M. Role of three cancer “master genes” p53, bcl2 and c-myc on the apoptotic process. Tumori 1996, 82, 205–209. [Google Scholar]
- Espinosa, E.; Zamora, P.; Feliu, J.; Baron, M.G. Classification of anticancer drugs - a new system based on therapeutic targets. Cancer Treat. Rev. 2003, 29, 515–523. [Google Scholar] [CrossRef]
- Sun, J.C.; Wei, Q.; Zhou, Y.B.; Wang, J.Q.; Liu, Q.; Xu, H. A systematic analysis of FDA-approved anticancer drugs. BMC Syst. Biol. 2017, 11, 27–43. [Google Scholar] [CrossRef]
- Xu, X.L.; Gu, H.; Wang, Y.; Wang, J.; Qin, P. Autoencoder Based Feature Selection Method for Classification of Anticancer Drug Response. Front Genet. 2019, 10, 233. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Harrington, D.; Belani, C.P.; Langer, C.; Sandler, A.; Krook, J.; Zhu, J.M.; Johnson, D.H.; Grp, E.C.O. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. New Engl. J. Med. 2002, 346, 92–98. [Google Scholar] [CrossRef]
- Hanna, N.; Shepherd, F.A.; Fossella, F.V.; Pereira, J.R.; De Marinis, F.; von Pawel, J.; Gatzemeier, U.; Tsao, T.C.Y.; Pless, M.; Muller, T.; et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J. Clin. Oncol. 2004, 22, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Winton, T.; Livingston, R.; Johnson, D.; Rigas, J.; Johnston, M.; Butts, C.; Cormier, Y.; Goss, G.; Inculet, R.; Vallieres, E.; et al. Vinorelbine plus cisplatin vs. observation in resected non-small-cell lung cancer. New Engl. J. Med. 2005, 352, 2589–2597. [Google Scholar] [CrossRef]
- Le Chevalier, T.; Arriagada, R.; Le Pechoux, C.; Grunenwald, D.; Dunant, A.; Pignon, J.P.; Tarayre, M.; Abratt, R.; Arriagada, R.; Bergman, B.; et al. Cisplatin-based adjuvant chemotherapy in patients with completely resected non-small-cell lung cancer. New Engl. J. Med. 2004, 350, 351–360. [Google Scholar]
- Sp, N.; Kang, D.Y.; Kim, D.H.; Park, J.H.; Lee, H.G.; Kim, H.J.; Darvin, P.; Park, Y.M.; Yang, Y.M. Nobiletin Inhibits CD36-Dependent Tumor Angiogenesis, Migration, Invasion, and Sphere Formation Through the Cd36/Stat3/Nf-Kb Signaling Axis. Nutrients 2018, 10, 772. [Google Scholar] [CrossRef]
- Kang, D.Y.; Darvin, P.; Yoo, Y.B.; Joung, Y.H.; Sp, N.; Byun, H.J.; Yang, Y.M. Methylsulfonylmethane inhibits HER2 expression through STAT5b in breast cancer cells. Int. J. Oncol. 2016, 48, 836–842. [Google Scholar] [CrossRef]
- Sp, N.; Darvin, P.; Yoo, Y.B.; Joung, Y.H.; Kang, D.Y.; Kim, D.N.; Hwang, T.S.; Kim, S.Y.; Kim, W.S.; Lee, H.K.; et al. The combination of methylsulfonylmethane and tamoxifen inhibits the Jak2/STAT5b pathway and synergistically inhibits tumor growth and metastasis in ER-positive breast cancer xenografts. BMC Cancer 2015, 15, 474. [Google Scholar] [CrossRef]
- Schlessinger, J. Receptor Tyrosine Kinases: Legacy of the First Two Decades. Cold Spring Harbor Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Sforza, V.; Martinelli, E.; Ciardiello, F.; Gambardella, V.; Napolitano, S.; Martini, G.; Della Corte, C.; Cardone, C.; Ferrara, M.L.; Reginelli, A.; et al. Mechanisms of resistance to anti-epidermal growth factor receptor inhibitors in metastatic colorectal cancer. World J. Gastroenterol. 2016, 22, 6345–6361. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, J.Y.; Zhou, W.L.; Ren, Y.; Wang, X.X.; Chen, H.P.; Zhang, J.Y.; Chen, J.L.; Sun, Y.H.; Cui, L.J.; et al. Activation of an AKT/FOXM1/STMN1 pathway drives resistance to tyrosine kinase inhibitors in lung cancer. Brit. J. Cancer 2017, 117, 974–983. [Google Scholar] [CrossRef]
- Jacobsen, K.; Bertran-Alamillo, J.; Molina, M.A.; Teixido, C.; Karachaliou, N.; Pedersen, M.H.; Castellvi, J.; Garzon, M.; Codony-Servat, C.; Codony-Servat, J.; et al. Convergent Akt activation drives acquired EGFR inhibitor resistance in lung cancer. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Sionov, R.V.; Haupt, Y. The cellular response to p53: The decision between life and death. Oncogene 1999, 18, 6145–6157. [Google Scholar] [CrossRef]
- Burns, T.F.; El-Deiry, W.S. The p53 pathway and apoptosis. J. Cell Physiol. 1999, 181, 231–239. [Google Scholar] [CrossRef]
- Matsuda, K.; Tanikawa, C. The transcriptional landscape of p53 signaling pathway. Cancer Res. 2017, 77, 109–119. [Google Scholar]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef]
- Zheng, L.; Ren, J.Q.; Ll, H.; Kong, Z.L.; Zhu, H.G. Downregulation of wild-type p53 protein by HER-2/neu mediated PI3K pathway activation in human breast cancer cells: Its effect on cell proliferation and implication for therapy. Cell Res. 2004, 14, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Z.; Han, C.Y.; Duan, F.G.; Fan, X.X.; Yao, X.J.; Parks, R.J.; Tang, Y.J.; Wang, M.F.; Liu, L.; Tsang, B.K.; et al. p53 sensitizes chemoresistant non-small cell lung cancer via elevation of reactive oxygen species and suppression of EGFR/PI3K/AKT signaling. Cancer Cell Int. 2019, 19, 188. [Google Scholar] [CrossRef] [PubMed]
- Porta, C.; Hadj-Slimane, R.; Nejmeddine, M.; Pampin, M.; Tovey, M.G.; Espert, L.; Alvarez, S.; Chelbi-Alix, M.K. Interferons alpha and gamma induce p53-dependent and p53-independent apoptosis, respectively. Oncogene 2005, 24, 605–615. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Takaoka, A.; Hayakawa, S.; Yanai, H.; Stoiber, D.; Negishi, H.; Kikuchi, H.; Sasaki, S.; Imai, K.; Shibue, T.; Honda, K.; et al. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 2003, 424, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef]
- Menendez, D.; Shatz, M.; Azzam, K.; Garantziotis, S.; Fessler, M.B.; Resnick, M.A. The Toll-Like Receptor Gene Family Is Integrated into Human DNA Damage and p53 Networks. PLos Genet. 2011, 7, e1001360. [Google Scholar] [CrossRef]
- Shatz, M.; Menendez, D.; Resnick, M.A. The Human TLR Innate Immune Gene Family Is Differentially Influenced by DNA Stress and p53 Status in Cancer Cells. Cancer Res. 2012, 72, 3948–3957. [Google Scholar] [CrossRef]
- Textor, S.; Fiegler, N.; Arnold, A.; Porgador, A.; Hofmann, T.G.; Cerwenka, A. Human NK Cells Are Alerted to Induction of p53 in Cancer Cells by Upregulation of the NKG2D Ligands ULBP1 and ULBP2. Cancer Res. 2011, 71, 5998–6009. [Google Scholar] [CrossRef]
- Quan, X.W.; Li, X.L.; Yin, Z.H.; Ren, Y.W.; Zhou, B.S. p53/miR-30a-5p/SOX4 feedback loop mediates cellular proliferation, apoptosis, and migration of non-small-cell lung cancer. J. Cell Physiol. 2019, 234, 22884–22895. [Google Scholar] [CrossRef]
- Biamonte, F.; Battaglia, A.M.; Zolea, F.; Oliveira, D.M.; Aversa, I.; Santamaria, G.; Giovannone, E.D.; Rocco, G.; Viglietto, G.; Costanzo, F. Ferritin heavy subunit enhances apoptosis of non-small cell lung cancer cells through modulation of miR-125b/p53 axis. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef]
- Wu, Y.L.; Chen, W.Y.; Xu, Z.P.; Gu, W.Y. PD-L1 Distribution and Perspective for Cancer Immunotherapy-Blockade, Knockdown, or Inhibition. Front. Immunol. 2019, 10, 2022. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kang, D.Y.; Sp, N.; Jo, E.S.; Rugamba, A.; Hong, D.Y.; Lee, H.G.; Yoo, J.-S.; Liu, Q.; Jang, K.-J.; Yang, Y.M. The Inhibitory Mechanisms of Tumor PD-L1 Expression by Natural Bioactive Gallic Acid in Non-Small-Cell Lung Cancer (NSCLC) Cells. Cancers 2020, 12, 727. https://doi.org/10.3390/cancers12030727
Kang DY, Sp N, Jo ES, Rugamba A, Hong DY, Lee HG, Yoo J-S, Liu Q, Jang K-J, Yang YM. The Inhibitory Mechanisms of Tumor PD-L1 Expression by Natural Bioactive Gallic Acid in Non-Small-Cell Lung Cancer (NSCLC) Cells. Cancers. 2020; 12(3):727. https://doi.org/10.3390/cancers12030727
Chicago/Turabian StyleKang, Dong Young, Nipin Sp, Eun Seong Jo, Alexis Rugamba, Dae Young Hong, Hong Ghi Lee, Ji-Seung Yoo, Qing Liu, Kyoung-Jin Jang, and Young Mok Yang. 2020. "The Inhibitory Mechanisms of Tumor PD-L1 Expression by Natural Bioactive Gallic Acid in Non-Small-Cell Lung Cancer (NSCLC) Cells" Cancers 12, no. 3: 727. https://doi.org/10.3390/cancers12030727
APA StyleKang, D. Y., Sp, N., Jo, E. S., Rugamba, A., Hong, D. Y., Lee, H. G., Yoo, J.-S., Liu, Q., Jang, K.-J., & Yang, Y. M. (2020). The Inhibitory Mechanisms of Tumor PD-L1 Expression by Natural Bioactive Gallic Acid in Non-Small-Cell Lung Cancer (NSCLC) Cells. Cancers, 12(3), 727. https://doi.org/10.3390/cancers12030727