1. Introduction
Lung cancer has the highest mortality rate in Korea, and has done so for more than 10 years (Korean Government e-nara index,
www.index.go.kr, 28 January 2020). Patients with lung cancer are typically diagnosed at a late stage, after too much progress, because there are no special symptoms at an early stage. Therefore, the methods for lung cancer treatment at the end stage are very limited. Recently, immunotherapy has been considered for patients with end-stage lung cancer. Furthermore, immune anticancer drugs are in the spotlight as a new treatment that compensates the shortcomings of chemotherapy and targeted anticancer drugs. In particular, the contribution of immunotherapeutic agents to non-small cell lung cancer (NSCLC) is increasing [
1]. However, despite the benefits of immunotherapy, its use is limited for many lung cancer patients because of the cost and the requirement for treatment standards. The expression levels of programmed cell death ligand 1 (PD-L1) and other relevant biomarkers in NSCLC should be compatible with therapeutic standard [
2]. At present, immuno-chemotherapy is attracting attention as a next-generation therapy that helps patients overcome through enhancement of immunity.
Macrophages are a major regulator of inflammatory responses to disease and infections, and a main immune component of the tumor microenvironment [
3,
4]. Macrophages are polarized into specific phenotypes, generally described as the classically activated macrophage (M1) and the alternatively activated macrophage (M2), depending on the type of stimulant [
5,
6]. Traditionally, M1 and M2 reflect Type 1 and Type 2 helper T cell (T
h1 and T
h2) polarization of immune responses, respectively [
5,
7], and are associated with inflammatory and anti-inflammatory reactions, respectively [
8]. With respect to cancer, macrophage subsets have been found to be highly involved in the cancer microenvironment. M1 exhibits anticancer effects, but M2-polarized tumor-associated macrophages (TAMs) promote tumor growth, progression, angiogenesis, and metastasis in malignant cancers [
5,
9,
10,
11]. Developing molecules or modulators that properly regulate macrophage polarization may help to achieve positive results in cancer treatment.
Several therapeutic agents, such as Gefitinib and Imatinib, suppress M2-like polarization in NSCLC [
12,
13]. Nuclear Factor-kappa B (NF-κB) and/or signal transducer and activator of transcription (STAT) 1 are activated for M1 polarization, while STAT3, STAT6, peroxisome proliferator-activated receptor (PPAR) δ, and/or PPARγ are activated for M2 polarization [
14]. Activation of p38 mitogen-activated protein kinase (MAPK) is also involved in M2 polarization [
15]. Compared to M1 polarization, signaling pathways regarding M2 polarization are not fully established. The number of molecules identified as inhibitors of M2 polarization is also small.
Sea hares secrete many kinds of bioactive molecules, which have various bioactivities [
16,
17]. The immunological activities of sea hares have already been demonstrated through our previous studies as follows: sea hare-derived glycosaminoglycans induce macrophage activation [
18], and sea hare hydrolysate (SHH), which has higher bioactivities compared to extracts, reduce asthmatic parameters in mouse models of allergic asthma [
19]. In addition, sea hare-derived glycosaminoglycans show cytotoxicity in stomach cancer cell AGS, but not in normal mouse ileum epithelial cells (patent KR20110132746A). These findings led us to study the hypothesis that SHH may be able to treat lung cancer through the regulation of macrophages. This study was performed to identify the effects of SHH on macrophage polarization and on growth of NSCLC using mouse and human macrophages, human TAMs, and human NSCLC cells.
3. Discussion
TAMs, which are abundant in most types of malignant tumors, show an M2 profile. In this study, in TAMs, the expression levels of M2 markers were high (
Figure 3E). High infiltrations of M2 in tumor islets and stroma are associated with reduced overall survival in NSCLC [
25]. NSCLCs are relatively insensitive to radiotherapy and/or chemotherapy compared to small cell lung cancers (SCLCs) [
26]. The development of anti-cancer agents that reduce M2 function is needed to treat NSCLCs with drug resistance and malignant tendencies.
SHH have many benefits for treating lung cancer, namely, M1 activation, M2 suppression, growth inhibition, migration inhibition, and cytotoxicity in cancer cells. This study demonstrates the effects of SHH on the regulating macrophages in mouse macrophage cell line RAW264.7 cells, mouse primary peritoneal macrophages, a human monocytic cell line THP-1 cell, and human TAMs. M1 markers (iNOS and TNF-α) were upregulated and M2 markers (Arg-1 and Ym-1) were downregulated in mouse macrophages. In human macrophages (THP-1-differentiated macrophages), similary, SHH induced an increase in M1 markers (IL-1β, IL-6, and TNF-α) and a decrease in M2 markers (CD206, CD209, fibronectin-1, and IL-10). However, the expression pattern of macrophage markers differed by species and cell type. The mouse M2 marker Arg-1 was not detected in human THP-1-differentiated M2. SHH intrincically modulated the expression of macrophage markers in human macrophages, but not in mouse macrophages. On the other hand, SHH reduced the expression level of the M2 markers upregulated by IL-4 in mouse primary peritoneal macrophages. RAW264.7 cells predominantly show an M1 phenotype, which does not respond to IL-4 treatment (
Figure 1E). As a result, SHH have a regulatory effect on both mouse and human macrophages.
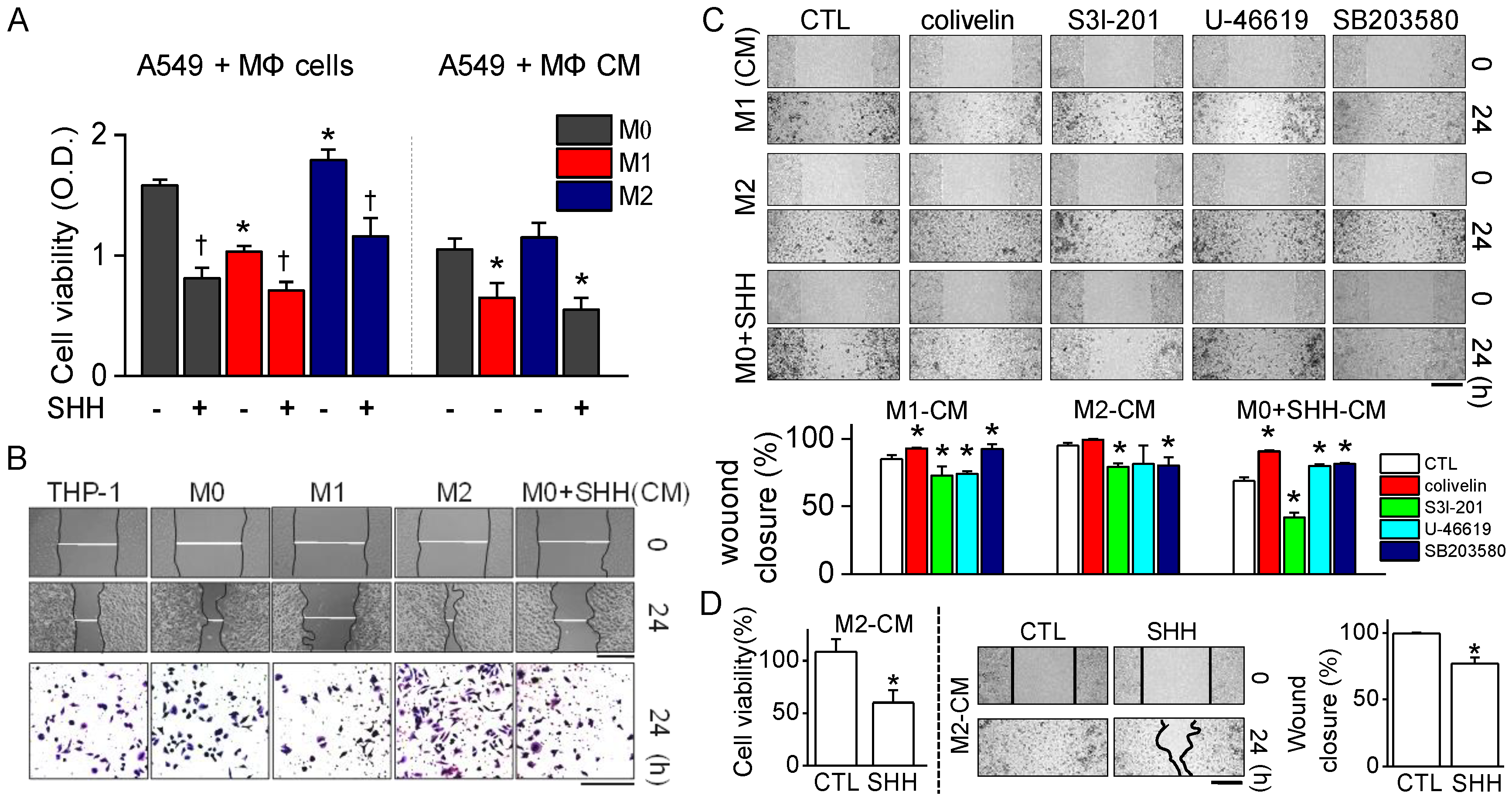
The anticancer effects of SHH on cell viability, colony formation, and cell cycle distribution were investigated in A549 cells. The effects were compared to macrophage conditioned medium and doxorubicin. In line with earlier studies, M1 reduced the cell viability and wound healing of A549 cells, whereas M2 enhanced them (
Figure 4). SHH reduced the cell viability and wound healing of A549 cells, like M1 did. The anticancer effect of SHH may be due to M1 activation, which shows high phagocytic ability toward cancer cells, oncolytic ability by protease secretion and inflammasome activation. SHH-activated M1 produced pro-inflammatory mediators such as IL-1β, IL-6, TNF-α, and NO. In addition, other molecules are also secreted from activated macrophages. These molecules, including pro-inflammatory cytokines, have the potential to induce lysis in cancer cells through STAT3 inhibition. SHH induces NF-κB activation for induction of M1. Activated STAT3 and NF-κB crosstalk to promote cancer progression [
27]. Activation of NF-κB by SHH may reduce the anticancer effect of SHH itself. Interestingly, however, SHH induces activation of NF-κB and inhibition of STAT3 in macrophages and NSCLC cells. As a result, various inflammatory cytokines secreted from M1 activated by SHH are likely to be involved in the anticancer effect of SHH through inhibition of STAT3 activation.
The cell viability and wound healing were regulated by p38 and STAT3 modulators; thus, p38 and STAT3 are promising targets for chemotherapy. Persistent phosphorylation of p38 and STAT3 has been reported in NSCLC. The constitutive activation of p38 and STAT3 is related to increase in cell proliferation and metastasis in NSCLC [
28,
29,
30], and inhibition of p38 and STAT3 reduces the metastatic potential of NSCLC [
30,
31]. In this study, however, there was no significant difference between the p38 activator and inhibitor treatments in the proliferation and migration of cancer cells. On the other hand, STAT3 activator recovered wound healing reduced by SHH, and its inhibitor enhanced SHH’s effect (
Figure 4). These results indicate that SHH’s anticancer effects are strongly mediated by low STAT3 activity.
In addition, the anticancer effect of SHH may be due to M2 suppression, which reduces tumor immunosurveillance escape, angiogenesis, and matrix remodelin, is also contributed. p38 inhbition downregulates the activation of M2 markers in mouse peritoneal macrophages [
15] and the human monocytic cell line U937 [
32]. STAT3 signaling polarizes macrophages toward M2 [
33], and inhibition of the IL-6/STAT3 signaling pathway turns macrophages into M1 [
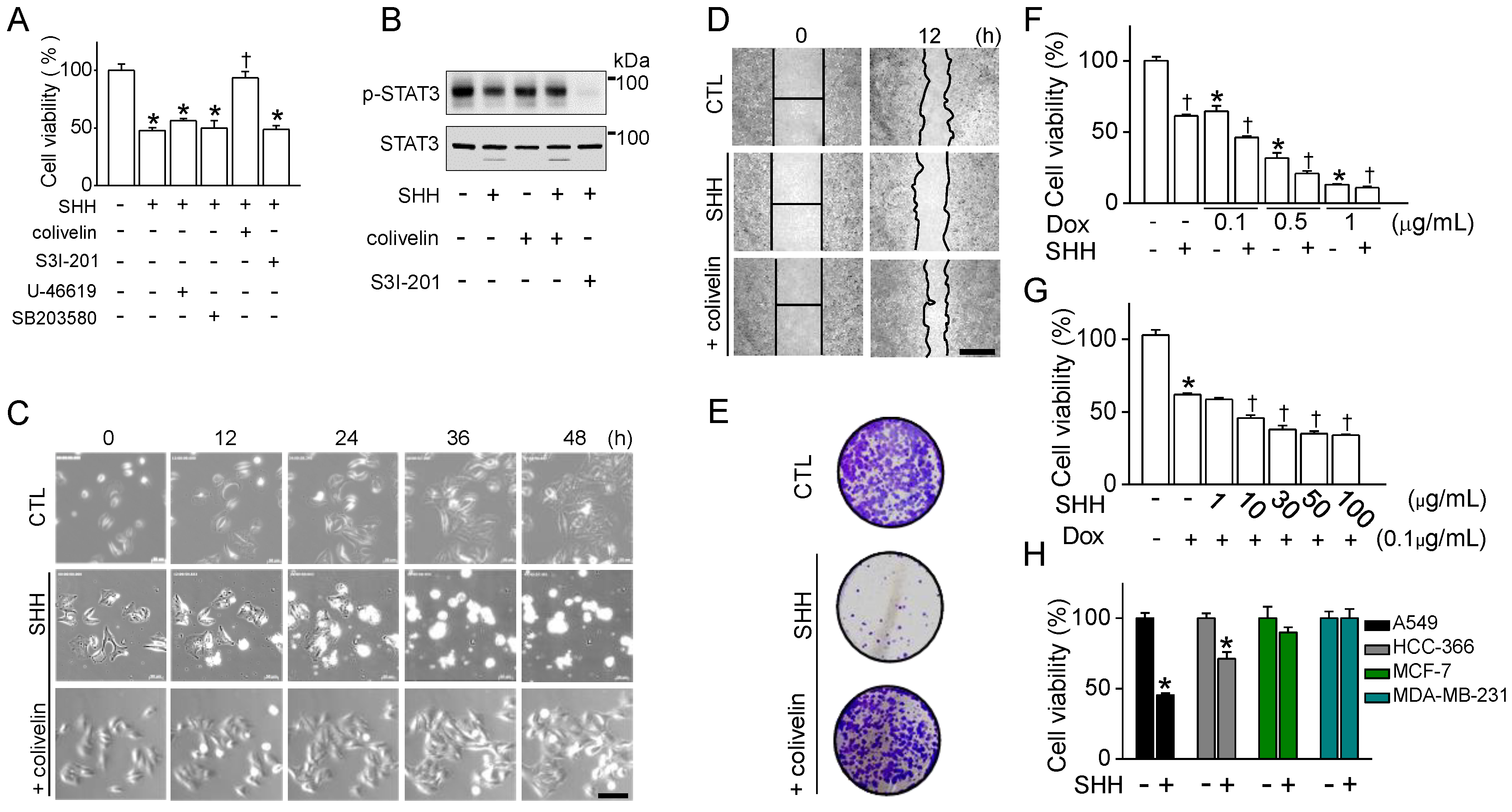
34]. In line with previous studies, p38 and STAT3 were involved in the regulation of mouse and human macrophage polarization. The expression levels of the M2 markers downregulated by SHH were restored by p38 and STAT3 activators in mouse, but not in human macrophages. In human macrophages, only STAT3 activator functions on M2 markers reduced by SHH. SHH reduced STAT3 activity in A549 cells, and the combination of the STAT3 activator and SHH reduced the effect of SHH on STAT3 phosphorylation, cell viability, wound healing, cell growth, colony formation, G2/M phase arrest, and caspase-1 activation (
Figure 4,
Figure 5,
Figure 6,
Figure 7 and
Figure 8). These results indicate that STAT3 is highly involved in SHH-induced suppression of human M2 and cancer cells.
SHH enhanced the cytotoxic effects of doxorubicin in A549 cells. The cytotoxic effect of SHH was more specific to NSCLC (A549 and HCC366) compared to breast cancer cells (MCF-7 and MDA-MB-231) (
Figure 6). In some NSCLC cells, blockade of STAT3 induces apoptois with the induction of Bax and the reduction of Bcl-2, which prevents cytochrome c release from mitochondria [
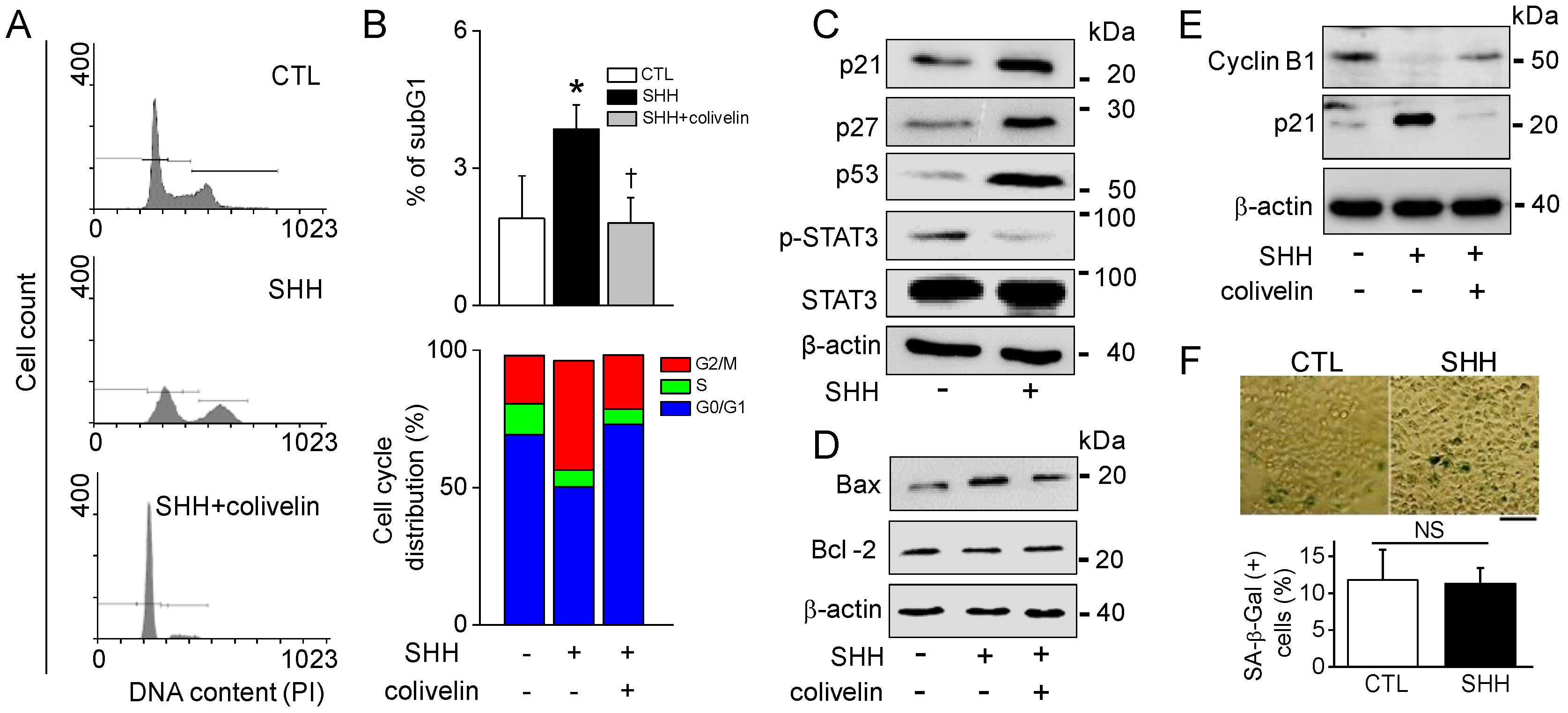
30]. In line with earlier studies, SHH reduced STAT3 activity, increased Bax expression, and decreased Bcl-2 (
Figure 7). In addition, SHH induced the upregulation of p21, p27, and p53 and the downregulation of cyclin B1. The STAT3 activator attenuated these effects. However, the caspase inhibitor failed to recover SHH-induced cell death. In addition, the apoptosis markers were not affected by SHH treatment; namely, Caspase 3/7 and 9 activity, PARP cleavage, cytochrome C release, and MMP changes. These results suggest that other mechanisms exist. Interestingly, however, SHH-treated cells showed positive signals in Annexin V/PI and TUNEL staining. Annexin V and TUNEL stainings are used for the monitoring of early and late apoptosis, respectively. SHH-treated macrophages showed M1 phenotypes with short spreading, vacuoles, and flat round shape. A549 cells also showed changed phenotypes with membrane blebbing and numerous vacuoles, like activated macrophages. Membrane damage and DNase release may explain Annexin V and TUNEL staining in the absence of changes in other apoptotic markers in SHH-treated A549 cells (
Figure 8). Necrotic cells stained by PI were also seen in the SHH-treated cells. In addition, SHH-treated A549 cells induced IL-1β secretion and caspase-1 activation (
Figure 8E). Senescence events and autophagy markers were not consistently detected in SHH-treated A549 cells (
Figure 8F).
Pyroptosis and necroptosis are mechanisms of regulated cell death distinct from apoptosis. Lytic cell death requires gasdermin D (GSDMD) for pyroptosis and mixed lineage kinase domain like pseudokinase (MLKL) for necroptosis. Both GSDMD and MLKL result in IL-1β-driven inflammation, which is from caspase-1 activation [
35]. Pyroptotic and necroptotic cell death show distinct morphological differences; reduced cell swelling and flattened cytoplasm in pyroptosis, and cell swelling and subsequent osmolysis in necroptosis. However, both cell death mechanisms have in common that they damage the plasma membrane [
36,
37]. Annexin V staining could not distinguish between apoptosis, pyroptosis, or necroptosis. In this study, SHH-treated cells showed lytic cell death with reduced cell volume, membrane alteration, Caspase-1 activation, and IL-1β secretion. The pyroptotic and necroptotic death pathways are thought to be crosstalked. Macrophages undergoing pyroptosis exhibit both apoptotic and necrotic morphological characteristics [
38]. Z-YVAD-FMK and necrostatin-1, which block pyroptosis and necroptosis, inhibited SHH-induced cell death. However, the chemicals were not fully recovered cell death induced by SHH (
Figure 8J), indicating that other mechanism may exist in SHH-induced cell death.
Several immunotherapies have been widely applied to cancer to inhibit malignant activity or to activate the immune system against the immune evasion mechanism of cancer [
39,
40,
41]. Most natural immune modulators induce M1 or M2 polarization in one direction, but SHH beneficially affect both M1 and M2 polarization in macrophages for treating cancer. Collectively, our results suggest that SHH may be applied as a therapeutic substance for the activation of the immunocompromised environment in relation to cancer progression. A better understanding of the mechanisms and physiological roles of pyroptosis and necroptosis will be able to explain the mechanism by which SHH cause cancer cell death and anti-cancer immune responses. In addition, the effect of SHH on PD-1 and PD-L1 expression should be investigated in further studies.
4. Materials and Methods
4.1. Ethical Approval and Chemicals
All experiments were performed with the approval of the Ethics Committee and in accordance with the guidelines of the Institutional Animal Care and Use Committee of Gyeongsang National University (GNU-160811-M0036). Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich (St Louis, MO, USA). Stock solutions of colivelin (378 µM; Tocris Bioscience, Bristol, UK), doxorubicin (2 mg/mL), and lipopolysaccharide (LPS; 5 mg/mL) were prepared in distilled water. The mouse IL-4 (0.1 mg/mL), human IL-4 (0.1 mg/mL), human IL-13 (0.1 mg/mL), and human IFN-γ (0.1 mg/mL), which were purchased from PeproTech (Rocky Hill, NJ, USA), were dissolved in distilled water. Bay 11-7085 (50 mM), necrostatin-1 (20 mg/mL; Cayman Chemical, Ann Arbor, MI, USA), PMA (1 mM), SB203580 (25 mM), S3l-201 (50 mM; Selleckchem, Houston, TX, USA), Z-VAD-FMK (10 mg/mL; InvivoGen, San Diego, CA, USA), and Z-YVAD-FMK (20 mM; Abcam, Cambridge, MA, USA) were dissolved in dimethyl sulfoxide (DMSO). U-46619 (28.53 mM) was prepared in methyl acetate. The solutions were then diluted in culture medium to the working concentration. An equivalent concentration or volume of solvents was added in the control group. The final concentrations of DMSO and methyl acetate were ~0.1% (v/v).
4.2. Preparation of Sea Hare Hydrolysates
The preparation of SHH was performed as previously described [
19]. Briefly, sea hare (
Aplysia kurodai) was washed, blanched, and minced. Flavourzyme (2%) was directly added to the minced sea hare. The minced sea hare in the enzyme solution was adjusted to pH 6.0 with citric acid powder, incubated at 60 °C for 15 h, boiled at 100 °C for 10 min to inactivate the enzyme reaction, and then filtered with a 200 mesh screen sieve. The filtered solution was concentrated to Brix 50 using a rotary vacuum evaporator (WCR-P6, Daihan Scientific Co., Ltd, Wonju, Korea). Three volumes of ethanol were added to the concentrated hydrolysates. The hydrolysate solution was stirred and left to stand for 12 h until precipitation. The precipitate was freeze-dried, ground into powder, and stored at −70 °C until further analysis. At the time of the experiment, the dried materials were dissolved in distilled water at the indicated concentrations.
4.3. Cell Culture and Isolation of Peritoneal Macrophages
The RAW264.7 macrophage cell line derived from Abelson murine leukemia virus-induced tumors was obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). Human non-small cell lung cancer (NSCLC) cell lines (A549 and HCC-366), human breast cancer cell lines (MCF-7 and MDA-MB-231), and the human monocytic THP-1 cell line were obtained from Korean Cell Line Bank (KCLB, Seoul, Korea). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco-BRL, Gaithersburg, MD, USA) or RPMI 1640 (Lonza, Walkersville, MD, USA; for THP-1), supplemented with 10% fetal bovine serum (FBS; Gibco-BRL) and 1% penicillin/streptomycin (100 μg/mL), in a humidified atmosphere with 5% CO2 at 37 °C. The medium was replaced every 2 days.
Peritoneal macrophages were isolated from C57BL/6 mice (6-week-old, male,
n = 10) purchased from Koatech Co. (Animal Breeding Center, Pyongtaec, Korea), as previously described [
42]. Briefly, 1 mL of 3% Brewer thioglycollate broth (BD Biosciences, Franklin Lakes, NJ, USA) was injected per mouse into the peritoneal cavity. After four days of injection, the peritoneal cells were collected by injecting and aspirating 10 mL PBS from the peritoneum. To evaluate the purity of the isolated macrophages, the peritoneal cells were examined using a flow cytometer, as detailed in
Section 4.13.
4.4. Macrophage Polarization and Generation of Tumor-Associated Macrophages (TAMs)
THP-1 cells (2 × 105 cells/mL) were seeded in a 6-well plate and cultured for 24 h in RPMI medium. The cultured cells were differentiated into a macrophage-like phenotype by adding 150 nM PMA. After 24 h, the macrophages were polarized into M1 in the presence of 100 ng/mL LPS and 20 ng/mL IFN-γ for 48 h, or into M2 by adding 20 ng/mL IL-4 and 20 ng/mL IL-13 for 72 h. SHH (100 μg/mL) was treated to M0 alone or in combination with M1 or M2 inducers for 48 h or 72 h. The supernatant of each group was harvested just before media change to prepare the conditioned medium. THP-1-differentiated macrophages were co-cultured with A549 cells at a 1:1 ratio for 4 days to generate TAMs.
4.5. Cell Viability and Real-time Live Cell Imaging
Cell viability was determined colorimetrically using the 3-(4, 5-dimethylthiazole-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT) reagent (Duchefa, Haarlem, The Netherlands). The MTT assay procedures were performed as previously described [
43]. Briefly, cells at the exponential phase were seeded (2 × 10
4 cells/mL) in a 96-well plate. After treatment with SHH, 10 μL of 5 mg/mL MTT solution was added to each well and incubated for 2 h. The supernatants were then aspirated, the formazan crystals in each well were dissolved in 100 μL of DMSO for 10 min at room temperature, and the plate was read at 570 nm using a microplate reader (Molecular Devices, San Jose, CA, USA). Data are expressed as the optical density or percentage of viable cells compared to the control.
Real-time live cell imaging was performed using Cell Observer (Carl Zeiss, Jena, Germany). Briefly, the cells were seeded at the density of 1.5 × 103 cells/well on Culture-Insert 2-well (ibidi GmbH, Martinsried, Germany) and incubated in a humidified atmosphere with 5% CO2 at 37 °C for 48 h. The cells were treated with 100 µg/mL SHH, colivelin was pretreated for 2 h, and then transferred to the Cell Observer. The cells were maintained at 37 °C in 5% CO2. Images were captured at 10 min intervals for 48 h. Cells cultured in the medium without adding SHH were taken as a control.
4.6. Live/dead Cell Staining
Live/dead cell staining was performed using calcein-AM (Thermo Fisher Scientific, Eugene, OR, USA) and propidium iodide (PI). Viable cells and disordered areas of dead cell membrane were stained with Calcein-AM and PI and appeared green and red, respectively. A549 cells at the density of 5 × 103 cells/100 μL were cultured on glass-bottomed culture dish (SPL, Pocheon, Korea) for 48 h. The cells were treated with 100 µg/mL of SHH for 24 h, 48 h, or 72 h. The cells were washed twice with free DMEM and then stained with 2 μM calcein-AM and 3 μg/mL PI for 10 min at 37 °C. The stained cells were washed three times and observed using a confocal laser scanning microscope (Olympus, Tokyo, Japan) with filter sets for fluorescein isothiocyanate (FITC) and Texas Red.
4.7. Reverse Transcriptase (RT)—Polymerase Chain Reaction (PCR) Analysis
Total RNA from the cells was extracted with Trizol reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. First strand cDNA was synthesized from the total RNA (3 μg) isolated from the RAW264.7 cells, the mouse peritoneal and human THP-1-differentiated macrophages, and the TAMs using oligo dT (DiaStar RT Kit, SolGent, Daejeon, Korea). The first-strand cDNA was quantified using a spectrophotometer (NanoDrop
® ND-1000, NanoDrop Technologies, Wilmington, DE, USA). The quantified cDNA was used as a template for PCR amplification with G-
Taq polymerase (Cosmogenetech, Seoul, Korea). PCR assay was performed with specific primers for M1 and M2 polarization markers. The specific primer sequences are listed in
Table 1. The PCR steps included initial denaturation at 94 °C for 5 min, then 32 cycles at 94 °C for 30 s, 55 °C to 60 °C (see
Table 1) for 30 sec, 72 °C for 30 sec, and a final extension step at 72 °C for 10 min. The amplified PCR products were separated in 1.5% agarose gel stained with ethidium bromide. The bands obtained by RT-PCR were extracted and directly sequenced with an ABI PRISM
® 3100-Avant Genetic Analyzer (Applied Biosystems, Carlsbad, CA, USA).
4.8. Western Blot Analysis
Peritoneal and THP-1-differentiated macrophage cells and A549 cells were lysed in RIPA buffer (25 mM Tris-HCl; pH 7.4, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS) containing protease and phosphatase inhibitors. The procedures of Western blot analysis were as described previously [
43]. Briefly, the cell lysates were incubated for 30 min on ice with intermittent vortexing and were clarified by centrifugation at 15,000 rpm (22,250×
g; Micro 17TR, Hanil, Incheon, Korea) for 20 min at 4 °C. After centrifugation, the supernatant was separated and stored at −70 °C until use. Protein concentration was quantified using a Pierce bicinchoninic acid (BCA) Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts (35 μg) of total protein were analyzed among experimenal groups. Equal volumes of the supernatant and 2× sodium dodecyl sulfate (SDS) sample buffer were mixed, loaded on 10% SDS-polyacrylamide gel, and separated by electrophoresis for 120 min at 120 V. Then, the gel was transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA, USA) for 1 h at 100 V using a wet transfer system (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% fat-free dry milk and then incubated with anti-phospho-p38 MAPK pThr180+Tyr182 (1:1000 dilution; Thermo Fisher Scientific), anti-p38 MAPK (1:1000 dilution; Thermo Fisher Scientific), anti-phospho-STAT3 (Tyr705, 1:2000 dilution; Cell Signaling, Danvers, MA, USA), anti-STAT3 (1:1000 dilution; Cell signaling), anti-caspase-1 (1:1000 dilution; Abcam), anti-p21 (1:1000 dilution; Santa Cruz Biotechnology, Dallas, TX, USA), anti-p27 (1:1000 dilution; Santa Cruz Biotechnology), anti-p53 (1:1000 dilution; Santa Cruz Biotechnology), anti-Cyclin B1 (1:1000 dilution; Cell signaling) or anti-β-actin (1:5000 dilution). The primary antibody incubation was followed by incubation with a secondary peroxidase conjugated anti-rabbit or anti-mouse antibody at 1:5000 (Assay designs, Ann Arbor, MI, USA). Immuno-positive bands were visualized by enhanced chemiluminescence reagent (Dogen, Seoul, Korea) according to the manufacturer’s instructions.
4.9. Phagocytosis Assay
The phagocytic ability of macrophages was analyzed using a phagocytosis assay kit (Cayman Chemical) according to a previously described method [
18]. Briefly, 5 × 10
4 cells/well RAW 264.7 cells were cultured in a 96-well black-bottomed plate. The cultured cells were stimulated with LPS (1 μg/mL) or SHH (1, 10, and 100 μg/mL). Then, 10 μL of the latex beads rabbit IgG FITC solution was added and incubated for 2 h. The cells were washed three times with the assay buffer. The washed cells were analyzed for fluorescence intensity using a fluorescence microplate reader (Tecan, Maennendorf, Switzerland). The phagocytic ability of macrophages was also analyzed through co-culture with A549 cells, which were transfected with green fluorescent protein (GFP) in pcDNA3.1 using LipofectAMINE2000 (Thermo Fisher Scientific) and Opti-MEM I (Thermo Fisher Scientific). The A549 cells were added to RAW264.7 cells cultured in a 35 mm cell imaging dish with a glass coverslip bottom, and SHH was added to the dish. Green fluorescence from cells expressing GFP was detected with a confocal laser scanning microscope equipped with a fluorescence system (IX70 Fluoview, Olympus).
4.10. Migration Assay
For wound-healing (two-dimensional (2D)) migration assays, cells were seeded onto a 24-well culture dish at a density of 2 × 105 cells/well. Cells were scratched with a sterile pipette tip, washed with PBS, and incubated in fresh medium for 24 h. Another assay was performed using Culture-Insert 2-well (ibidi GmbH) according to the manufacturer’s protocol. Briefly, A549 cell suspension at the density of 66 × 104 cells/mL (70 µL) was seeded into each well, and the cells were incubated for 24 h, then washed three times with free medium. After washing, the cells were captured as the initial images at 0 h. Macrophage conditioned media were added to the cells and incubated for 12 or 24 h at 37 °C. Photomicrographs were taken after incubation for the periods indicated. Relative cell migration distance (wound closure) was determined by measuring the wound width on the monolayer using AxioVision 4.5 software under a microscope (Axiovert 40C, Carl Zeiss MicroImaging, Göttingen, Germany) and by dividing this value from the initial value (i.e., the initial wound width at 0 h). The data acquired from the three scratches on each well were converted to the percentage of wound closure at a given time.
The transwell (three-dimensional (3D)) migration assay was performed using the CytoSelect™ Cell Migration Assay Kit containing polycarbonate membrane inserts (0.8 μm pore membrane; Cell Biolabs, Inc., CA, USA) according to the manufacturer’s instructions. The cell suspension (2 × 104 cells/300 μL) in macrophage conditioned media was added to the upper chamber of each insert. The lower chamber contained medium with 10% FBS to allow cell migration toward the lower face of the transwell culture inserts. Cells were incubated for 24 h at 37 °C in a 95% air—5% CO2 gas mixture. Non-migrating cells on the inner side of the transwell culture inserts were gently removed with a cotton-tipped swab. Migrated cells remaining on the bottom surface were stained with a cell-staining solution (0.4% crystal violet) for 10 min at room temperature. Photomicrographs of five individual fields per insert were taken using a microscope (Axiovert 40C).
4.11. Colony Formation Assay
A549 cells at the density of 5 × 103/well were seeded in a 6-well plate and grown in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin. After 24 h, different concentrations of SHH were added to the cells for 48 h. The medium was replaced with a fresh medium every 2 days for 14 days. On the last day, the colonies were fixed in methanol for 10 min. The methanol was removed and the colonies were air-dried for 30 min until white in color. After that, the colonies were stained with 0.1% crystal violet solution for 30 min. The stain solution was removed, and the colonies were washed five times with distilled water. Finally, the colonies were air-dried and captured.
4.12. Tumor Spheroid Assay
The preparation of 1.5% agarose and spheroid formation was followed as previously described [
44]. Agarose/DMEM mixture (1.5%) was added to a 96-well plate to generate agarose-coated well. The prepared A549 single cell suspension was added into each well of the agarose-coated plate (2.5 × 10
3/200 μL/well). The cell suspensions were incubated for 4 days in a humidified atmosphere with 5% CO
2 at 37 °C. After 4 days, the supernatant of 100 μL was replaced with SHH-supplemented media, and then cultured for 10 days. Finally, the spheroid images were captured using AxioVision 4.5 software under a microscope (Axiovert 40C).
4.13. Flow Cytometric Analysis and Staining
Flow cytometric analysis was performed using a BD FACSAria II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA) with FACSDiva software version 6.1.2 (BD Biosciences). The analysis of mouse peritoneal macrophages was performed using the following antibody: PE-conjugated anti-mouse CD14 (Biolegend, San Diego, CA, USA). The cells and 1 μL of anti-CD14 PE antibody were incubated for 40 min at 4 °C in the dark. The cells were then washed twice to remove unbound antibodies using washing buffer (PBS with 1% BSA). Subsequently, the cells were analyzed by flow cytometry with fixation (4% paraformaldehyde).
A549 cells were cultured at a density of 8 × 104 cells/well in a 60 mm dish for 48 h. The cells were treated with 100 μg/mL SHH for 48 h, trypsinized, fixed with cold 70% ethanol, and incubated for 1 h at −20 °C. The cells were pelleted and resuspended in 1 mL PBS containing PI (1 mg/mL) and RNase A (1 mg/mL). Following incubation at 25 °C for 30 min, cell cycle and apoptosis (subG1) were determined by a flow cytometer (FACscan, BD Biosciences) with CXP 2.2 software (Beckman Coulter, Brea, CA, USA).
4.14. Cellular Senescence Assay
Cellular senescence assay was performed according to the manufacturer’s protocol (BiMilpitas, CA, USA). Briefly, A549 cells at density of 9 × 103 cells/mL were seeded in a 24-well plate and incubated at 37 °C and 5% CO2 for 24 h, and the cells were further incubated in the presence of SHH treatment for 48 h. The cells were washed once with 1mL of 1 × PBS, and were fixed with 0.5 mL of a fixative solution for 15 min at room temperature. The fixed cells were washed twice with 1 mL of 1× PBS. Finally, a staining solution mixture (470 µL of staining solution + 5 µL of staining supplement + 25 µL of 20 mg/mL X-gal in DMF) was added to each well, and the plate was incubated overnight at 37 °C. The images were captured using AxioVision 4.5 software under a microscope (Axiovert 40C).
4.15. Caspase 3/7 Assay
A549 cells at a density of 3.5 × 104/mL were seeded in a 96-well black-bottomed plate. The cells were grown in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin for 48 h. The cells were treated with 100 µg/mL of SHH for 48 h. To detect caspase-3 and -7 activity in the cell-based assay, caspase-Glo® 3/7 reagent (Caspase-Glo® 3/7 substrate + Caspase-Glo® buffer; Promega, Madison, WI, USA) was added to the cells in the medium (100 μL of medium + 100 μL of reagent), mixed by orbital shaking (300–500 rpm) for 30 sec, and incubated for 1 h at room temperature. The caspase activity was analyzed by measuring the luminescence of each sample in the GloMax® microplate reader (Promega).
4.16. TUNEL Staining
TUNEL assay was performed using the DeadEnd™ Fluorometric TUNEL System (Promega) according to the manufacturer’s protocol. Briefly, cells grown on poly-L-lysine-coated cover glass were fixed in 4% paraformaldehyde in PBS for 25 min at 4 °C, washed twice in PBS, and permeabilized in 0.2% Triton X-100 in PBS for 5 min. After the two washes in PBS, the cells were equilibrated in an equilibration buffer for 10 min. The cells were labeled with TdT reaction mix and incubated for 60 min at 37 °C in a dark humidified chamber. The reaction was stopped with 2× saline sodium citrate (SSC) solution, followed by washing three time in PBS. Counter staining was carried out by incubation with 2 μg/mL DAPI for 10 min at room temperature in the dark. TUNEL-positive green fluorescent cells were observed using a confocal laser scanning microscope (Olympus).
4.17. Annexin V/PI Staining
Annexin V staining was performed using the Annexin-V-FLUOS staing kit (Roche, Mannheim, Germany) according to the manufacturer’s protocol. Briefly, cells grown on a glass-bottomed culture dish (SPL) were labeled with Annexin-V-Fluos labeling reagent and PI, and then incubated for 15 min at room temperature in the dark. Following incubation, the cells were analyzed using a confocal laser scanning microscope (Olympus).
4.18. Caspase-1 Assay
Active caspase-1 was detected using the caspase-1 staining kit (Abcam) according to the manufacturer’s protocol. Briefly, cells grown on poly-L-lysine-coated cover glass were treated with chemicals for 48 h and washed twice with a washing buffer. The FAM-YVAD-FMK was added to the cells in 150 μL of washing buffer at a 1:150 ratio, and the cells were incubated for 60 min at room temperature with 0.3 µL of 500X Hoechst solution. The stained cells were washed and were mounted with permount mounting medium (Fisher Chemical, Geel, Belgium), and then analyzed using a confocal laser scanning microscope (Olympus).
4.19. IL-1β Immunoassay
The level of IL-1β in the medium was measured using an enzyme-linked immunosorbent assay (ELISA) kit (R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s protocol. Biefly, cells (5 × 105 cells/mL) were cultured in a 60 mm culture dish, incubated overnight, and stimulated with SHH (100 μg/mL) for 48 h. Cell supernatants were then collected. Cell supernatants, 50 μL of assay diluent, and the standard were added to the a 96-well plate, which was pre-coated with anti-human IL-1β antibody. The plates were covered with an adhesive strip, incubated for 2 h at room temperature, and washed three times with wash buffer. Then, 100 μL of human IL-1β conjugate was added, incubated at room temperature for 2 h, and washed three times. The reaction was quenched by the addition of 100 μL stop solution, and the absorbance of the plates was read at 450 nm with a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
4.20. Data Analysis and Statistical Analysis
A LAS-4000 (Fujifilm Corp, Tokyo, Japan) and MaXidoc Gel Imaging System (Daihan Scientific, Wonju, Korea) were used to capture images of the Western blots and agarose gel, respectively. The bands were quantified by ImageJ software (version 1.49, National Institute of Health, Bethesda, MD, USA). Data are presented as the mean ± standard deviation (SD). The one-way ANOVA/Bonferroni test or the Kruskal-Wallis/Mann-Whitney test was chosen after the normality test to analyze the differences among groups (OriginPro2020, OriginLab Corp. Northampton, MA, USA). Significance was set at p < 0.05.

 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}