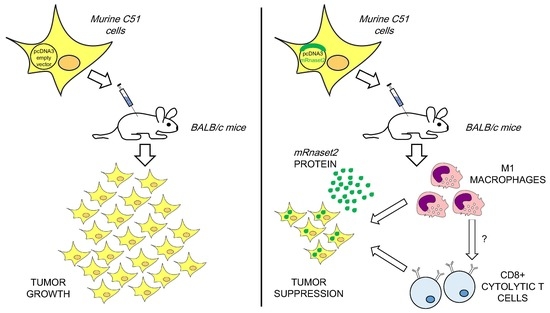
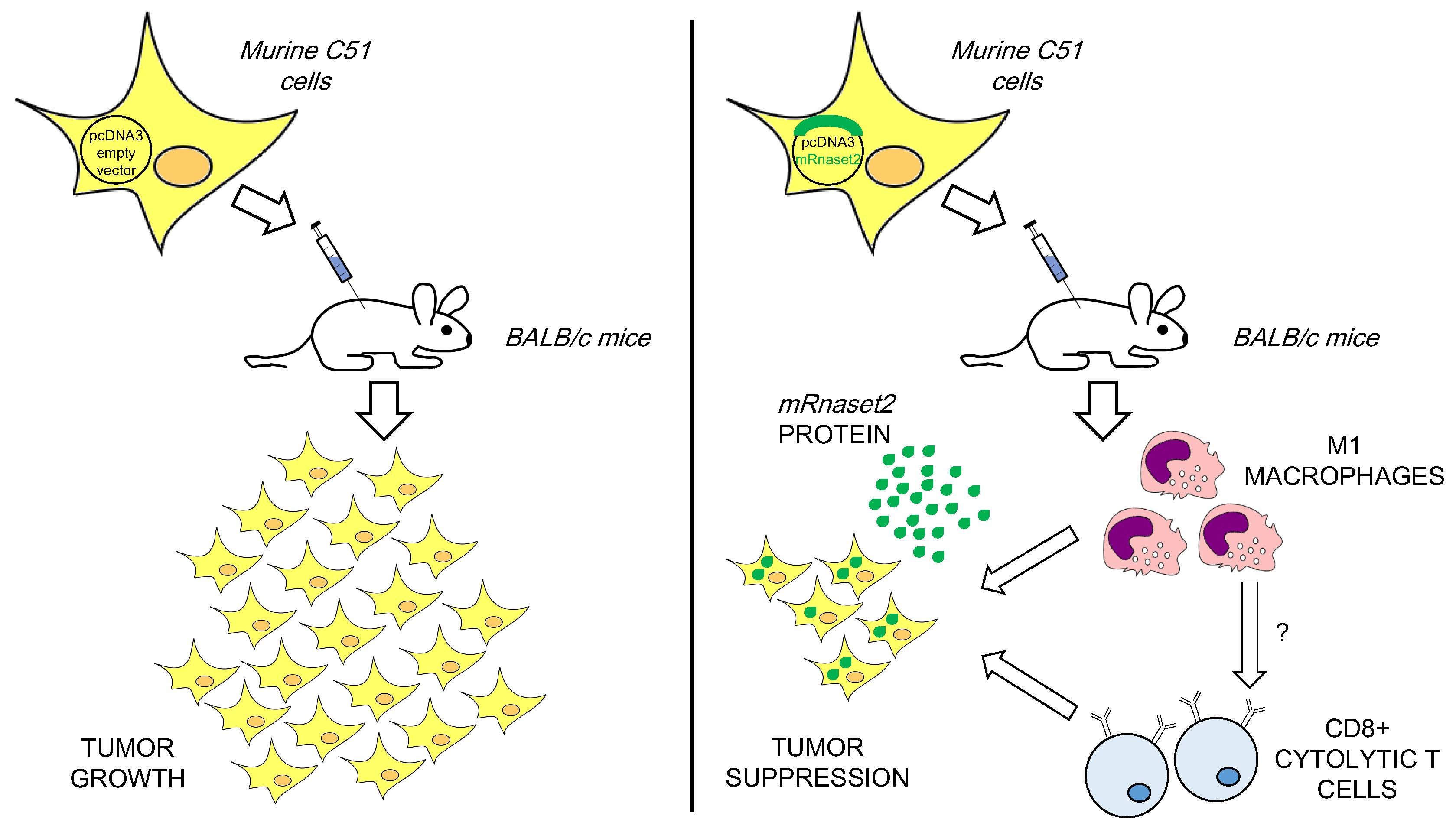
Overexpression of Murine Rnaset2 in a Colon Syngeneic Mouse Carcinoma Model Leads to Rebalance of Intra-Tumor M1/M2 Macrophage Ratio, Activation of T Cells, Delayed Tumor Growth, and Rejection

 ,
, 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
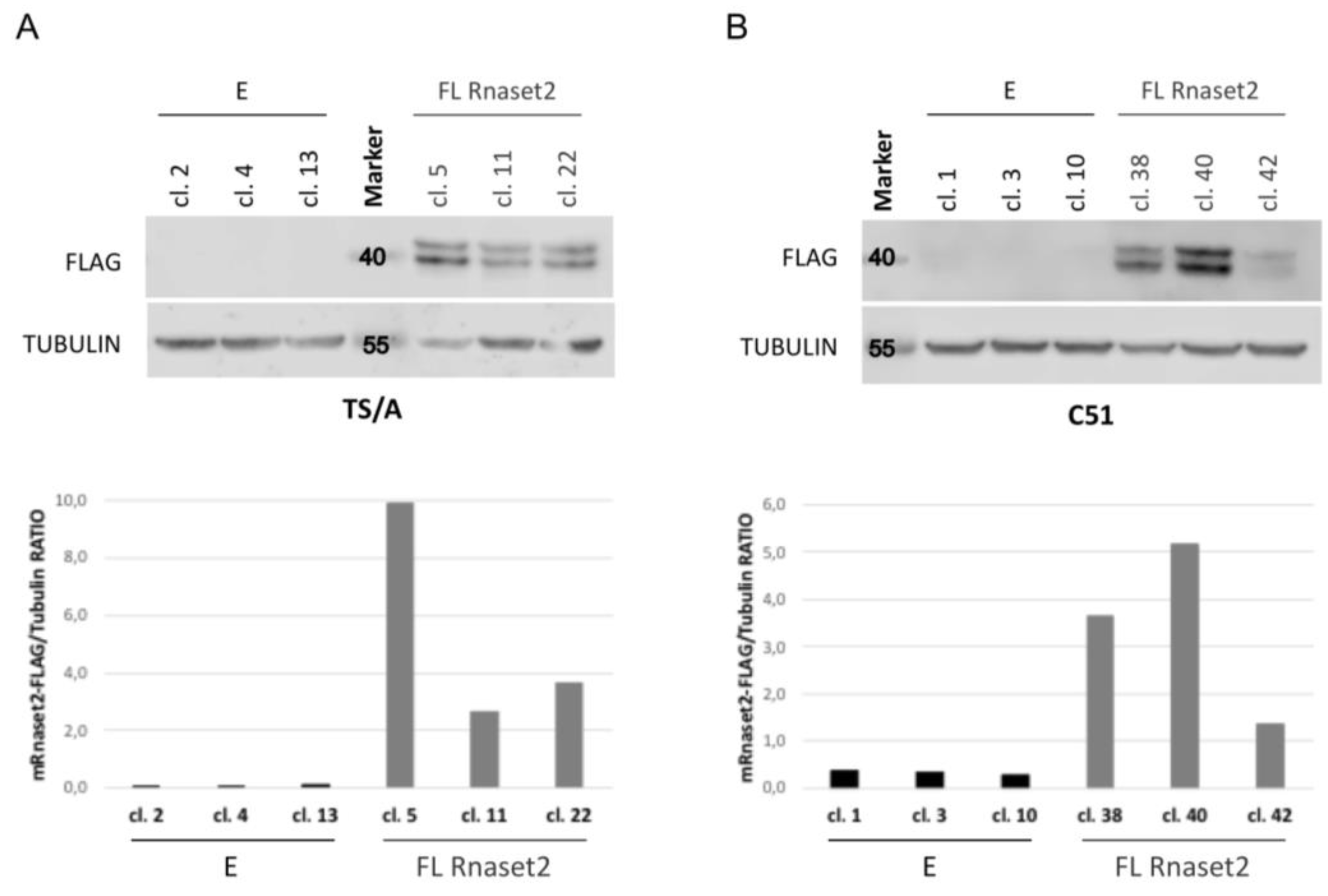
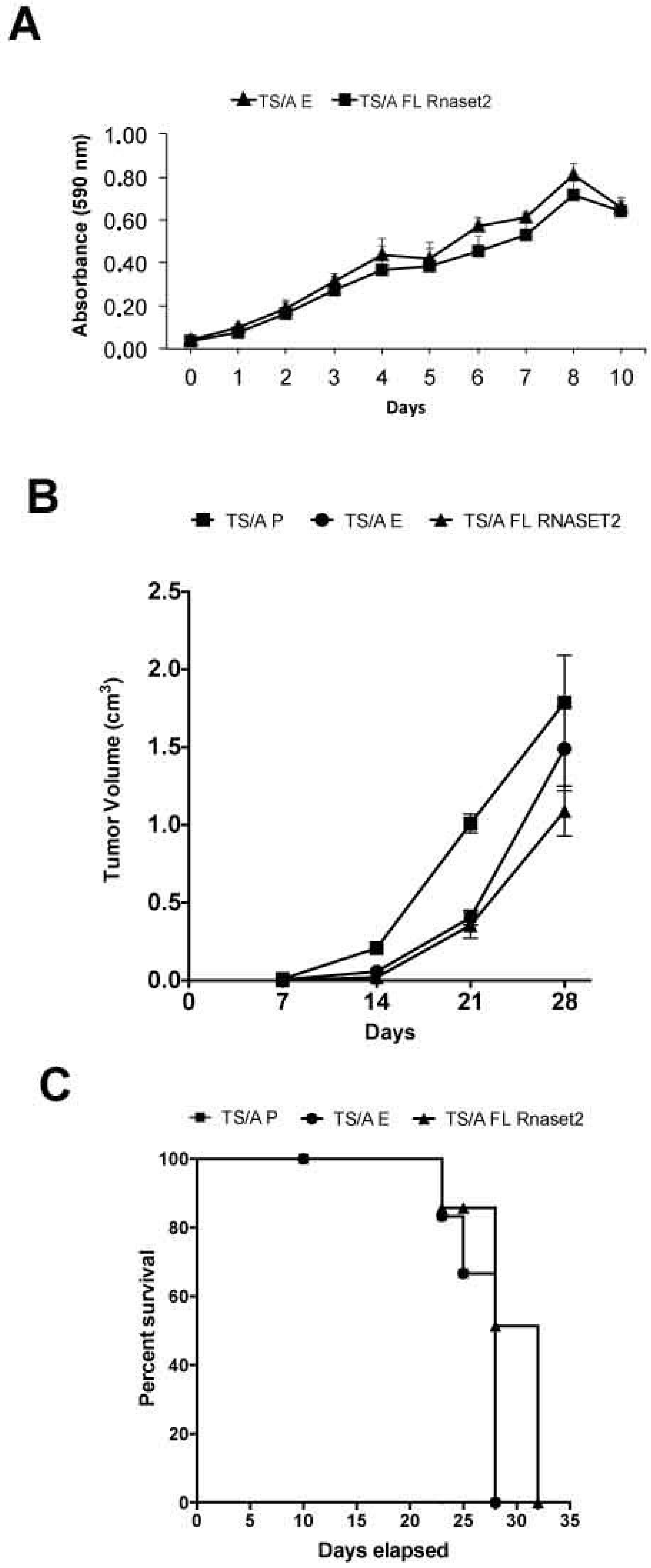
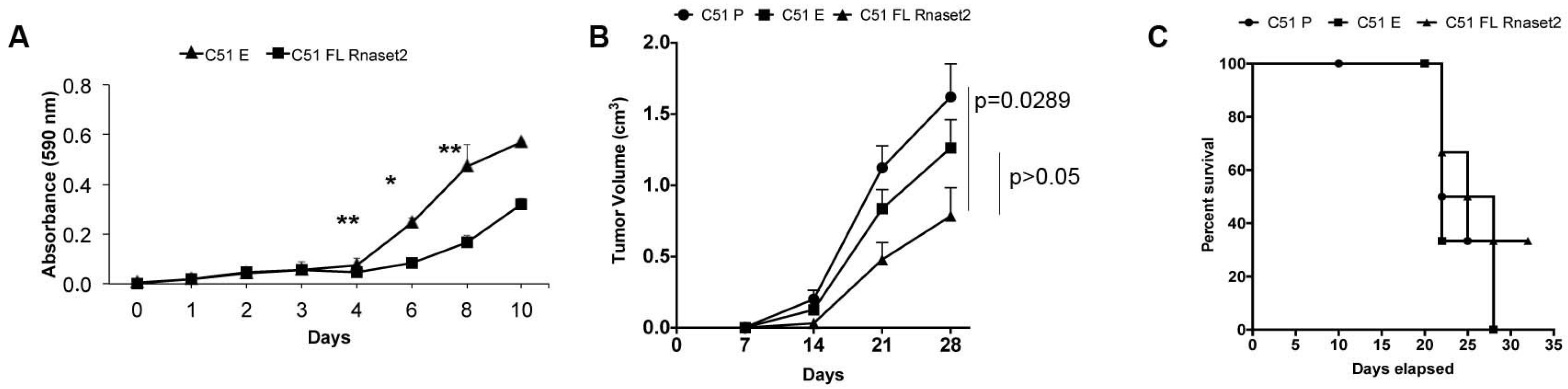
2.1. Establishment of Mouse Rnaset2-Overexpressing TS/A and C51 Cell Clones and Assessment of Their Proliferation Behavior In Vitro and In Vivo
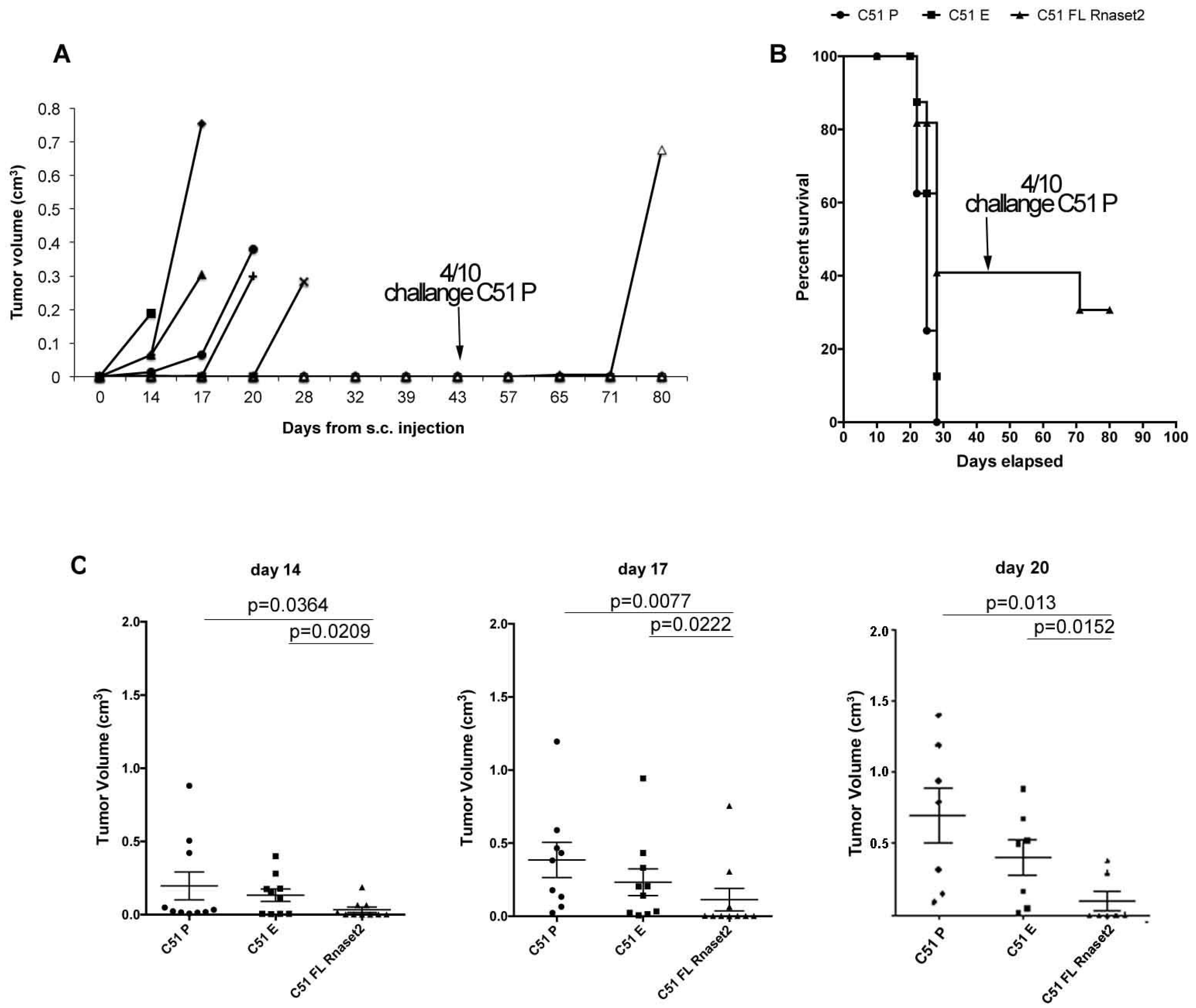
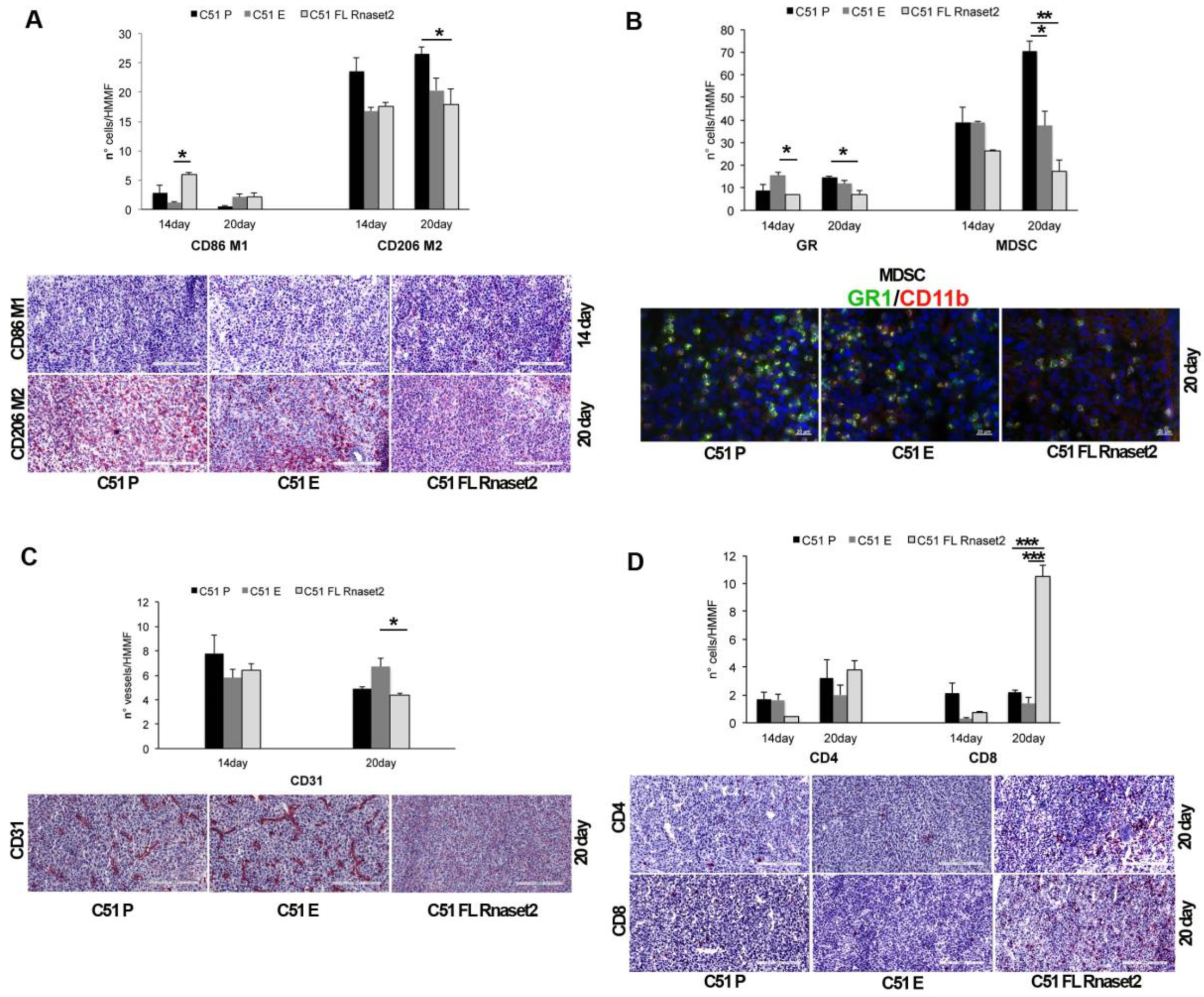
2.2. Murine FL Rnaset2 Overexpression in C51 Cells Correlates with Partial Tumor Rejection, with Tumor Microenvironment’s Immune Cells Modulation and Tumor Vessel Inhibition
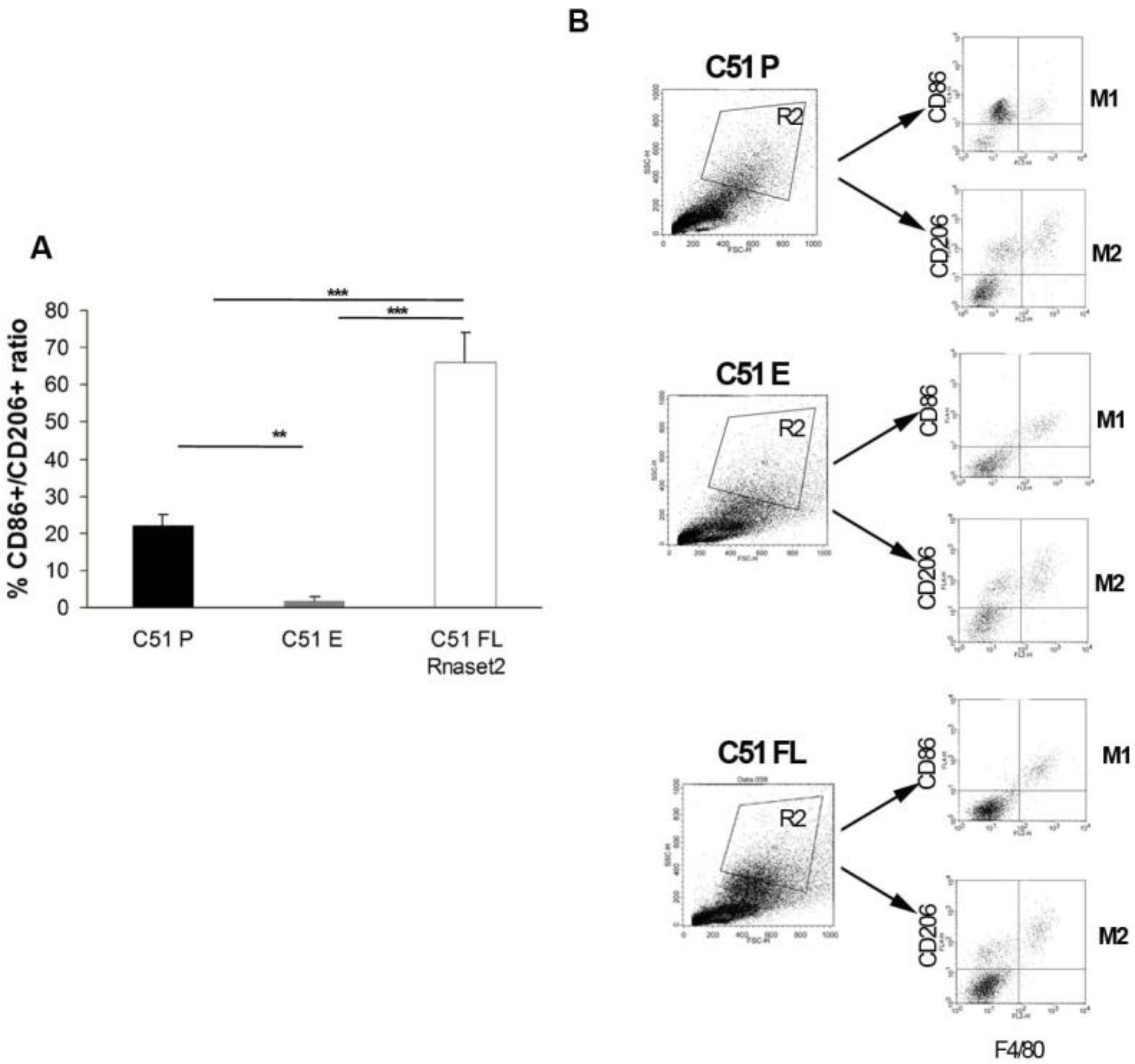
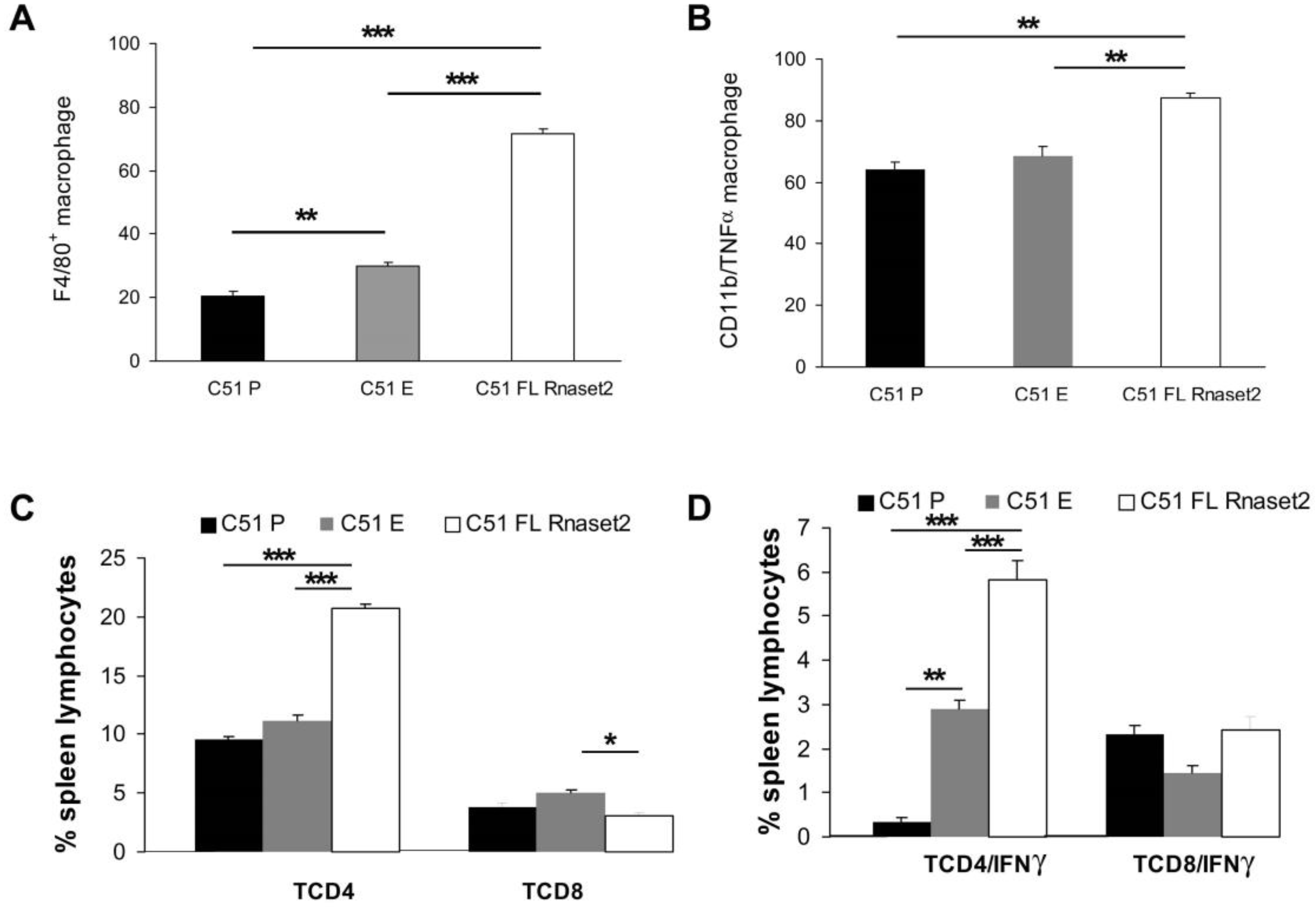
2.3. Systemic Immune Cell Investigation Revealed that Rnaset2-Associated Rejection Correlates with Expansion of M1 Macrophages and Triggering of CD4+ TH1 Cells
3. Discussion
4. Materials and Methods
4.1. Cloning of the Murine Rnaset2 cDNA in pcDNA3 Plasmid
- 3xFLAG_version2 Rev: 5′ AGAGGTTCTAGACTACTTGTCATCGTCATCCTTGTAATC 3′
- 3xFLAG _version2 Fw: 5′ AACTGGCTCGAGGACTACAAAGACCATGACGGTGA 3′
- mRnaset2A Fw: 5′ TGCTAAGGATCCACCATGGCGCCG 3′
- mRnaset2A Rev: 5′ ACCTGAGAATTCATGTTGGGTCTTTGTAGGTGGA 3′
- mRnaset2A overlap Fw: 5′ AGCCGGGGGAGCAGCTGTCCTCCAGGCAGGAA 3′
- mRnaset2A overlap Rev: 5′ CTGCTTGGGGGACGGCTGCTCCCCTGGCTCA 3′
4.2. Animal Tumor Models
4.3. Cell Proliferation Assay
4.4. Western Blot Analysis
4.5. Lymphoid and Myeloid Organ Isolation
4.6. Flow Cytometry Analysis
4.7. Intracellular Staining Analysis
4.8. Immunohistochemistry of Tumor Samples
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Acquati, F.; Bertilaccio, S.; Grimaldi, A.; Monti, L.; Cinquetti, R.; Bonetti, P.; Lualdi, M.; Vidalino, L.; Fabbri, M.; Sacco, M.G.; et al. Microenvironmental control of malignancy exerted by RNASET2, a widely conserved extracellular RNase. Proc. Natl. Acad. Sci. USA 2011, 108, 1104–1109. [Google Scholar] [CrossRef] [PubMed]
- Acquati, F.; Lualdi, M.; Bertilaccio, S.; Monti, L.; Turconi, G.; Fabbri, M.; Grimaldi, A.; Anselmo, A.; Inforzato, A.; Collotta, A.; et al. Loss of function of Ribonuclease T2, an ancient and phylogenetically conserved RNase, plays a crucial role in ovarian tumorigenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 8140–8145. [Google Scholar] [CrossRef] [PubMed]
- Lualdi, M.; Pedrini, E.; Rea, K.; Monti, L.; Scaldaferri, D.; Gariboldi, M.; Camporeale, A.; Ghia, P.; Monti, E.; Tomassetti, A.; et al. Pleiotropic modes of action in tumor cells of RNASET2, an evolutionary highly conserved extracellular RNase. Oncotarget 2015, 6, 7851–7865. [Google Scholar] [CrossRef] [PubMed]
- Roggiani, F.; Riva, C.; Raspagliesi, F.; Porta, G.; Valli, R.; Taramelli, R.; Acquati, F.; Mezzanzanica, D.; Tomassetti, A. A Cell-Autonomous Oncosuppressive Role of Human RNASET2 Affecting ECM-Mediated Oncogenic Signaling. Cancers 2019, 11, 255. [Google Scholar] [CrossRef] [PubMed]
- Smirnoff, P.; Roiz, L.; Angelkovitch, B.; Schwartz, B.; Shoseyov, O. A recombinant human RNASET2 glycoprotein with antitumorigenic and antiangiogenic characteristics: Expression, purification, and characterization. Cancer 2006, 107, 2760–2769. [Google Scholar] [CrossRef]
- Schwartz, B.; Shoseyov, O.; Melnikova, V.O.; McCarty, M.; Leslie, M.; Roiz, L.; Smirnoff, P.; Hu, G.F.; Lev, D.; Bar-Eli, M. ACTIBIND, a T2 RNase, competes with angiogenin and inhibits human melanoma growth, angiogenesis, and metastasis. Cancer Res. 2007, 67, 5258–5266. [Google Scholar] [CrossRef]
- Acquati, F.; Mortara, L.; De Vito, A.; Baci, D.; Albini, A.; Cippitelli, M.; Taramelli, R.; Noonan, D.M. Innate Immune Response Regulation by the Human RNASET2 Tumor Suppressor Gene. Front. Immunol. 2019, 10, 2587–2596. [Google Scholar] [CrossRef]
- Wang, Q.; Jiang, M.; Wu, J.; Ma, Y.; Li, T.; Chen, Q.; Zhang, X.; Xiang, L. Stress-induced RNASET2 overexpression mediates melanocyte apoptosis via the TRAF2 pathway in vitro. Cell Death Dis. 2014, 5, e1022. [Google Scholar] [CrossRef]
- Caputa, G.; Zhao, S.; Criado, A.E.; Ory, D.S.; Duncan, J.G.; Schaffer, J.E. RNASET2 is required for ROS propagation during oxidative stress-mediated cell death. Cell Death Differ. 2016, 23, 347–357. [Google Scholar] [CrossRef]
- Baranzini, N.; Pedrini, E.; Girardello, R.; Tettamanti, G.; De Eguileor, M.; Taramelli, R.; Acquati, F.; Grimaldi, A. Human recombinant RNASET2-induced inflammatory response and connective tissue remodeling in the medicinal leech. Cell Tissue Res. 2017, 368, 337–351. [Google Scholar] [CrossRef]
- Teng, M.W.L.; Galon, J.; Fridman, W.-H.; Smyth, M.J. From mice to humans: Developments in cancer immunoediting. J. Clin. Invest. 2015, 125, 3338–3346. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.; Li, Y.; Lin, Y.; Liu, E.T.; Patnaik, A. Mouse Models for Cancer Immunotherapy. Res. Cancer Discov. 2018, 8, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Aragon-Sanabria, V.; Kim, G.B.; Dong, C. From Cancer Immunoediting to New Strategies in Cancer Immunotherapy: The Roles of Immune Cells and Mechanics in Oncology. Adv. Exp. Med. Biol. 2018, 1092, 113–138. [Google Scholar] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef]
- Buchbinder, E.; Hodi, F.S. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J. Clin. Investig. 2015, 125, 3377–3383. [Google Scholar] [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti- PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Braendstrup, P.; Levine, B.L.; Ruella, M. The long road to the first FDA-approved gene therapy: Chimeric antigen receptor T cells targeting CD19. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [PubMed]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed]
- Gordon, G. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S.; Locati, M.; Mantovani, A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006, 177, 7303–7311. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Nielsen, S.R.; Schmid, M.C. Macrophages as key drivers of cancer progression and metastasis. Mediators of Inflammation. Mediat. Inflamm. 2017, 2017, 9624760. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 phosphatase inhibits STAT3 transcription factor activity in myeloid cells and promotes tumor-associated macrophage differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef]
- Chen, F.; Zhuang, X.; Lin, L.; Yu, P.; Wang, Y.; Shi, Y.; Hu, G.; Sun, Y. New horizons in tumor microenvironment biology: Challenges and opportunities. BMC Med. 2015, 13, 45. [Google Scholar] [CrossRef]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 28, 61–68. [Google Scholar] [CrossRef]
- Datta, M.; Coussens, L.M.; Nishikawa, H.; Hodi, F.S.; Jain, R.K. Reprogramming the Tumor Microenvironment to Improve Immunotherapy: Emerging Strategies and Combination Therapies. Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 165–174. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Scaldaferri, D.; Bosi, A.; Fabbri, M.; Pedrini, E.; Inforzato, A.; Valli, R.; Frattini, A.; De Vito, A.; Noonan, D.M.; Taramelli, R.; et al. The human RNASET2 protein affects the polarization pattern of human macrophages in vitro. Immunol. Lett. 2018, 203, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Noonan, D.M.; Mortara, L. Contribution to Tumor Angiogenesis from Innate Immune Cells within the Tumor Microenvironment: Implications for Immunotherapy. Front. Immunol. 2018, 5, 527. [Google Scholar] [CrossRef] [PubMed]
- Mortara, L.; Benest, A.V.; Bates, D.O.; Noonan, D.M. Can the co-dependence of the immune system and angiogenesis facilitate pharmacological targeting of tumours? Curr. Opin. Pharmacol. 2017, 35, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Nesiel-Nuttman, L.; Doron, S.; Schwartz, B.; Shoseyov, O. Human RNASET2 derivatives as potential anti-angiogenic agents: Actin binding sequence identification and characterization. Oncoscience 2014, 2, 31–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roiz, L.; Smirnoff, P.; Lewin, I.; Shoseyov, O.; Schwartz, B. Human recombinant RNASET2: A potential anti-cancer drug. Oncoscience 2016, 3, 71–84. [Google Scholar]
- Zheng, X.; Turkowski, K.; Mora, J.; Brüne, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef]
- Yuan, R.; Li, S.; Geng, H.; Wang, X.; Guan, Q.; Li, X.; Ren, C.; Yuan, X. Reversing the polarization of tumor-associated macrophages inhibits tumor metastasis. Int. Immunopharmacol. 2017, 49, 30–37. [Google Scholar] [CrossRef]
- Ostroumov, D.; Fekete-Drimusz, N.; Saborowski, M.; Kühnel, F.; Woller, N. CD4 and CD8 T lymphocyte interplay in controlling tumor growth. Cell Mol. Life Sci. 2018, 75, 689–713. [Google Scholar] [CrossRef] [PubMed]
- Borst, J.; Ahrends, T.; Bąbała, N.; Melief, C.J.M.; Kastenmüller, W. CD4+ T cell help in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2018, 18, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Mortara, L.; Castellani, P.; Meazza, R.; Tosi, G.; De Lerma Barbaro, A.; Procopio, F.A.; Comes, A.; Zardi, L.; Ferrini, S.; Accolla, R.S. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. Clin. Cancer Res. 2006, 12, 3435–3443. [Google Scholar] [CrossRef] [PubMed]
- Mortara, L.; Frangione, V.; Castellani, P.; De Lerma Barbaro, A.; Accolla, R.S. Irradiated CIITA-positive mammary adenocarcinoma cells act as a potent anti-tumor-preventive vaccine by inducing tumor-specific CD4+ T cell priming and CD8+ T cell effector functions. Int. Immunol. 2009, 21, 655–665. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Giermasz, A.S.; Urban, J.A.; Nakamura, Y.; Watchmaker, P.; Cumberland, R.L.; Gooding, W.; Kalinski, P. Type-1 polarized dendritic cells primed for high IL-12 production show enhanced activity as cancer vaccines. Cancer Immunol. Immunother. 2009, 58, 1329–1336. [Google Scholar] [CrossRef]
- Morandi, B.; Mortara, L.; Chiossone, L.; Accolla, R.S.; Mingari, M.C.; Moretta, L.; Moretta, A.; Ferlazzo, F. Dendritic cell editing by activated natural killer cells results in a more protective cancer-specific immune response. PLoS ONE 2012, 7, e39170. [Google Scholar] [CrossRef]
- Balza, E.; Zanellato, S.; Poggi, A.; Reverberi, D.; Rubartelli, A.; Mortara, L. The therapeutic T-cell response induced by tumor delivery of TNF and melphalan is dependent on early triggering of natural killer and dendritic cells. Eur. J. Immunol. 2017, 47, 743–753. [Google Scholar] [CrossRef][Green Version]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Vito, A.; Orecchia, P.; Balza, E.; Reverberi, D.; Scaldaferri, D.; Taramelli, R.; Noonan, D.M.; Acquati, F.; Mortara, L. Overexpression of Murine Rnaset2 in a Colon Syngeneic Mouse Carcinoma Model Leads to Rebalance of Intra-Tumor M1/M2 Macrophage Ratio, Activation of T Cells, Delayed Tumor Growth, and Rejection. Cancers 2020, 12, 717. https://doi.org/10.3390/cancers12030717
De Vito A, Orecchia P, Balza E, Reverberi D, Scaldaferri D, Taramelli R, Noonan DM, Acquati F, Mortara L. Overexpression of Murine Rnaset2 in a Colon Syngeneic Mouse Carcinoma Model Leads to Rebalance of Intra-Tumor M1/M2 Macrophage Ratio, Activation of T Cells, Delayed Tumor Growth, and Rejection. Cancers. 2020; 12(3):717. https://doi.org/10.3390/cancers12030717
Chicago/Turabian StyleDe Vito, Annarosaria, Paola Orecchia, Enrica Balza, Daniele Reverberi, Debora Scaldaferri, Roberto Taramelli, Douglas M. Noonan, Francesco Acquati, and Lorenzo Mortara. 2020. "Overexpression of Murine Rnaset2 in a Colon Syngeneic Mouse Carcinoma Model Leads to Rebalance of Intra-Tumor M1/M2 Macrophage Ratio, Activation of T Cells, Delayed Tumor Growth, and Rejection" Cancers 12, no. 3: 717. https://doi.org/10.3390/cancers12030717
APA StyleDe Vito, A., Orecchia, P., Balza, E., Reverberi, D., Scaldaferri, D., Taramelli, R., Noonan, D. M., Acquati, F., & Mortara, L. (2020). Overexpression of Murine Rnaset2 in a Colon Syngeneic Mouse Carcinoma Model Leads to Rebalance of Intra-Tumor M1/M2 Macrophage Ratio, Activation of T Cells, Delayed Tumor Growth, and Rejection. Cancers, 12(3), 717. https://doi.org/10.3390/cancers12030717