tRNA Queuosine Modification Enzyme Modulates the Growth and Microbiome Recruitment to Breast Tumors

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
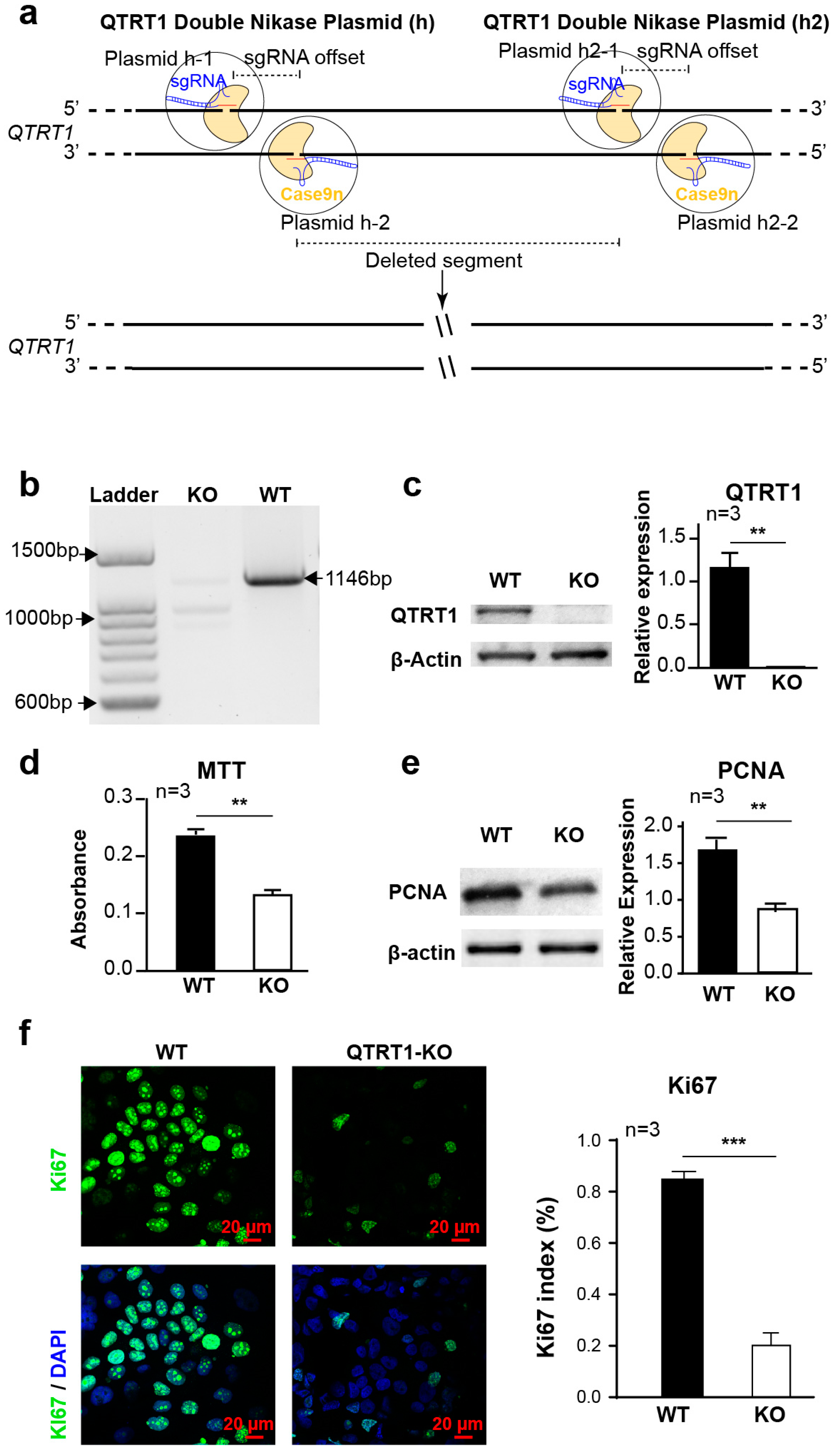
2.1. Establishment of QTRT1 Knockout (KO) Clone in Breast Cancer Cell Line
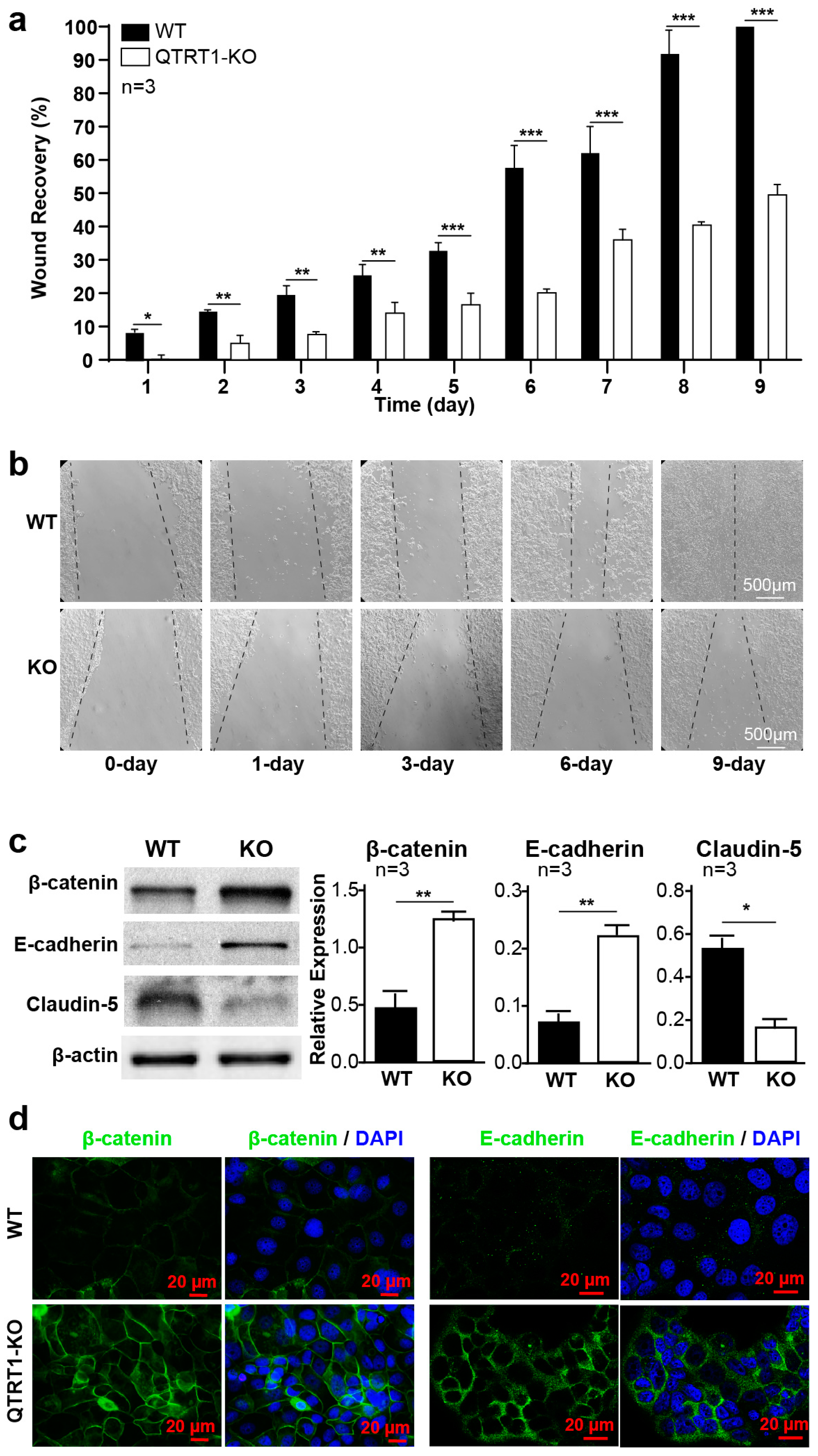
2.2. Knockout of QTRT1 Suppressed Cell Proliferation and Migration
2.3. Knockout of QTRT1 Altered Cell Adhesion and Tight Junctions
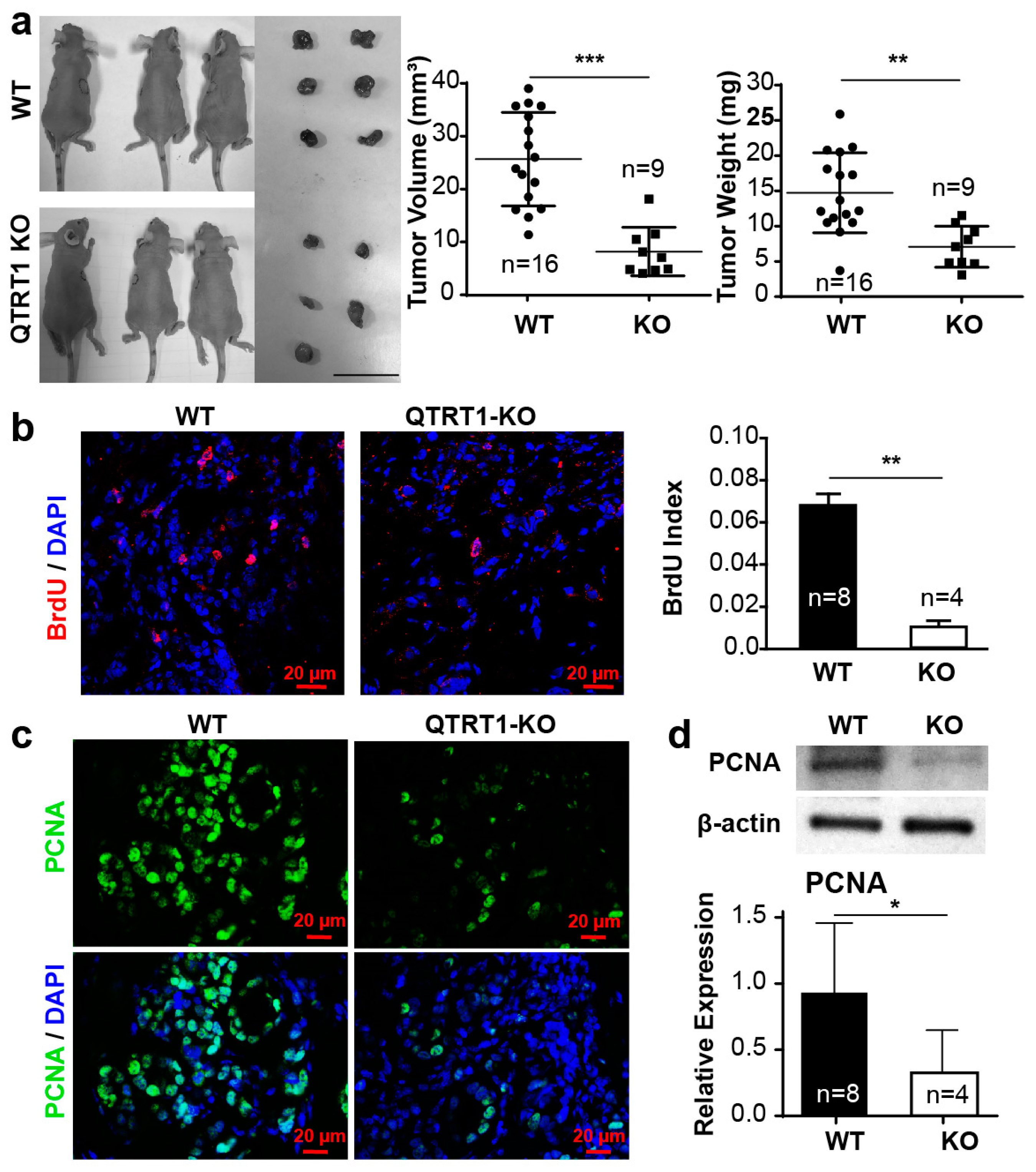
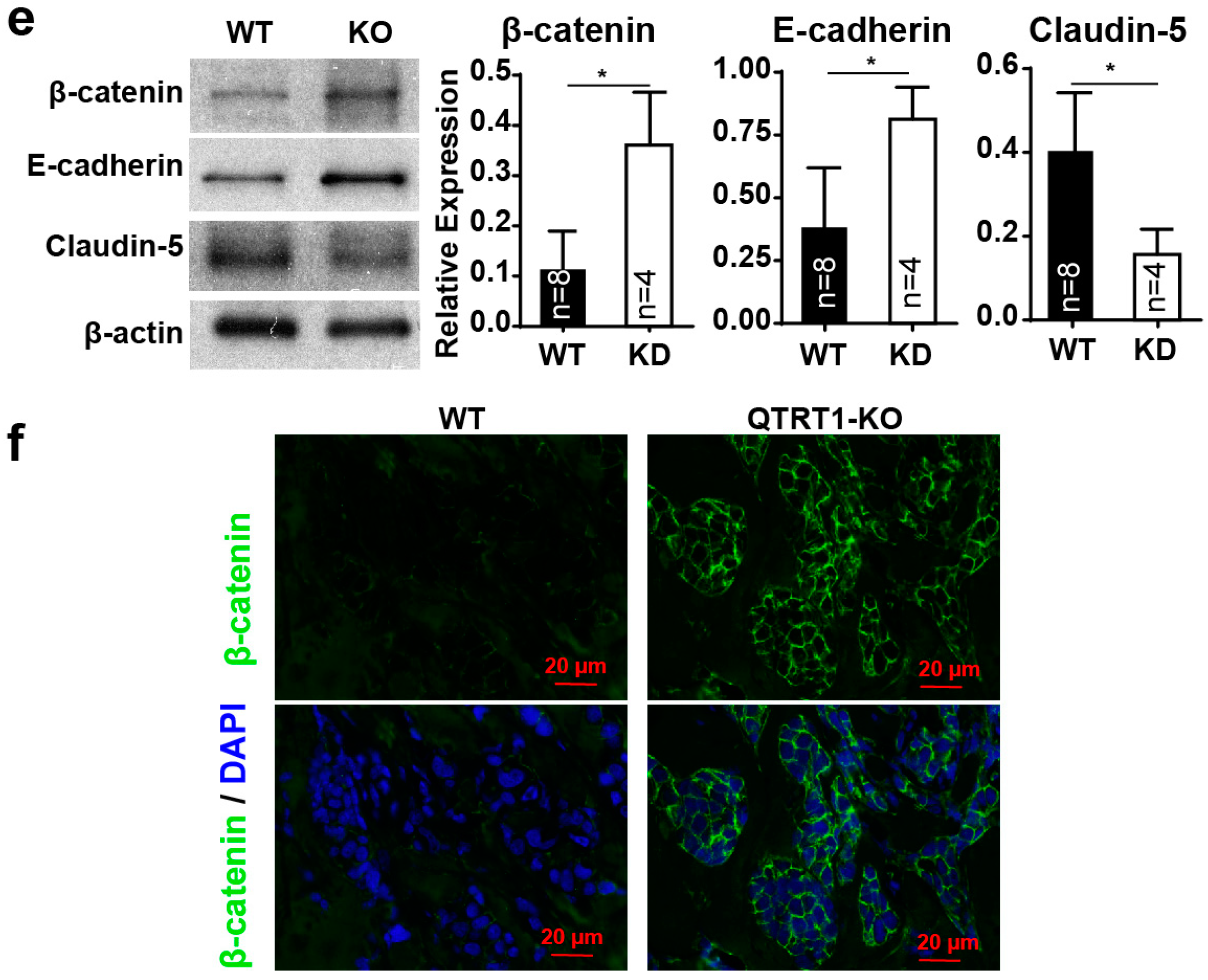
2.4. Knockout of QTRT1 Decreased Tumor Growth and Altered Tight Junctions Regulators In Vivo
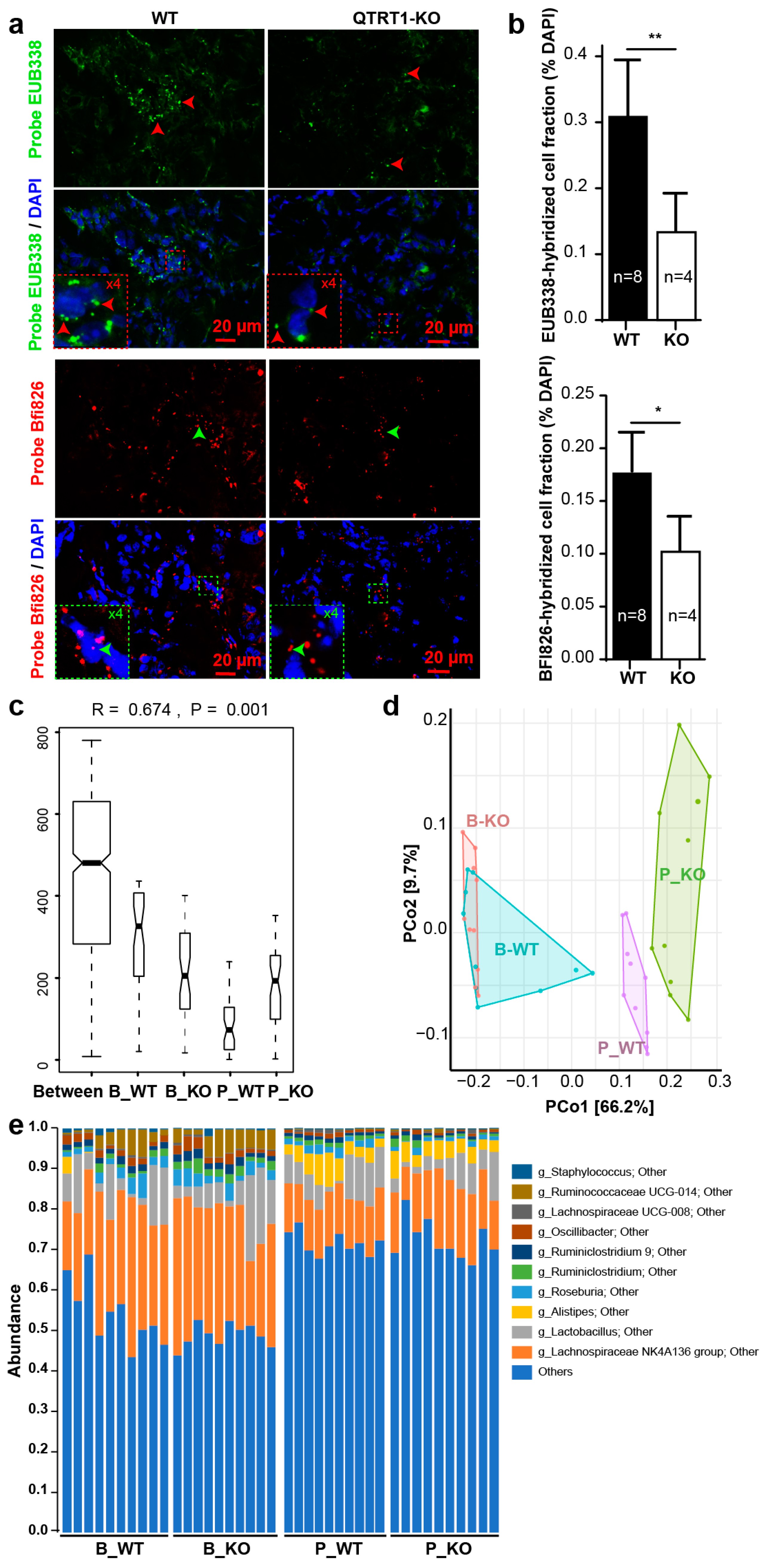
2.5. Bacteria Were Detected in Tumors from the Nude Mice
2.6. Altered Gut Microbiome in the Nude Mice Injected with WT MCF7 Cells
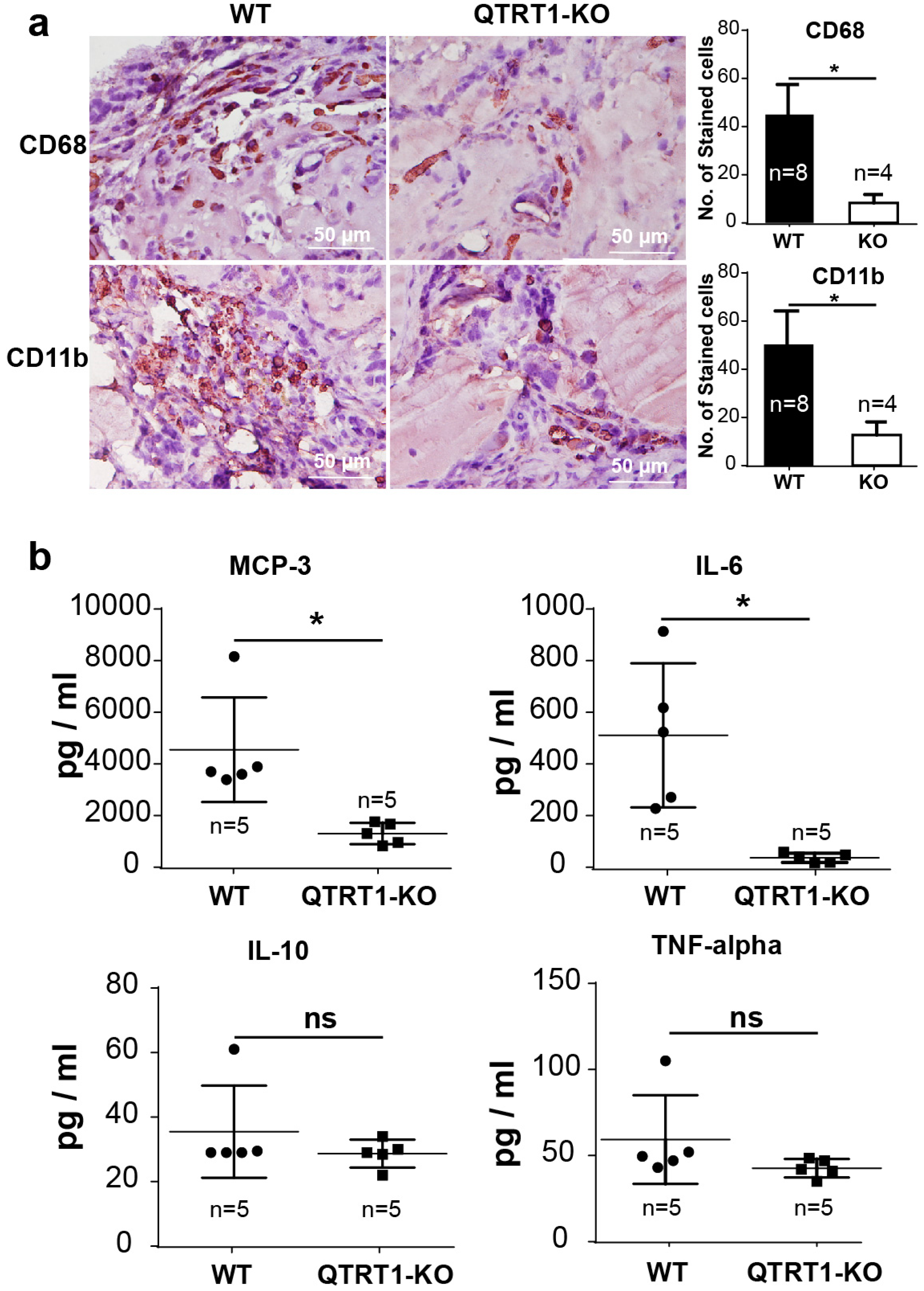
2.7. Inflammation Enhanced the Bacteria in Tumors of Nude Mice
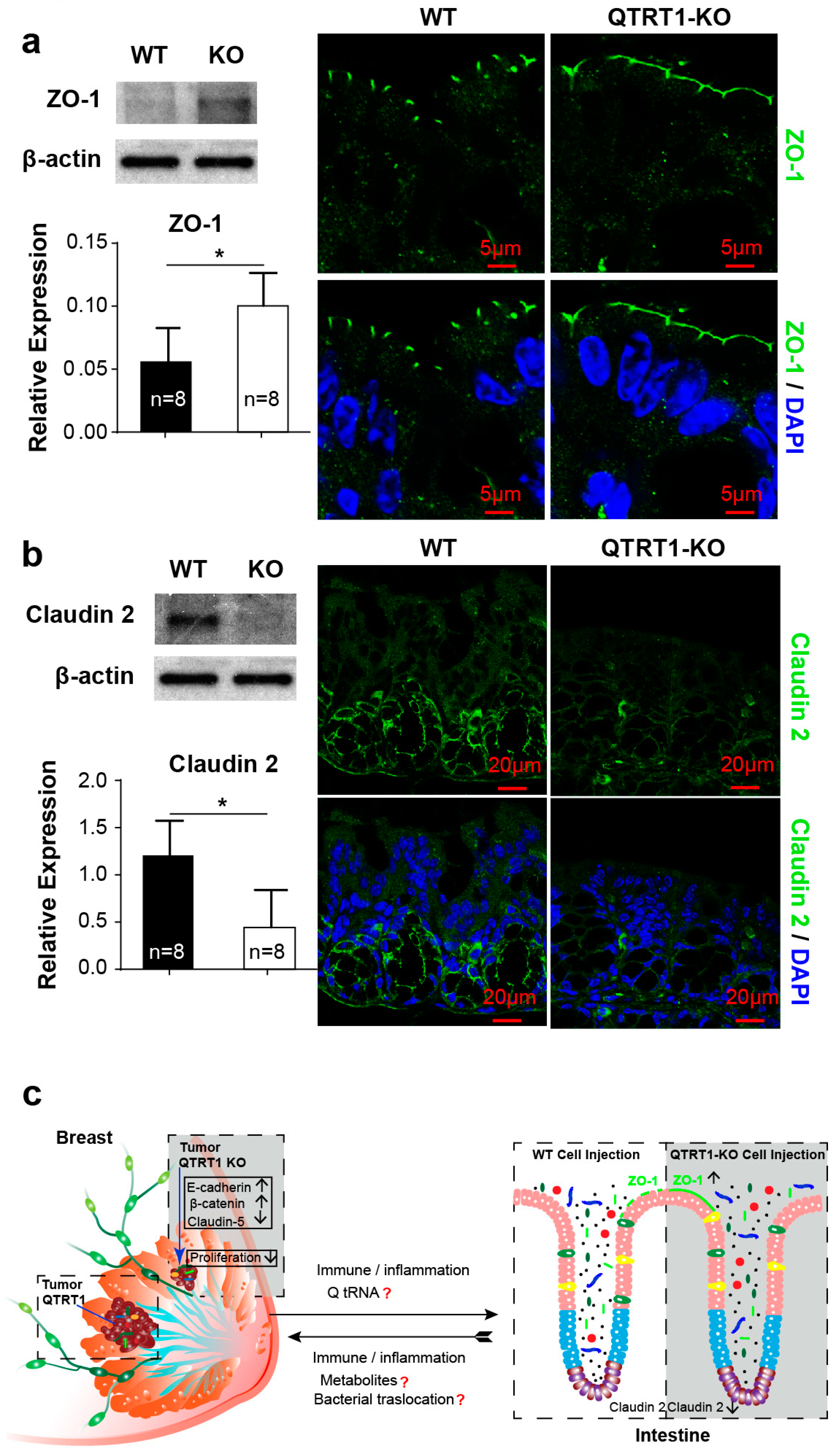
2.8. Disrupted Tight Junctions in the Colon of Mice Injected with WT MCF7 Cells
3. Discussion
4. Methods
4.1. Regents and Cell Lines
4.2. Transfection and Selection
4.3. Clone Validation
4.4. Cell Proliferation Assay
4.5. Wound Healing Assay
4.6. In Vivo Nude Mice Model
4.7. Western Blot Analysis
4.8. The Northern Blot Analysis
- tRNAHis:5′-TGCCGTGACTCGGATTCGAACCGAGGTTGCTGCGGCCACAACGCAGAGTACTAA CCACTATACGATCACGGC;
- tRNAAsn:5′-CGTCCCTGGGTGGGCTCGAACCACCAACCTTTCGGTTAACAGCCGAACGCGCTA ACCGATTGCGCCACAGAGAC.
4.9. Immunofluorescence and Confocal Imaging
4.10. Immunohistochemistry
4.11. Luminex Immunoassays
4.12. Microbial Sampling and Sequencing
4.13. Sequencing Bioinformatics
4.14. Statistical Analysis
4.15. Availability of Data and Material
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Honda, S.; Loher, P.; Shigematsu, M.; Palazzo, J.P.; Suzuki, R.; Imoto, I.; Rigoutsos, I.; Kirino, Y. Sex hormone-dependent tRNA halves enhance cell proliferation in breast and prostate cancers. Proc. Natl. Acad. Sci. USA 2015, 112, E3816–E3825. [Google Scholar] [CrossRef] [PubMed]
- Guzman, N.; Agarwal, K.; Asthagiri, D.; Yu, L.; Saji, M.; Ringel, M.D.; Paulaitis, M.E. Breast Cancer-Specific miR Signature Unique to Extracellular Vesicles Includes “microRNA-like” tRNA Fragments. Mol. Cancer Res. 2015, 13, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Dhahbi, J.M.; Spindler, S.R.; Atamna, H.; Boffelli, D.; Martin, D.I. Deep Sequencing of Serum Small RNAs Identifies Patterns of 5’ tRNA Half and YRNA Fragment Expression Associated with Breast Cancer. Biomark. Cancer 2014, 6, 37–47. [Google Scholar] [CrossRef]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous tRNA-Derived Fragments Suppress Breast Cancer Progression via YBX1 Displacement. Cell 2015, 161, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Fergus, C.; Barnes, D.; Alqasem, M.A.; Kelly, V.P. The queuine micronutrient: Charting a course from microbe to man. Nutrients 2015, 7, 2897–2929. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.C.; Brown, K.G.; Elliott, M.S. The effect of queuosine on tRNA structure and function. J. Biomol. Struct. Dyn. 1999, 16, 757–774. [Google Scholar] [CrossRef]
- Ames, B.N. Prolonging healthy aging: Longevity vitamins and proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 10836–10844. [Google Scholar] [CrossRef]
- Wang, X.; Matuszek, Z.; Huang, Y.; Parisien, M.; Dai, Q.; Clark, W.; Schwartz, M.H.; Pan, T. Queuosine modification protects cognate tRNAs against ribonuclease cleavage. Rna 2018, 24, 1305–1313. [Google Scholar] [CrossRef]
- Muller, M.; Legrand, C.; Tuorto, F.; Kelly, V.P.; Atlasi, Y.; Lyko, F.; Ehrenhofer-Murray, A.E. Queuine links translational control in eukaryotes to a micronutrient from bacteria. Nucleic Acids Res. 2019, 47, 3711–3727. [Google Scholar] [CrossRef]
- Tuorto, F.; Legrand, C.; Cirzi, C.; Federico, G.; Liebers, R.; Muller, M.; Ehrenhofer-Murray, A.E.; Dittmar, G.; Grone, H.J.; Lyko, F. Queuosine-modified tRNAs confer nutritional control of protein translation. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Matsunaga, T.; Iyoda, T.; Fukai, F. Adhesion-dependent cell Regulation via Adhesion molecule, integrin: Therapeutic application of integrin activation-modulating factors. In Colloid and Interface Science in Pharmaceutical Research and Development; Elsevier: Amsterdam, The Netherlands, 2014; pp. 243–260. [Google Scholar]
- Sugimoto, H.; Nagahara, M.; Bae, Y.; Nakagawa, T.; Ishikawa, T.; Sato, T.; Uetake, H.; Eishi, Y.; Sugihara, K. Clinicopathologic relevance of claudin 5 expression in breast cancer. Am. J. Clin. Pathol. 2015, 143, 540–546. [Google Scholar] [CrossRef]
- Meier, F.; Suter, B.; Grosjean, H.; Keith, G.; Kubli, E. Queuosine modification of the wobble base in tRNAHis influences ‘in vivo’ decoding properties. EMBO J. 1985, 4, 823–827. [Google Scholar] [CrossRef]
- Zaborske, J.M.; DuMont, V.L.; Wallace, E.W.; Pan, T.; Aquadro, C.F.; Drummond, D.A. A nutrient-driven tRNA modification alters translational fidelity and genome-wide protein coding across an animal genus. PLoS Biol. 2014, 12, e1002015. [Google Scholar] [CrossRef] [PubMed]
- Di Cello, F.; Shin, J.; Harbom, K.; Brayton, C. Knockdown of HMGA1 inhibits human breast cancer cell growth and metastasis in immunodeficient mice. Biochem. Biophys. Res. Commun. 2013, 434, 70–74. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ooms, L.M.; Binge, L.C.; Davies, E.M.; Rahman, P.; Conway, J.R.; Gurung, R.; Ferguson, D.T.; Papa, A.; Fedele, C.G.; Vieusseux, J.L.; et al. The Inositol Polyphosphate 5-Phosphatase PIPP Regulates AKT1-Dependent Breast Cancer Growth and Metastasis. Cancer Cell 2015, 28, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Stengel, C.; Newman, S.P.; Leese, M.P.; Thomas, M.P.; Potter, B.V.; Reed, M.J.; Purohit, A.; Foster, P.A. The In Vitro and In Vivo Activity of the Microtubule Disruptor STX140 Is Mediated by Hif-1 Alpha and CAIX Expression. Anticancer Res. 2015, 35, 5249–5261. [Google Scholar] [PubMed]
- Yano, S.; Takehara, K.; Miwa, S.; Kishimoto, H.; Tazawa, H.; Urata, Y.; Kagawa, S.; Bouvet, M.; Fujiwara, T.; Hoffman, R.M. In Vivo Isolation of a Highly-aggressive Variant of Triple-negative Human Breast Cancer MDA-MB-231 Using Serial Orthotopic Transplantation. Anticancer Res. 2016, 36, 3817–3820. [Google Scholar] [CrossRef] [PubMed]
- Khaled, M.; Belaaloui, G.; Jiang, Z.Z.; Zhu, X.; Zhang, L.Y. Antitumor effect of Deoxypodophyllotoxin on human breast cancer xenograft transplanted in BALB/c nude mice model. J. Infect. Chemother. 2016, 22, 692–696. [Google Scholar] [CrossRef]
- Nicolas, A.; Kenna, K.P.; Renton, A.E.; Ticozzi, N.; Faghri, F.; Chia, R.; Dominov, J.A.; Kenna, B.J.; Nalls, M.A.; Keagle, P.; et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron 2018, 97, 1268–1283. [Google Scholar] [CrossRef]
- Bhat, A.A.; Uppada, S.; Achkar, I.W.; Hashem, S.; Yadav, S.K.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Uddin, S. Tight Junction Proteins and Signaling Pathways in Cancer and Inflammation: A Functional Crosstalk. Front. Physiol. 2018, 9, 1942. [Google Scholar] [CrossRef]
- Meehan, C.J.; Beiko, R.G. A phylogenomic view of ecological specialization in the Lachnospiraceae, a family of digestive tract-associated bacteria. Genome Biol. Evol. 2014, 6, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Collard, M.; Austin, N.; Tallant, A.; Gallagher, P. Muscadine Grape Extract Reduces Lung and Liver Metastasis in Mice with Triple Negative Breast Cancer in Association with Changes in the Gut Microbiome (P05-017-19). Curr. Dev. Nutr. 2019, 3. [Google Scholar] [CrossRef]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Heeney, D.D.; Gareau, M.G.; Marco, M.L. Intestinal Lactobacillus in health and disease, a driver or just along for the ride? Curr. Opin. Biotechnol. 2018, 49, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Jafari, S.; Froushani, S.M.A.; Tokmachi, A. Combined Extract of Heated 4T1 and a Heat-Killed Preparation of Lactobacillus Casei in a Mouse Model of Breast Cancer. Iran. J. Med. Sci. 2017, 42, 457–464. [Google Scholar] [PubMed]
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Cocconi, D.; van Sinderen, D.; Ventura, M. Identification of universal gut microbial biomarkers of common human intestinal diseases by meta-analysis. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Jobin, C. Novel insights into microbiome in colitis and colorectal cancer. Curr. Opin. Gastroenterol. 2017, 33, 422–427. [Google Scholar] [CrossRef]
- Chan, A.A.; Bashir, M.; Rivas, M.N.; Duvall, K.; Sieling, P.A.; Pieber, T.R.; Vaishampayan, P.A.; Love, S.M.; Lee, D.J. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci. Rep. 2016, 6, 28061. [Google Scholar] [CrossRef]
- Frank, J.A.; Reich, C.I.; Sharma, S.; Weisbaum, J.S.; Wilson, B.A.; Olsen, G.J. Critical evaluation of two primers commonly used for amplification of bacterial 16S rRNA genes. Appl. Environ. Microbiol. 2008, 74, 2461–2470. [Google Scholar] [CrossRef]
- Kennedy, N.A.; Walker, A.W.; Berry, S.H.; Duncan, S.H.; Farquarson, F.M.; Louis, P.; Thomson, J.M. The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS ONE 2014, 9, e88982. [Google Scholar] [CrossRef]
- Zhang, Y.-g.; Wu, S.; Yi, J.; Xia, Y.; Jin, D.; Zhou, J.; Sun, J. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin. Ther. 2017, 39, 322–336. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Gallo, V.; Bueno-De-Mesquita, H.B.; Vermeulen, R.; Andersen, P.M.; Kyrozis, A.; Linseisen, J.; Kaaks, R.; Allen, N.E.; Roddam, A.W.; Boshuizen, H.C.; et al. Smoking and Risk for Amyotrophic Lateral Sclerosis: Analysis of the EPIC Cohort. Ann. Neurol. 2009, 65, 378–385. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Wu, S.; Lu, R.; Zhou, D.; Zhou, J.; Carmeliet, G.; Petrof, E.; Claud, E.C.; Sun, J. Tight junction CLDN2 gene is a direct target of the vitamin D receptor. Sci. Rep. 2015, 5, 10642. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Wu, S.; Xia, Y.; Sun, J. Salmonella infection upregulates the leaky protein claudin-2 in intestinal epithelial cells. PLoS ONE 2013, 8, e58606. [Google Scholar] [CrossRef]
- Clapp, M.; Aurora, N.; Herrera, L.; Bhatia, M.; Wilen, E.; Wakefield, S. Gut microbiota’s effect on mental health: The gut-brain axis. Clin. Pract. 2017, 7, 987. [Google Scholar] [CrossRef]
- Terada, N.; Karim, M.R.; Izawa, T.; Kuwamura, M.; Yamate, J. Immunolocalization of beta-catenin, E-cadherin and N-cadherin in neonate and adult rat kidney. J. Vet. Med. Sci. 2017, 79, 1785–1790. [Google Scholar] [CrossRef]
- Jia, W.; Lu, R.; Martin, T.A.; Jiang, W.G. The role of claudin-5 in blood-brain barrier (BBB) and brain metastases (review). Mol. Med. Rep. 2014, 9, 779–785. [Google Scholar] [CrossRef]
- Fernandez, M.F.; Reina-Perez, I.; Astorga, J.M.; Rodriguez-Carrillo, A.; Plaza-Diaz, J.; Fontana, L. Breast Cancer and Its Relationship with the Microbiota. Int. J. Environ. Res. Public Health 2018, 15, 1747. [Google Scholar] [CrossRef]
- Rodriguez Hernaez, J.; Ceron Cucchi, M.E.; Cravero, S.; Martinez, M.C.; Gonzalez, S.; Puebla, A.; Dopazo, J.; Farber, M.; Paniego, N.; Rivarola, M. The first complete genomic structure of Butyrivibrio fibrisolvens and its chromid. Microb. Genom. 2018, 4. [Google Scholar] [CrossRef]
- Chassaing, B.; Etienne-Mesmin, L.; Gewirtz, A.T. Microbiota-liver axis in hepatic disease. Hepatology 2014, 59, 328–339. [Google Scholar] [CrossRef]
- Ma, H.D.; Wang, Y.H.; Chang, C.; Gershwin, M.E.; Lian, Z.X. The intestinal microbiota and microenvironment in liver. Autoimmun. Rev. 2015, 14, 183–191. [Google Scholar] [CrossRef]
- Lee, S.H. Intestinal permeability regulation by tight junction: Implication on inflammatory bowel diseases. Intest. Res. 2015, 13, 11–18. [Google Scholar] [CrossRef]
- Shang, M.; Sun, J. Vitamin D/VDR, Probiotics, and Gastrointestinal Diseases. Curr. Med. Chem. 2017, 24, 876–887. [Google Scholar] [CrossRef]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef]
- Sun, J.; Kato, I. Gut microbiota, inflammation and colorectal cancer. Genes Dis. 2016, 3, 130–143. [Google Scholar] [CrossRef]
- Lechuga, S.; Ivanov, A.I. Disruption of the epithelial barrier during intestinal inflammation: Quest for new molecules and mechanisms. Biochim. Et Biophys. Acta. Mol. Cell Res. 2017, 1864, 1183–1194. [Google Scholar] [CrossRef]
- Rosean, C.B.; Bostic, R.R.; Ferey, J.C.; Feng, T.-Y.; Azar, F.N.; Tung, K.S.; Dozmorov, M.G.; Smirnova, E.; Bos, P.D.; Rutkowski, M.R. Pre-existing commensal dysbiosis is a host-intrinsic regulator of tissue inflammation and tumor cell dissemination in hormone receptor-positive breast cancer. Cancer Res. 2019, 79, 3662–3675. [Google Scholar] [CrossRef]
- Hieken, T.J.; Chen, J.; Hoskin, T.L.; Walther-Antonio, M.; Johnson, S.; Ramaker, S.; Xiao, J.; Radisky, D.C.; Knutson, K.L.; Kalari, K.R.; et al. The Microbiome of Aseptically Collected Human Breast Tissue in Benign and Malignant Disease. Sci. Rep. 2016, 6, 30751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, K.; Zhang, Y.; Lu, R.; Wu, S.; Tang, J.; Xia, Y.; Sun, J. Deletion of sorting nexin 27 suppresses proliferation in highly aggressive breast cancer MDA-MB-231 cells in vitro and in vivo. BMC Cancer 2019, 19, 555. [Google Scholar] [CrossRef] [PubMed]
- Aad, G.; Abbott, B.; Abbott, D.C.; Abdinov, O.; Abed Abud, A.; Abeling, K.; Abhayasinghe, D.K.; Abidi, S.H.; AbouZeid, O.S.; Abraham, N.L.; et al. Search for Magnetic Monopoles and Stable High-Electric-Charge Objects in 13 Tev Proton-Proton Collisions with the ATLAS Detector. Phys. Rev. Lett. 2020, 124, 031802. [Google Scholar] [CrossRef]
- Kocaturk, B.; Versteeg, H.H. Orthotopic injection of breast cancer cells into the mammary fat pad of mice to study tumor growth. J. Vis. Exp. Jove 2015. [Google Scholar] [CrossRef] [PubMed]
- Faustino-Rocha, A.; Oliveira, P.A.; Pinho-Oliveira, J.; Teixeira-Guedes, C.; Soares-Maia, R.; da Costa, R.G.; Colaco, B.; Pires, M.J.; Colaco, J.; Ferreira, R.; et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. 2013, 42, 217–224. [Google Scholar] [CrossRef]
- Lu, R.; Wu, S.; Liu, X.; Xia, Y.; Zhang, Y.G.; Sun, J. Chronic effects of a Salmonella type III secretion effector protein AvrA in vivo. PLoS ONE 2010, 5, e10505. [Google Scholar] [CrossRef]
- Tack, D.M.; Marder, E.P.; Griffin, P.M.; Cieslak, P.R.; Dunn, J.; Hurd, S.; Scallan, E.; Lathrop, S.; Muse, A.; Ryan, P.; et al. Preliminary incidence and trends of infections with pathogens transmitted commonly through food—Foodborne Diseases Active Surveillance Network, 10 U.S. sites, 2015–2018. Am. J. Transplant. 2019, 19, 1859–1863. [Google Scholar] [CrossRef]
- Lu, R.; Wu, S.; Zhang, Y.G.; Xia, Y.; Zhou, Z.; Kato, I.; Dong, H.; Bissonnette, M.; Sun, J. Salmonella Protein AvrA Activates the STAT3 Signaling Pathway in Colon Cancer. Neoplasia 2016, 18, 307–316. [Google Scholar] [CrossRef]
- Kong, Y.; He, M.; McAlister, T.; Seviour, R.; Forster, R. Quantitative fluorescence in situ hybridization of microbial communities in the rumens of cattle fed different diets. Appl. Environ. Microbiol. 2010, 76, 6933–6938. [Google Scholar] [CrossRef]
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef]
- Glockner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Shannon, C.E. A mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–423, 623–656. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois Press: Urbana, IL, USA, 1949. [Google Scholar]
- Chao, A. Nonparametric estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Bray, J.R.; Curtis, J.T. An ordination of upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 325–349. [Google Scholar] [CrossRef]
- Xia, Y.; Sun, J.; Chen, D.-G. Statistical Analysis of Microbiome Data with R; Springer: Singapore, 2018. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Lu, R.; Zhang, Y.; Matuszek, Ż.; Zhang, W.; Xia, Y.; Pan, T.; Sun, J. tRNA Queuosine Modification Enzyme Modulates the Growth and Microbiome Recruitment to Breast Tumors. Cancers 2020, 12, 628. https://doi.org/10.3390/cancers12030628
Zhang J, Lu R, Zhang Y, Matuszek Ż, Zhang W, Xia Y, Pan T, Sun J. tRNA Queuosine Modification Enzyme Modulates the Growth and Microbiome Recruitment to Breast Tumors. Cancers. 2020; 12(3):628. https://doi.org/10.3390/cancers12030628
Chicago/Turabian StyleZhang, Jilei, Rong Lu, Yongguo Zhang, Żaneta Matuszek, Wen Zhang, Yinglin Xia, Tao Pan, and Jun Sun. 2020. "tRNA Queuosine Modification Enzyme Modulates the Growth and Microbiome Recruitment to Breast Tumors" Cancers 12, no. 3: 628. https://doi.org/10.3390/cancers12030628
APA StyleZhang, J., Lu, R., Zhang, Y., Matuszek, Ż., Zhang, W., Xia, Y., Pan, T., & Sun, J. (2020). tRNA Queuosine Modification Enzyme Modulates the Growth and Microbiome Recruitment to Breast Tumors. Cancers, 12(3), 628. https://doi.org/10.3390/cancers12030628