Serine Biosynthesis Pathway Supports MYC–miR-494–EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells

 , , , , , , ,
, , , , , , ,  , , , , , , add
Show full author list
, , , , , , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
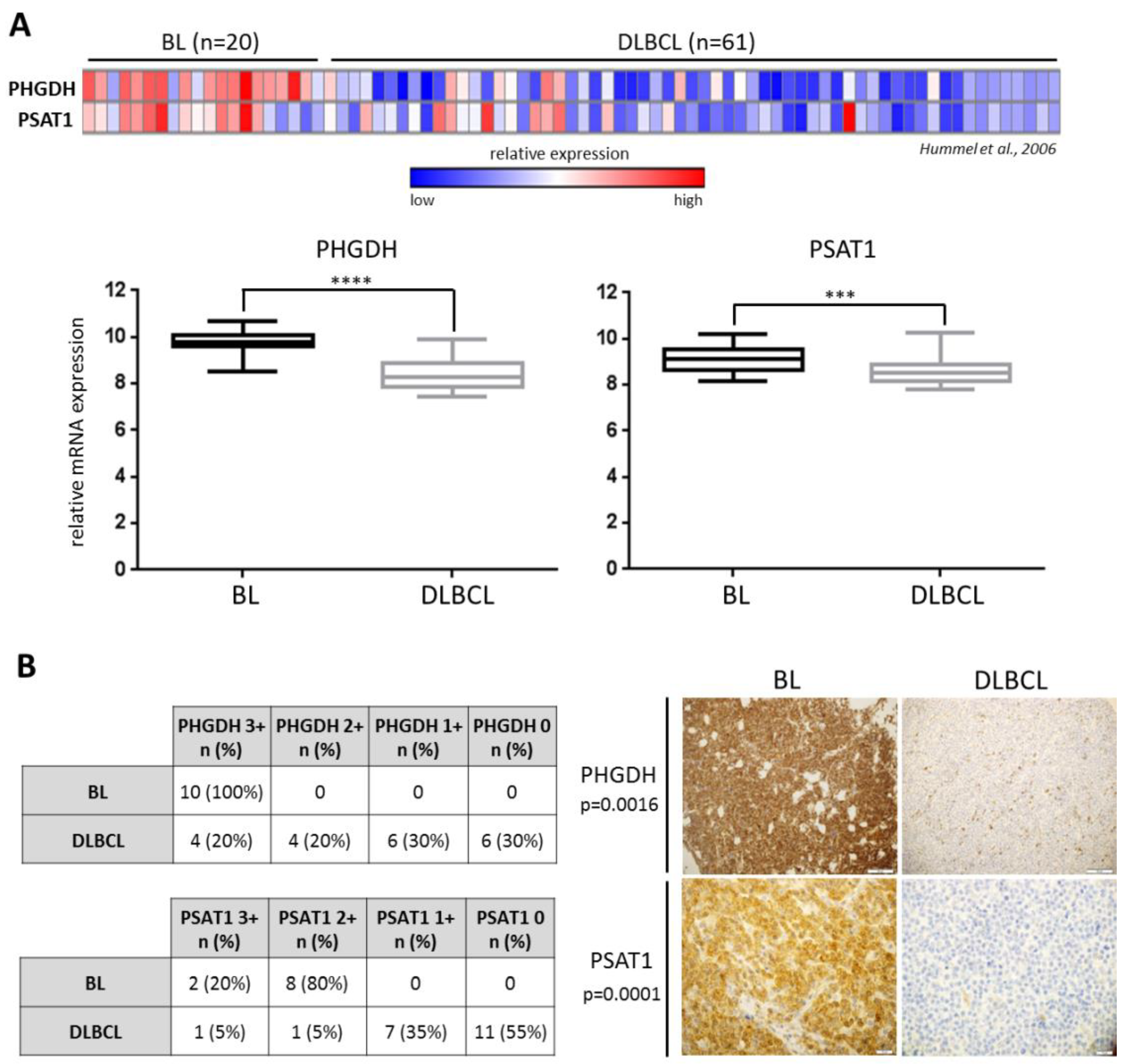
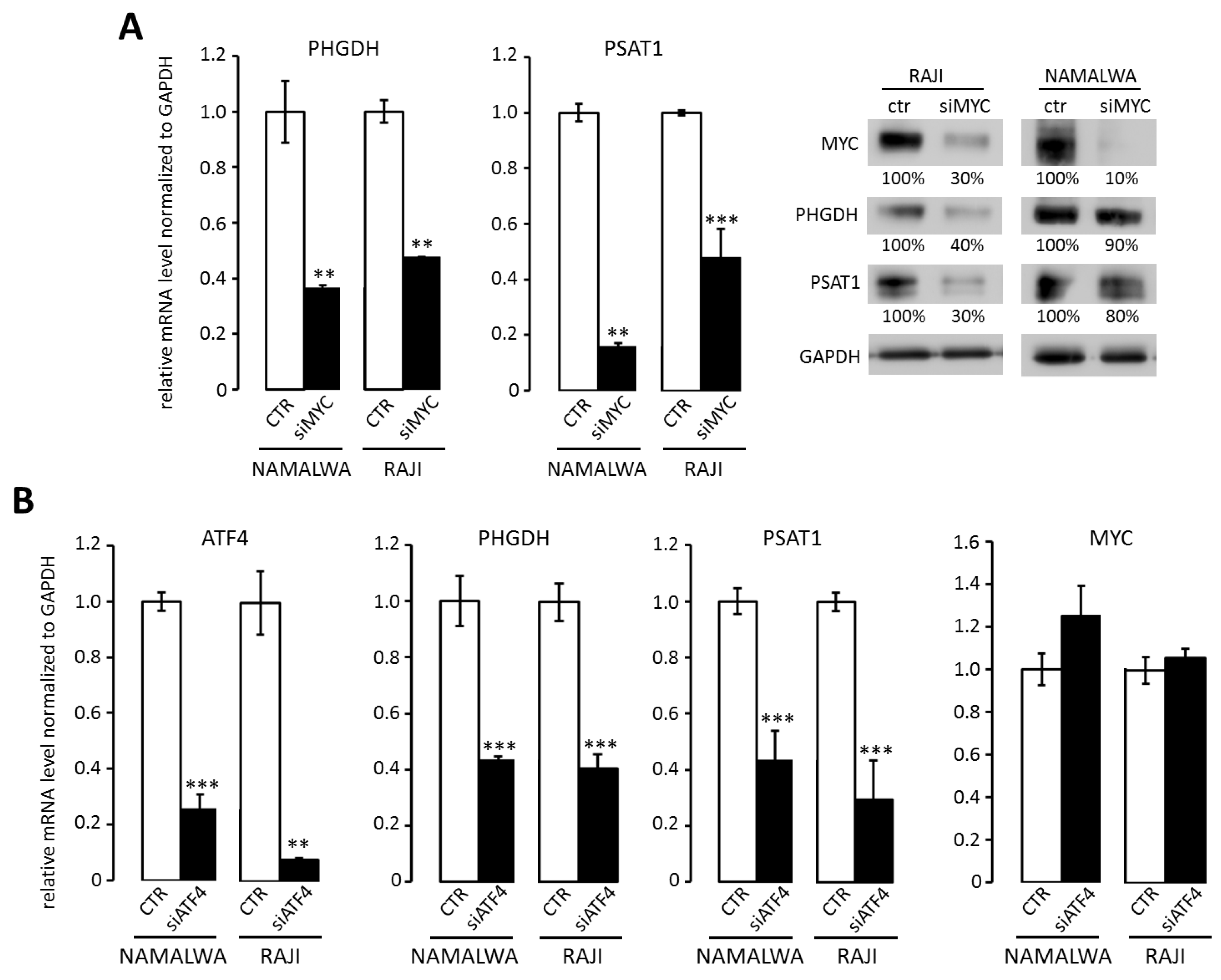
2.1. Overexpression of PHGDH and PSAT1 in BL Cells is Driven by MYC and ATF4
2.2. Knockdown of PSAT1 and PHGDH Impairs BL Cell Proliferation and Clonogenicity In Vitro
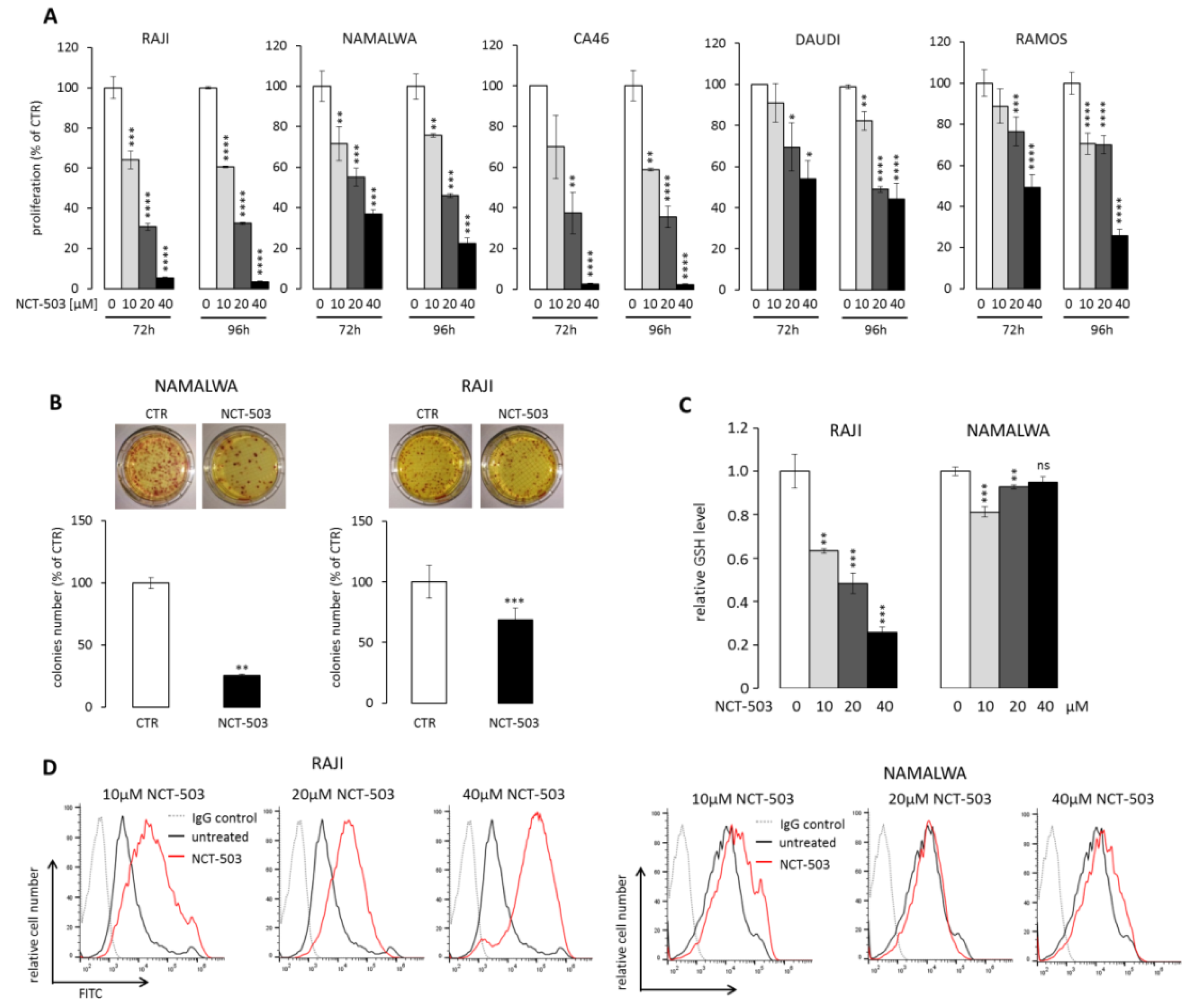
2.3. PHGDH Inhibitor NCT-503 Decreases Proliferation and Clonogenicity of BL Cells, Evokes Oxidative Stress, and Induces Apoptosis
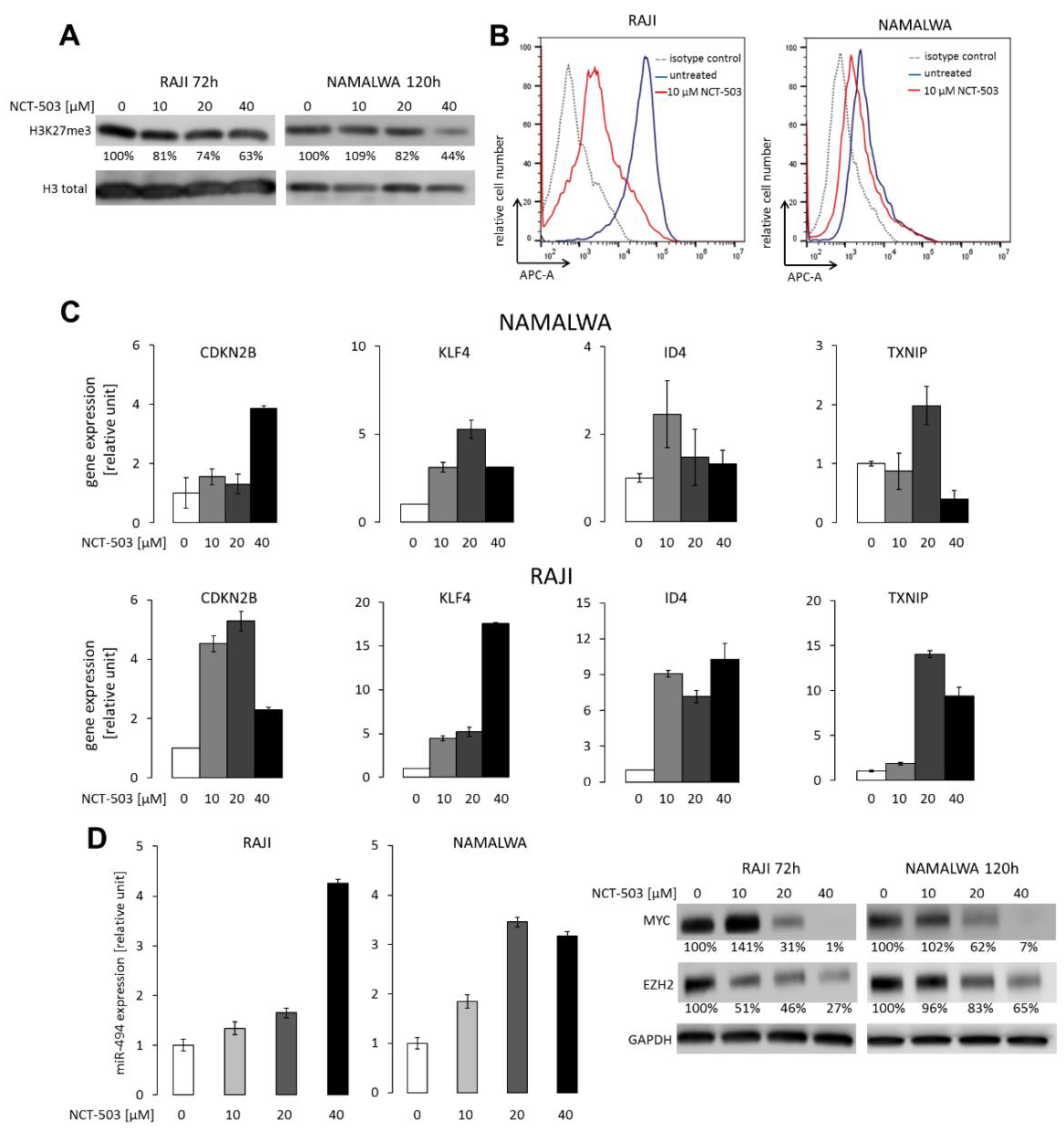
2.4. NCT-503 Decreases Histone and DNA Methylation and Causes Re-expression of Tumor Suppressor Genes
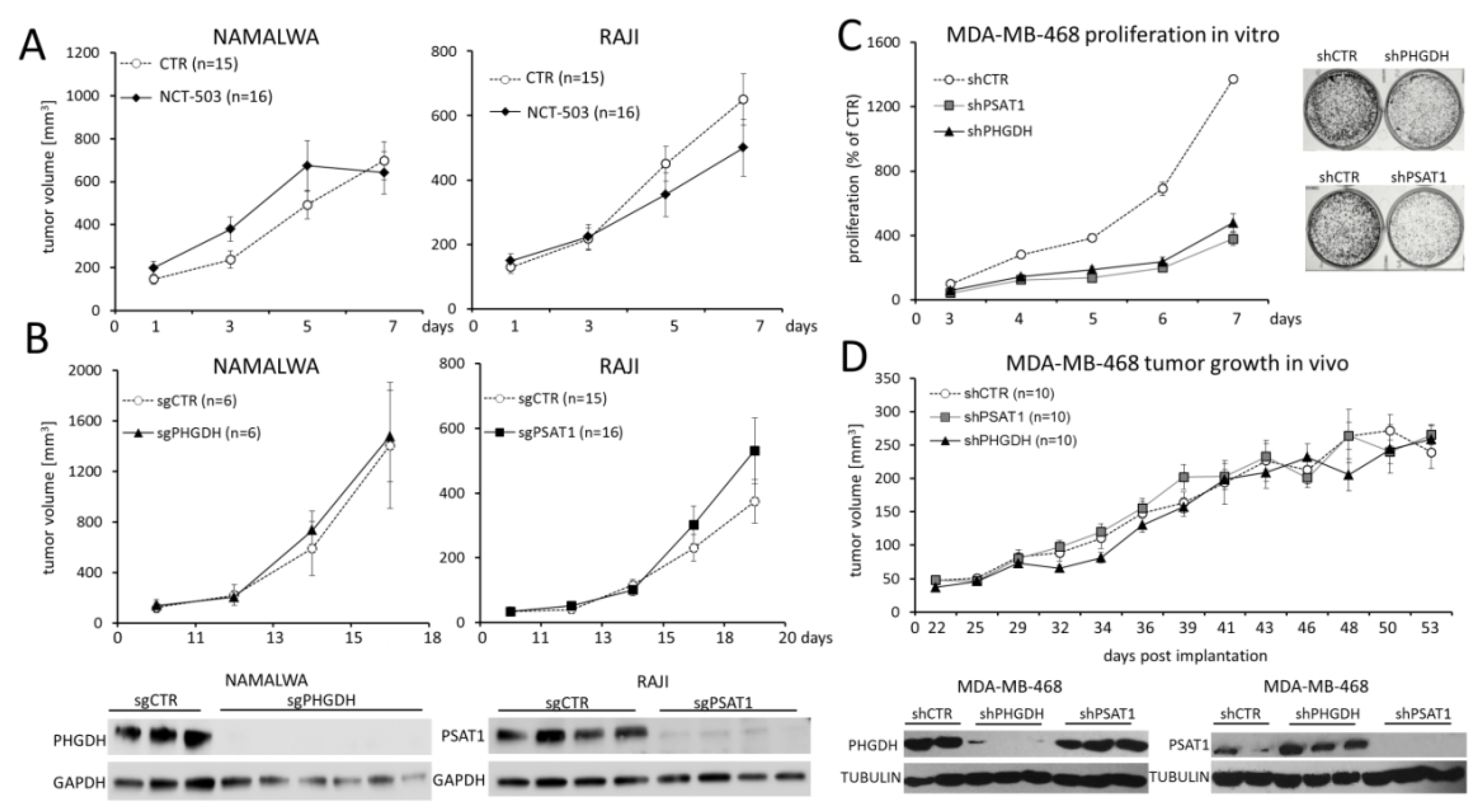
2.5. PHGDH and PSAT1 Inhibition Does Not Impair BL and Breast Cancer Growth In Vivo
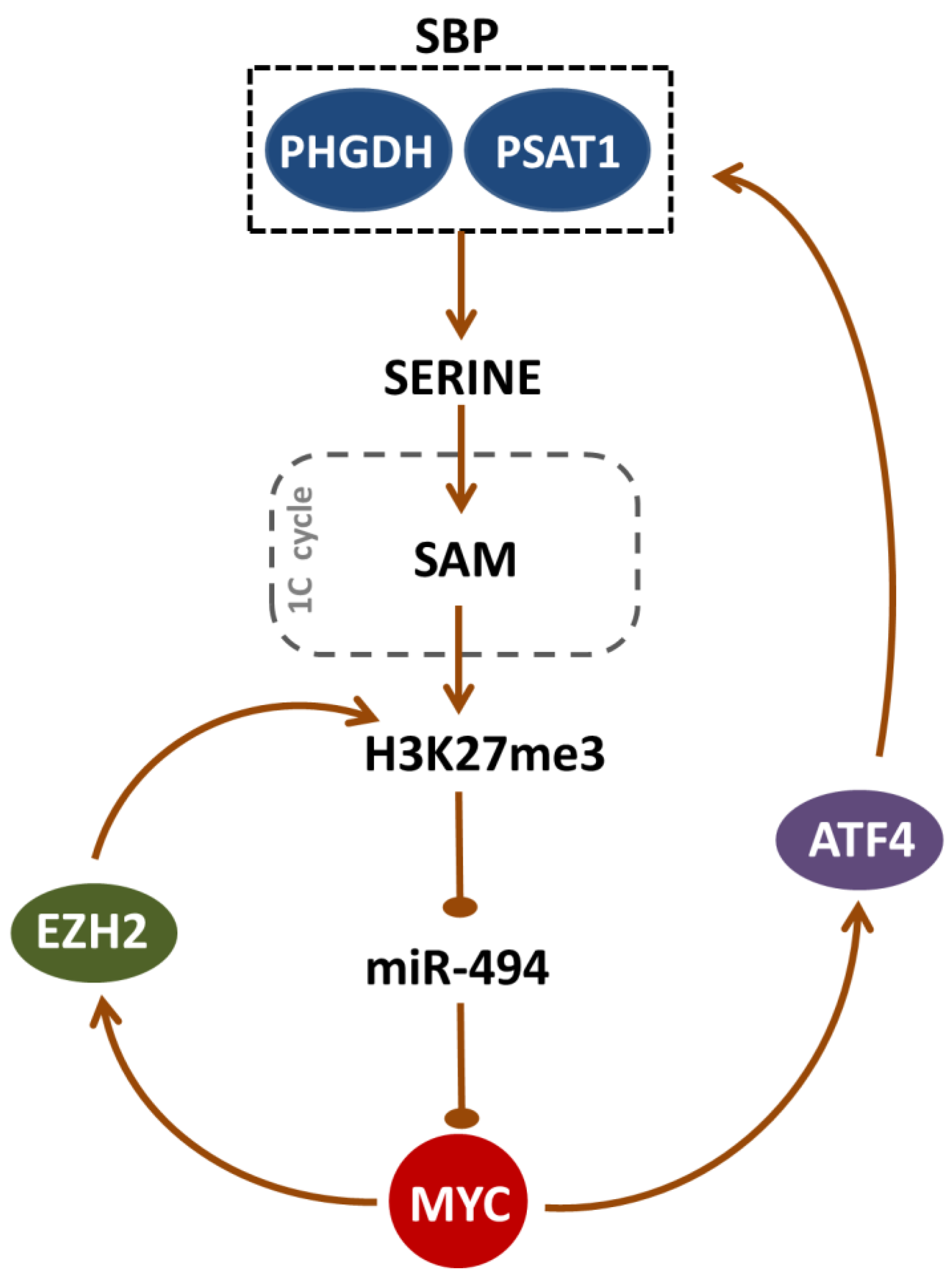
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Cell Culture and Chemicals
4.2. CRISPR/Cas9 and RNAi Gene Targeting
4.3. Real-Time Quantitative PCR (RQ-PCR)
4.4. Immunoblotting
4.5. Patient Samples and Immunohistochemistry
4.6. Cell Viability, Clonogenicity, Apoptosis, and ROS Quantification
4.7. DNA Methylation
4.8. Tumor Xenografts
4.9. Bioinformatic and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
References
- Dozzo, M.; Carobolante, F.; Donisi, P.M.; Scattolin, A.; Maino, E.; Sancetta, R.; Viero, P.; Bassan, R. Burkitt lymphoma in adolescents and young adults: Management challenges. Adolesc. Health Med. Ther. 2017, 8, 11–29. [Google Scholar] [CrossRef]
- Molyneux, E.M.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.J.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Linch, D.C. Burkitt lymphoma in adults. Br. J. Haematol. 2012, 156, 693–703. [Google Scholar] [CrossRef]
- Mbulaiteye, S.M.; Biggar, R.J.; Bhatia, K.; Linet, M.S. Sporadic childhood Burkitt lymphoma incidence in the United States during 1992–2005. Pediatr Blood Cancer 2009, 53, 366–370. [Google Scholar] [CrossRef]
- Kalisz, K.; Alessandrino, F.; Beck, R.; Smith, D.; Kikano, E.; Ramaiya, N.H.; Tirumani, S.H. An update on Burkitt lymphoma: A review of pathogenesis and multimodality imaging assessment of disease presentation, treatment response, and recurrence. Insights Imaging 2019, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Alberto, P.; Sancho, S.J.-M.; Olga, G.; Jordi, E.; Mar, T.; Pilar, M.; Ferran, V.-l.; Rodrigo, M.; Pau, M.; Jose, G.-C.; et al. Incidence, Treatment and Prognosis of Patients with Relapsed Burkitt Lymphoma/Leukemia Treated with Specific Chemotherapy or Immunochemotherapy in Spain. Blood 2015, 126, 2723. [Google Scholar]
- Cairo, M.S.; Sposto, R.; Gerrard, M.; Auperin, A.; Goldman, S.C.; Harrison, L.; Pinkerton, R.; Raphael, M.; McCarthy, K.; Perkins, S.L.; et al. Advanced stage, increased lactate dehydrogenase, and primary site, but not adolescent age (≥15 years), are associated with an increased risk of treatment failure in children and adolescents with mature B-cell non-Hodgkin’s lymphoma: Results of the FAB LMB 96 study. J. Clin. Oncol. 2012, 30, 387–393. [Google Scholar] [PubMed]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic mechanisms in Burkitt lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4, a014282. [Google Scholar] [CrossRef]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in Burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef]
- Sewastianik, T.; Prochorec-Sobieszek, M.; Chapuy, B.; Juszczyński, P. MYC deregulation in lymphoid tumors: Molecular mechanisms, clinical consequences and therapeutic implications. Biochim. Biophys. Acta 2014, 1846, 457–467. [Google Scholar] [CrossRef]
- Chen, H.; Liu, H.; Qing, G. Targeting oncogenic Myc as a strategy for cancer treatment. Signal Transduct. Target. Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Schwarzfischer, P.; Reinders, J.; Dettmer, K.; Kleo, K.; Dimitrova, L.; Hummel, M.; Feist, M.; Kube, D.; Szczepanowski, M.; Klapper, W.; et al. Comprehensive Metaboproteomics of Burkitt’s and Diffuse Large B-Cell Lymphoma Cell Lines and Primary Tumor Tissues Reveals Distinct Differences in Pyruvate Content and Metabolism. J. Proteome Res. 2017, 16, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, T.; Timmermans, M.A.; Sieuwerts, A.M.; Smid, M.; Look, M.P.; Grebenchtchikov, N.; Sweep, F.C.G.J.; Smits, J.G.; Magdolen, V.; van Deurzen, C.H.M.; et al. Phosphoserine aminotransferase 1 is associated to poor outcome on tamoxifen therapy in recurrent breast cancer. Sci. Rep. 2017, 7, 2099. [Google Scholar] [CrossRef]
- Qian, C.; Xia, Y.; Ren, Y.; Yin, Y.; Deng, A. Identification and validation of PSAT1 as a potential prognostic factor for predicting clinical outcomes in patients with colorectal carcinoma. Oncol. Lett. 2017, 14, 8014–8020. [Google Scholar] [CrossRef]
- Jia, X.Q.; Zhang, S.; Zhu, H.J.; Wang, W.; Zhu, J.H.; Wang, X.D.; Qiang, J.F. Increased Expression of PHGDH and Prognostic Significance in Colorectal Cancer. Transl. Oncol. 2016, 9, 191–196. [Google Scholar] [CrossRef]
- Pacold, M.E.; Brimacombe, K.R.; Chan, S.H.; Rohde, J.M.; Lewis, C.A.; Swier, L.J.; Possemato, R.; Chen, W.W.; Sullivan, L.B.; Fiske, B.P.; et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 2016, 12, 452–458. [Google Scholar] [CrossRef]
- Mullarky, E.; Mattaini, K.R.; Vander Heiden, M.G.; Cantley, L.C.; Locasale, J.W. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 2011, 24, 1112–1115. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Chen, P.H.; Mullarky, E.; Sudderth, J.A.; Hu, Z.; Wu, D.; Tang, H.; Xie, Y.; Asara, J.M.; Huffman, K.E.; et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 2015, 47, 1475–1481. [Google Scholar] [CrossRef]
- Hummel, M.; Bentink, S.; Berger, H.; Klapper, W.; Wessendorf, S.; Barth, T.F.; Bernd, H.W.; Cogliatti, S.B.; Dierlamm, J.; Feller, A.C.; et al. A biologic definition of Burkitt’s lymphoma from transcriptional and genomic profiling. N Engl. J. Med. 2006, 354, 2419–2430. [Google Scholar] [CrossRef] [PubMed]
- Klapper, W.; Szczepanowski, M.; Burkhardt, B.; Berger, H.; Rosolowski, M.; Bentink, S.; Schwaenen, C.; Wessendorf, S.; Spang, R.; Möller, P.; et al. Molecular profiling of pediatric mature B-cell lymphoma treated in population-based prospective clinical trials. Blood 2008, 112, 1374–1381. [Google Scholar] [CrossRef]
- Gao, S.; Ge, A.; Xu, S.; You, Z.; Ning, S.; Zhao, Y.; Pang, D. PSAT1 is regulated by ATF4 and enhances cell proliferation via the GSK3β/β-catenin/cyclin D1 signaling pathway in ER-negative breast cancer. J. Exp. Clin. Cancer Res. 2017, 36, 179. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Z.; Zhao, W.G.; Wang, G.N.; Cui, M.Y.; Zhang, Y.R.; Li, W.C. Inhibitor of DNA binding 4 functions as a tumor suppressor and is targetable by 5-aza-2′-deoxycytosine with potential therapeutic significance in Burkitt’s lymphoma. Mol. Med. Rep. 2016, 13, 1269–1274. [Google Scholar] [CrossRef]
- Robaina, M.C.; Faccion, R.S.; Arruda, V.O.; de Rezende, L.M.; Vasconcelos, G.M.; Apa, A.G.; Bacchi, C.E.; Klumb, C.E. Quantitative analysis of CDKN2A methylation, mRNA, and p16(INK4a) protein expression in children and adolescents with Burkitt lymphoma: Biological and clinical implications. Leuk. Res. 2015, 39, 248–256. [Google Scholar] [CrossRef]
- Guan, H.; Xie, L.; Leithäuser, F.; Flossbach, L.; Möller, P.; Wirth, T.; Ushmorov, A. KLF4 is a tumor suppressor in B-cell non-Hodgkin lymphoma and in classic Hodgkin lymphoma. Blood 2010, 116, 1469–1478. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, X.; Fiskus, W.; Lin, J.; Lwin, T.; Rao, R.; Zhang, Y.; Chan, J.C.; Fu, K.; Marquez, V.E.; et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell 2012, 22, 506–523. [Google Scholar] [CrossRef]
- Sander, S.; Bullinger, L.; Klapproth, K.; Fiedler, K.; Kestler, H.A.; Barth, T.F.; Möller, P.; Stilgenbauer, S.; Pollack, J.R.; Wirth, T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 2008, 112, 4202–4212. [Google Scholar] [CrossRef]
- Possemato, R.; Marks, K.M.; Shaul, Y.D.; Pacold, M.E.; Kim, D.; Birsoy, K.; Sethumadhavan, S.; Woo, H.K.; Jang, H.G.; Jha, A.K.; et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 2011, 476, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, A.L.; Walton, Z.E.; Altman, B.J.; Stine, Z.E.; Dang, C.V. MYC and metabolism on the path to cancer. Semin. Cell Dev. Biol. 2015, 43, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058. [Google Scholar] [CrossRef]
- Song, M.; Kim, S.H.; Im, C.Y.; Hwang, H.J. Recent Development of Small Molecule Glutaminase Inhibitors. Curr. Top. Med. Chem. 2018, 18, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, M.A.; Chandriani, S.; O’Connell, B.; Petrenko, O.; Kotenko, I.; Beavis, A.; Sedivy, J.M.; Cole, M.D. A functional screen for Myc-responsive genes reveals serine hydroxymethyltransferase, a major source of the one-carbon unit for cell metabolism. Mol. Cell. Biol. 2002, 22, 5793–5800. [Google Scholar] [CrossRef]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef]
- Fan, J.; Teng, X.; Liu, L.; Mattaini, K.R.; Looper, R.E.; Vander Heiden, M.G.; Rabinowitz, J.D. Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem. Biol. 2015, 10, 510–516. [Google Scholar] [CrossRef]
- Mayers, J.R.; Vander Heiden, M.G. Famine versus feast: Understanding the metabolism of tumors in vivo. Trends Biochem. Sci. 2015, 40, 130–140. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting autophagy in cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef]
- Ghislat, G.; Patron, M.; Rizzuto, R.; Knecht, E. Withdrawal of essential amino acids increases autophagy by a pathway involving Ca2+/calmodulin-dependent kinase kinase-β (CaMKK-β). J. Biol. Chem. 2012, 287, 38625–38636. [Google Scholar] [CrossRef] [PubMed]
- Sharif, T.; Martell, E.; Dai, C.; Ghassemi-Rad, M.S.; Lee, K.; Singh, S.K.; Weaver, I.C.G.; Gujar, S. Phosphoglycerate dehydrogenase inhibition induces p-mTOR-independent autophagy and promotes multilineage differentiation in embryonal carcinoma stem-like cells. Cell Death Dis. 2018, 9, 990. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chung, F.; Yang, G.; Pu, M.; Gao, H.; Jiang, W.; Yin, H.; Capka, V.; Kasibhatla, S.; Laffitte, B.; et al. Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget 2013, 4, 2502–2511. [Google Scholar] [CrossRef]
- Sewastianik, T.; Szydlowski, M.; Bialopiotrowicz, E.; Jablonska, E.; Kiliszek, P.; Polak, A.; Prochorec-Sobieszek, M.; Szumera-Cieckiewicz, A.; Kaminski, T.S.; Gorniak, P.; et al. FOXO1-p300-Txn Circuit Regulates Oxidative Stress Responses in Diffuse Large B-Cell Lymphomas Characterized By Enhanced Oxidative Phosphorylation. Blood 2015, 126, 466. [Google Scholar] [CrossRef]
- Mucha, O.; Podkalicka, P.; Czarnek, M.; Biela, A.; Mieczkowski, M.; Kachamakova-Trojanowska, N.; Stepniewski, J.; Jozkowicz, A.; Dulak, J.; Loboda, A. Pharmacological versus genetic inhibition of heme oxygenase-1—The comparison of metalloporphyrins, shRNA and CRISPR/Cas9 system. Acta Biochim. Pol. 2018, 65, 277–286. [Google Scholar] [CrossRef]
- Szydlowski, M.; Kiliszek, P.; Sewastianik, T.; Jablonska, E.; Bialopiotrowicz, E.; Gorniak, P.; Polak, A.; Markowicz, S.; Nowak, E.; Grygorowicz, M.A.; et al. FOXO1 activation is an effector of SYK and AKT inhibition in tonic BCR signal-dependent diffuse large B-cell lymphomas. Blood 2016, 127, 739–748. [Google Scholar] [CrossRef]
- Bialopiotrowicz, E.; Gorniak, P.; Pula, B.; Noyszewska-Kania, M.; Makuch-Lasica, H.; Nowak, G.; Szydlowski, M.; Jablonska, E.; Sewastianik, T.; Polak, A.; et al. Microenvironment-Induced Expression of PIM Kinases Supports Chronic Lymphocytic Leukemia Cells Survival and Promotes CXCR4-mTOR Pathway Dependent Migration. Blood 2016, 128, 3239. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. World Health Organization Classification of Tumours of Hematopoietic and Lymphoid, 4th ed.; IARC Press: Lyon, France, 2017. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Białopiotrowicz, E.; Noyszewska-Kania, M.; Kachamakova-Trojanowska, N.; Łoboda, A.; Cybulska, M.; Grochowska, A.; Kopczyński, M.; Mikula, M.; Prochorec-Sobieszek, M.; Firczuk, M.; et al. Serine Biosynthesis Pathway Supports MYC–miR-494–EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells. Cancers 2020, 12, 580. https://doi.org/10.3390/cancers12030580
Białopiotrowicz E, Noyszewska-Kania M, Kachamakova-Trojanowska N, Łoboda A, Cybulska M, Grochowska A, Kopczyński M, Mikula M, Prochorec-Sobieszek M, Firczuk M, et al. Serine Biosynthesis Pathway Supports MYC–miR-494–EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells. Cancers. 2020; 12(3):580. https://doi.org/10.3390/cancers12030580
Chicago/Turabian StyleBiałopiotrowicz, Emilia, Monika Noyszewska-Kania, Neli Kachamakova-Trojanowska, Agnieszka Łoboda, Magdalena Cybulska, Aleksandra Grochowska, Michał Kopczyński, Michał Mikula, Monika Prochorec-Sobieszek, Małgorzata Firczuk, and et al. 2020. "Serine Biosynthesis Pathway Supports MYC–miR-494–EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells" Cancers 12, no. 3: 580. https://doi.org/10.3390/cancers12030580
APA StyleBiałopiotrowicz, E., Noyszewska-Kania, M., Kachamakova-Trojanowska, N., Łoboda, A., Cybulska, M., Grochowska, A., Kopczyński, M., Mikula, M., Prochorec-Sobieszek, M., Firczuk, M., Graczyk-Jarzynka, A., Zagożdżon, R., Ząbek, A., Młynarz, P., Dulak, J., Górniak, P., Szydłowski, M., Pyziak, K., Martyka, J., ... Juszczyński, P. (2020). Serine Biosynthesis Pathway Supports MYC–miR-494–EZH2 Feed-Forward Circuit Necessary to Maintain Metabolic and Epigenetic Reprogramming of Burkitt Lymphoma Cells. Cancers, 12(3), 580. https://doi.org/10.3390/cancers12030580