Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma: A Review of the Current Status of Literature

 ,
,
Abstract
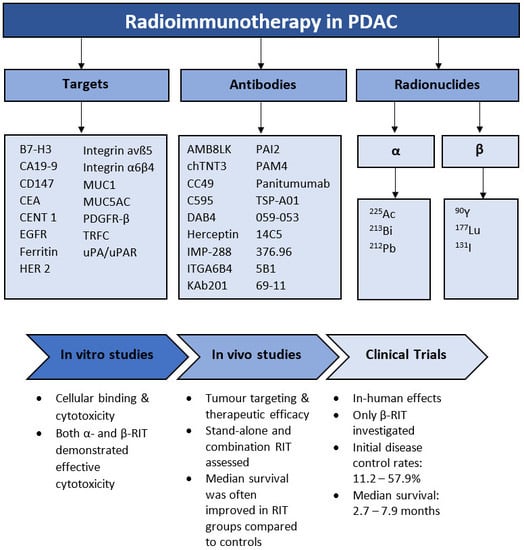
1. Introduction
2. Methods
3. Results and Discussion
3.1. Literature Analysis
3.2. Radionuclides
3.3. Targets
3.4. In Vitro Studies
3.5. In Vivo Studies
3.6. Clinical Trials
3.7. Limitations of Review
3.8. Overall Discussion
4. Conclusions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Search Terms | |
|---|---|
| 1 | Pancreatic Neoplasms/ |
| 2 | Carcinoma, Pancreatic Ductal/ |
| 3 | (Pancrea * adj3 (neoplasm * or cancer * or carcinoma * or tumo?r *)).mp. [mp = title, abstract, original title, name of substance word, subject heading word, floating sub-heading word, keyword heading word, organism supplementary concept word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier, synonyms] |
| 4 | 1 or 2 or 3 |
| 5 | A Particles/tu [Therapeutic Use] |
| 6 | Radioimmunotherapy/ |
| 7 | Β particles/tu [Therapeutic use] |
| 8 | (immuno?radio?therap * or radio?immuno?therap * or α therap * or α particle? therap * or α radio?therap * or β therap * or β particle? therap * or β particle? radio?therap * or β radio?therap * or radio?labelled anti?bod * or α immuno?conjugat * or β immuno?conjugat * or radio?immuno?conjugat *).mp. [mp = title, abstract, original title, name of substance word, subject heading word, floating sub-heading word, keyword heading word, organism supplementary concept word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier, synonyms] |
| 9 | 5 or 6 or 7 or 8 |
| 10 | 4 or 9 |
| 11 | Limit 10 to (English language and year = “2000–current”) |
Appendix B

References
- Australian Institute of Health and Welfare. Cancer in Australia: 2019; Government of Australia: Canberra, Australia, 2019.
- Muniraj, T.; Barve, P. Laparoscopic staging and surgical treatment of pancreatic cancer. N. Am. J. Med. Sci. 2013, 5, 1–9. [Google Scholar] [CrossRef]
- Yoshii, Y.; Matsumoto, H.; Yoshimoto, M.; Oe, Y.; Zhang, M.R.; Nagatsu, K.; Sugyo, A.; Tsuji, A.B.; Higashi, T. 64Cu-intraperitoneal radioimmunotherapy: A novel approach for adjuvant treatment in a clinically relevant preclinical model of pancreatic cancer. J. Nucl. Med. 2019, 60, 1437–1443. [Google Scholar] [CrossRef]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. World J. Gastroenterol. 2018, 24, 4846–4861. [Google Scholar] [CrossRef]
- Mollberg, N.; Rahbari, N.N.; Koch, M.; Hartwig, W.; Hoeger, Y.; Büchler, M.W.; Weitz, J. Arterial resection during pancreatectomy for pancreatic cancer: A systematic review and meta-analysis. Ann. Surg. 2011, 254, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Al-Ejeh, F.; Pajic, M.; Shi, W.; Kalimutho, M.; Miranda, M.; Nagrial, A.M.; Chou, A.; Biankin, A.V.; Grimmond, S.M.; Brown, M.P.; et al. Gemcitabine and Chk1 inhibition potentiate EGFR-directed radioimmunotherapy against pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2014, 20, 3187–3197. [Google Scholar] [CrossRef] [PubMed]
- Pasternack, J.B.; Domogauer, J.D.; Khullar, A.; Akudugu, J.M.; Howell, R.W. The advantage of antibody cocktails for targeted alpha therapy depends on specific activity. J. Nucl. Med. 2014, 55, 2012–2019. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, J.; Rajasekaran, A.K. Biological impediments to monoclonal antibody–based cancer immunotherapy. Mol. Cancer Ther. 2004, 3, 1493–1501. [Google Scholar] [PubMed]
- Gharibi, A.; Adamian, Y.; Kelber, J.A. Cellular and molecular aspects of pancreatic cancer. Acta Histochem. 2016, 118, 305–316. [Google Scholar] [CrossRef]
- Vaupel, P.; Höckel, M.; Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox. Sign. 2007, 9, 1221–1236. [Google Scholar] [CrossRef]
- Marcu, L.; Bezak, E.; Allen, B.J. Global comparison of targeted alpha vs. targeted beta therapy for cancer: In vitro, in vivo and clinical trials. Crit. Rev. Oncol. Hemat. 2018, 123, 7–20. [Google Scholar] [CrossRef]
- Marcu, L.; Bezak, E.; Allen, B. Biomedical Physics in Radiotherapy for Cancer; CSIRO: Canberra, Australia, 2012; Volume 1.
- Ryschich, E.; Huszty, G.; Knaebel, H.P.; Hartel, M.; Büchler, M.W.; Schmidt, J. Transferrin receptor is a marker of malignant phenotype in human pancreatic cancer and in neuroendocrine carcinoma of the pancreas. Eur. J. Cancer 2004, 40, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Roots, R.; Okada, S. Estimation of life times and diffusion distances of radicals involved in x-ray-induced DNA strand breaks or killing of mammalian cells. Radiat. Res. 1975, 64, 306–320. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.; Marcu, L.; Bezak, E. Targeted alpha therapy for cancer. In Advances in Medical Physics; Godfrey, D., Van Dyk, J., Das, S., Curran, B., Wolbarst, A., Eds.; Medical Physics Publishing: Madison, WI, USA, 2016; Volume 6, pp. 177–202. [Google Scholar]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.J. A comparative evaluation of Ac225 vs. Bi213 as therapeutic radioisotopes for targeted alpha therapy for cancer. Australas. Phys. Eng. Sci. Med. 2017, 40, 369–376. [Google Scholar] [CrossRef]
- Pouget, J.P.; Lozza, C.; Deshayes, E.; Boudousq, V.; Navarro-Teulon, I. Introduction to radiobiology of targeted radionuclide therapy. Front. Med. 2015, 2, 1–11. [Google Scholar] [CrossRef]
- Abbas Rizvi, S.M.; Sarkar, S.; Goozee, G.; Allen, B. Radioimmunoconjugates for targeted α therapy of malignant melanoma. Melanoma Res. 2000, 10, 281–289. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Georgakilas, A.G.; Ravanat, J.-L. Targeted and off-target (bystander and abscopal) effects of radiation therapy: Redox mechanisms and risk/benefit analysis. Antioxid. Redox. Sign. 2018, 29, 1447–1487. [Google Scholar] [CrossRef]
- Brady, D.; O’Sullivan, J.M.; Prise, K.M. What is the role of the bystander response in radionuclide therapies? Front. Oncol. 2013, 3, 215. [Google Scholar] [CrossRef]
- Boyd, M.; Ross, S.C.; Dorrens, J.; Fullerton, N.E.; Tan, K.W.; Zalutsky, M.R.; Mairs, R.J. Radiation-induced biologic bystander effect elicited in vitro by targeted radiopharmaceuticals labeled with alpha-, beta-, and auger electron-emitting radionuclides. J. Nucl. Med. 2006, 47, 1007–1015. [Google Scholar]
- Liu, Y.; Dong, Y.; Kong, L.; Shi, F.; Zhu, H.; Yu, J. Abscopal effect of radiotherapy combined with immune checkpoint inhibitors. J. Hematol. Oncol. 2018, 11, 104. [Google Scholar] [CrossRef]
- Loos, M.; Hedderich, D.M.; Ottenhausen, M.; Giese, N.A.; Laschinger, M.; Esposito, I.; Kleeff, J.; Friess, H. Expression of the costimulatory molecule B7-H3 is associated with prolonged survival in human pancreatic cancer. BMC Cancer 2009, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Viola-Villegas, N.T.; Rice, S.L.; Carlin, S.; Wu, X.; Evans, M.J.; Sevak, K.K.; Drobjnak, M.; Ragupathi, G.; Sawada, R.; Scholz, W.W.; et al. Applying PET to broaden the diagnostic utility of the clinically validated CA19.9 serum biomarker for oncology. J. Nucl. Med. 2013, 54, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.; Merchant, N.; Newsome, G.; Goldenberg, D.M.; Gold, D.V. Differentiation of pancreatic ductal adenocarcinoma from chronic pancreatitis by PAM4 immunohistochemistry. Arch. Pathol. Lab. Med. 2014, 138, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Riethdorf, S.; Reimers, N.; Assmann, V.; Kornfeld, J.W.; Terracciano, L.; Sauter, G.; Pantel, K. High incidence of EMMPRIN expression in human tumors. Int. J. Cancer 2006, 119, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Enjoji, M.; Tsuneyoshi, M. Pancreatoduodenal carcinoma: A clinicopathologic study of 304 patients and immunohistochemical observation for CEA and CA19-9. J. Surg. Oncol. 1991, 47, 148–154. [Google Scholar] [CrossRef]
- Jiao, R.; Allen, K.J.H.; Malo, M.E.; Helal, M.; Jiang, Z.; Smart, K.; Buhl, S.V.; Rickles, D.; Bryan, R.A.; Dadachova, E. Evaluation of novel highly specific antibodies to cancer testis antigen centrin-1 for radioimmunoimaging and radioimmunotherapy of pancreatic cancer. Cancer Med. 2019, 8, 5289–5300. [Google Scholar] [CrossRef]
- Sabbah, E.N.; Kadouche, J.; Ellison, D.; Finucane, C.; Decaudin, D.; Mather, S.J. In vitro and in vivo comparison of DTPA- and DOTA-conjugated antiferritin monoclonal antibody for imaging and therapy of pancreatic cancer. Nucl. Med. Biol. 2007, 34, 293–304. [Google Scholar] [CrossRef]
- Harder, J.; Ihorst, G.; Heinemann, V.; Hofheinz, R.; Moehler, M.; Buechler, P.; Kloeppel, G.; Röcken, C.; Bitzer, M.; Boeck, S.; et al. Multicentre phase II trial of trastuzumab and capecitabine in patients with HER2 overexpressing metastatic pancreatic cancer. Brit. J. Cancer 2012, 106, 1033–1038. [Google Scholar] [CrossRef]
- Vervoort, L.; Burvenich, I.; Staelens, S.; Dumolyn, C.; Waegemans, E.; Van Steenkiste, M.; Baird, S.K.; Scott, A.M.; De Vos, F. Preclinical evaluation of monoclonal antibody 14C5 for targeting pancreatic cancer. Cancer Biother. Radio 2010, 25, 193–205. [Google Scholar] [CrossRef]
- Aung, W.; Tsuji, A.B.; Sudo, H.; Sugyo, A.; Furukawa, T.; Ukai, Y.; Kurosawa, Y.; Saga, T. Immunotargeting of integrin α6β4 for single-photon emission computed tomography and near-infrared fluorescence imaging in a pancreatic cancer model. Mol. Imaging 2016, 15, 1–11. [Google Scholar] [CrossRef]
- Cruz-Monserrate, Z.; Qiu, S.; Evers, B.; O’Connor, K. Upregulation and redistribution of integrin α6β4 expression occurs at an early stage in pancreatic adenocarcinoma progression. Mod. Pathol. 2007, 20, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Li, Y.; Song, Y.; Rizvi, S.; Raja, C.; Zhang, D.; Samra, J.; Smith, R.; Perkins, A.C.; Apostolidis, C.; et al. MUC1 expression in primary and metastatic pancreatic cancer cells for in vitro treatment by 213Bi-C595 radioimmunoconjugate. Brit. J. Cancer 2004, 91, 2086–2093. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Yokoi, K.; Bucana, C.D.; Tsan, R.; Killion, J.J.; Evans, D.B.; Fidler, I.J. Inhibition of platelet-derived growth factor receptor phosphorylation by STI571 (Gleevec) reduces growth and metastasis of human pancreatic carcinoma in an orthotopic nude mouse model. Clin. Cancer Res. 2003, 9, 6534. [Google Scholar] [PubMed]
- Qu, C.; Song, E.Y.; Li, Y.; Rizvi, S.M.A.; Raja, C.; Smith, R.; Morgenstern, A.; Apostolidis, C.; Allen, B.J. Pre-clinical study of 213Bi labeled PAI2 for the control of micrometastatic pancreatic cancer. Clin. Exp. Metastasis 2005, 22, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.; Pillai, K.; Morris, D.L. Mucins in pancreatic cancer: Biological role, implications in carcinogenesis and applications in diagnosis and therapy. Am. J. Cancer Res. 2017, 7, 1372–1383. [Google Scholar]
- Bartel, M.J.; Chakraborty, S.; Raimondo, M. Molecular testing in pancreatic cancer. In Diagnostic Molecular Pathology; Coleman, W.B., Tsongalis, G.J., Eds.; Academic Press: Cambridge, MA, USA, 2017; pp. 349–359. [Google Scholar]
- Teng, D.; Wu, K.; Sun, Y.; Zhang, M.; Wang, D.; Wu, J.; Yin, T.; Gong, W.; Ding, Y.; Xiao, W.; et al. Significant increased CA199 levels in acute pancreatitis patients predicts the presence of pancreatic cancer. Oncotarget 2018, 9, 12745–12753. [Google Scholar] [CrossRef]
- Willett, C.G.; Daly, W.J.; Warshaw, A.L. CA 19-9 is an index of response to neoadjunctive chemoradiation therapy in pancreatic cancer. Am. J. Surg. 1996, 172, 350–352. [Google Scholar] [CrossRef]
- Engle, D.D.; Tiriac, H.; Rivera, K.D.; Pommier, A.; Whalen, S.; Oni, T.E.; Alagesan, B.; Lee, E.J.; Yao, M.A.; Lucito, M.S.; et al. The glycan CA19-9 promotes pancreatitis and pancreatic cancer in mice. Science 2019, 364, 1156–1162. [Google Scholar] [CrossRef]
- Houghton, J.L.; Abdel-Atti, D.; Scholz, W.W.; Lewis, J.S. Preloading with unlabeled CA19.9 targeted human monoclonal antibody leads to improved PET imaging with 89Zr-5B1. Mol. Pharm. 2017, 14, 908–915. [Google Scholar] [CrossRef]
- Kim, J.J.; Rajagopalan, K.; Hussain, B.; Williams, B.H.; Kulkarni, P.; Mooney, S.M. CENT1 is a cancer testis antigen with expression in prostate and pancreatic cancers. Biomark. Res. 2013, 1, 22. [Google Scholar] [CrossRef]
- Saeidnia, S.; Manayi, A.; Abdollahi, M. From in vitro experiments to in vivo and clinical studies; pros and cons. Curr. Drug Discov. Technol. 2015, 12, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Kasten, B.B.; Gangrade, A.; Kim, H.; Fan, J.; Ferrone, S.; Ferrone, C.R.; Zinn, K.R.; Buchsbaum, D.J. 212Pb-labeled B7-H3-targeting antibody for pancreatic cancer therapy in mouse models. Nucl. Med. Biol. 2018, 58, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Songl, Y.J.; Rizvi, S.M.A.; Li, Y.; Smith, R.; Perkins, A.C.; Morgenstern, A.; Brechbiel, M.; Allen, B.J. In vivo and in vitro inhibition of pancreatic cancer growth by targeted alpha therapy using 213Bi-CHX.A”-C595. Cancer Biol. Ther. 2005, 4, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Sugyo, A.; Tsuji, A.B.; Sudo, H.; Koizumi, M.; Ukai, Y.; Kurosawa, G.; Kurosawa, Y.; Saga, T.; Higashi, T. Efficacy evaluation of combination treatment using gemcitabine and radioimmunotherapy with 90Y-labeled fully human anti-CD147 monoclonal antibody 059-053 in a BxPC-3 xenograft mouse model of refractory pancreatic cancer. Int. J. Mol. Sci. 2018, 19, 2979. [Google Scholar] [CrossRef] [PubMed]
- Aghevlian, S.; Cai, Z.; Lu, Y.; Hedley, D.W.; Winnik, M.A.; Reilly, R.M. Radioimmunotherapy of PANC-1 human pancreatic cancer xenografts in nrg mice with panitumumab modified with metal-chelating polymers complexed to 177Lu. Mol. Pharm. 2019, 16, 768–778. [Google Scholar] [CrossRef]
- Aung, W.; Tsuji, A.B.; Sudo, H.; Sugyo, A.; Ukai, Y.; Kouda, K.; Kurosawa, Y.; Furukawa, T.; Saga, T.; Higashi, T. Combined treatment of pancreatic cancer xenograft with 90Y-ITGA6B4-mediated radioimmunotherapy and PI3K/mTOR inhibitor. World J. Gastroenterol. 2017, 23, 7551–7562. [Google Scholar] [CrossRef]
- Sugyo, A.; Tsuji, A.B.; Sudo, H.; Okada, M.; Koizumi, M.; Satoh, H.; Kurosawa, G.; Kurosawa, Y.; Saga, T. Evaluation of efficacy of radioimmunotherapy with 90Y-labeled fully human anti-transferrin receptor monoclonal antibody in pancreatic cancer mouse models. PLoS ONE 2015, 10, e0123761. [Google Scholar] [CrossRef]
- International Atomic Energy Agency. Alpha Emitting Radionuclides and Radiopharmaceuticals for Therapy; International Atomic Energy Agency: Vienna, Austria, 2013; pp. 1–75. [Google Scholar]
- Katt, M.E.; Placone, A.L.; Wong, A.D.; Xu, Z.S.; Searson, P.C. In vitro tumor models: Advantages, disadvantages, variables, and selecting the right platform. Front. Bioeng. Biotechnol. 2016, 4, 12. [Google Scholar] [CrossRef]
- Mehta, G.; Hsiao, A.Y.; Ingram, M.; Luker, G.D.; Takayama, S. Opportunities and challenges for use of tumor spheroids as models to test drug delivery and efficacy. J. Contol. Release 2012, 164, 192–204. [Google Scholar] [CrossRef]
- Sharkey, R.M.; Karacay, H.; Govindan, S.V.; Goldenberg, D.M. Combination radioimmunotherapy and chemoimmunotherapy involving different or the same targets improves therapy of human pancreatic carcinoma xenograft models. Mol. Cancer Ther. 2011, 10, 1072–1081. [Google Scholar] [CrossRef]
- Karacay, H.; Sharkey, R.M.; Gold, D.V.; Ragland, D.R.; McBride, W.J.; Rossi, E.A.; Chang, C.H.; Goldenberg, D.M. Pretargeted radioimmunotherapy of pancreatic cancer xenografts: TF10-90Y-IMP-288 alone and combined with gemcitabine. J. Nucl. Med. 2009, 50, 2008–2016. [Google Scholar] [CrossRef] [PubMed]
- Poty, S.; Carter, L.M.; Mandleywala, K.; Membreno, R.; Abdel-Atti, D.; Ragupathi, A.; Scholz, W.W.; Zeglis, B.M.; Lewis, J.S. Leveraging bioorthogonal click chemistry to improve 225Ac-radioimmunotherapy of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2019, 25, 868. [Google Scholar] [CrossRef] [PubMed]
- Milenic, D.E.; Garmestani, K.; Brady, E.D.; Albert, P.S.; Ma, D.; Abdulla, A.; Brechbiel, M.W. Targeting of HER2 antigen for the treatment of disseminated peritoneal disease. Clin. Cancer Res. 2004, 10, 7834–7841. [Google Scholar] [CrossRef] [PubMed]
- Bryan, R.A.; Jiang, Z.; Jandl, T.; Strauss, J.; Koba, W.; Onyedika, C.; Morgenstern, A.; Bruchertseifer, F.; Epstein, A.L.; Dadachova, E. Treatment of experimental pancreatic cancer with 213-bismuth-labeled chimeric antibody to single-strand DNA. Expert Rev. Anticancer Ther. 2014, 14, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Song, E.Y.; Rizvi, S.M.A.; Qu, C.F.; Raja, C.; Brechbiel, M.W.; Morgenstern, A.; Apostolidis, C.; Allen, B.J. Pharmacokinetics and toxicity of 213Bi-labeled PAI2 in preclinical targeted alpha therapy for cancer. Cancer Biol. Ther. 2007, 6, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Houghton, J.L.; Membreno, R.; Abdel-Atti, D.; Cunanan, K.M.; Carlin, S.; Scholz, W.W.; Zanzonico, P.B.; Lewis, J.S.; Zeglis, B.M. Establishment of the in vivo efficacy of pretargeted radioimmunotherapy utilizing inverse electron demand Diels-Alder click chemistry. Mol. Cancer Ther. 2017, 16, 124–133. [Google Scholar] [CrossRef]
- Aung, W.; Tsuji, A.B.; Sudo, H.; Sugyo, A.; Ukai, Y.; Kouda, K.; Kurosawa, Y.; Furukawa, T.; Saga, T. Radioimmunotherapy of pancreatic cancer xenografts in nude mice using 90Y-labeled anti-α6β4 integrin antibody. Oncotarget 2016, 7, 38835–38844. [Google Scholar] [CrossRef]
- Aghevlian, S.; Lu, Y.; Winnik, M.A.; Hedley, D.W.; Reilly, R.M. Panitumumab modified with metal-chelating polymers (MCP) complexed to 111In and 177Lu—An EGFR-targeted theranostic for pancreatic cancer. Mol. Pharm. 2018, 15, 1150–1159. [Google Scholar]
- Al-Ejeh, F.; Darby, J.M.; Brown, M.P. Chemotherapy synergizes with radioimmunotherapy targeting La autoantigen in tumors. PLoS ONE 2009, 4, e4630. [Google Scholar] [CrossRef]
- Baranowska-Kortylewicz, J.; Abe, M.; Nearman, J.; Enke, C.A. Emerging role of platelet-derived growth factor receptor-beta inhibition in radioimmunotherapy of experimental pancreatic cancer. Clin. Cancer Res. 2007, 13, 299–306. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Ying, Z.; Gold, D.V. Therapeutic advantage of 90Yttrium- versus 131Iodine-labeled PAM4 antibody in experimental pancreatic cancer. Clin. Cancer Res. 2001, 17, 3186–3192. [Google Scholar]
- Gold, D.V.; Cardillo, T.; Goldenberg, D.M.; Sharkey, R.M. Localization of pancreatic cancer with radiolabeled monoclonal antibody PAM4. Crit. Rev. Oncol. Hemat. 2001, 39, 147–154. [Google Scholar] [CrossRef]
- Cardillo, T.M.; Blumenthal, R.; Ying, Z.; Gold, D.V. Combined gemcitabine and radioimmunotherapy for the treatment of pancreatic cancer. Int. J. Cancer 2002, 97, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Gold, D.V.; Schutsky, K.; Modrak, D.; Cardillo, T.M. Low-dose radioimmunotherapy (90Y-PAM4) combined with gemcitabine for the treatment of experimental pancreatic cancer. Clin. Cancer Res. 2003, 9, 3929s–3937s. [Google Scholar] [PubMed]
- Gold, D.V.; Modrak, D.E.; Schutsky, K.; Cardillo, T.M. Combined 90Yttrium-DOTA-labeled PAM4 antibody radioimmunotherapy and gemcitabine radiosensitization for the treatment of a human pancreatic cancer xenograft. Int. J. Cancer 2004, 109, 618–626. [Google Scholar] [CrossRef]
- Pauwels, B.; Korst, A.E.C.; Lardon, F.; Vermorken, J.B. Combined modality therapy of gemcitabine and radiation. Oncologist 2005, 10, 34–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Wei, M.F.; Wang, C.W.; Lee, H.W.; Pan, S.L.; Gao, M.; Kuo, S.H.; Cheng, A.L.; Teng, C.M. Dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitor is an effective radiosensitizer for colorectal cancer. Cancer Lett. 2015, 357, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.; Jaggi, J.S.; O’Donoghue, J.A.; Ruan, S.; McDevitt, M.; Larson, S.M.; Scheinberg, D.A.; Humm, J.L. Renal uptake of bismuth-213 and its contribution to kidney radiation dose following administration of actinium-225-labeled antibody. Phys. Med. Biol. 2011, 56, 721–733. [Google Scholar] [CrossRef]
- de Kruijff, R.M.; Raavé, R.; Kip, A.; Molkenboer-Kuenen, J.; Morgenstern, A.; Bruchertseifer, F.; Heskamp, S.; Denkova, A.G. The in vivo fate of 225Ac daughter nuclides using polymersomes as a model carrier. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef]
- de Kruijff, R.M.; Wolterbeek, H.T.; Denkova, A.G. A critical review of alpha radionuclide therapy-how to deal with recoiling daughters? Pharmaceuticals 2015, 8, 321–336. [Google Scholar] [CrossRef]
- International Commission on Radiological Protection (ICRP). Limits for Intakes of Radionuclides by Workers; ICRP: Oxford, UK, 1980. [Google Scholar]
- Sultana, A.; Shore, S.; Raraty, M.G.; Vinjamuri, S.; Evans, J.E.; Smith, C.T.; Lane, S.; Chauhan, S.; Bosonnet, L.; Garvey, C.; et al. Randomised phase I/II trial assessing the safety and efficacy of radiolabelled anti-carcinoembryonic antigen I(131) KAb201 antibodies given intra-arterially or intravenously in patients with unresectable pancreatic adenocarcinoma. BMC Cancer 2009, 25, 66. [Google Scholar]
- Picozzi, V.J.; Ramanathan, R.K.; Lowery, M.A.; Ocean, A.J.; Mitchel, E.P.; O’Neil, B.H.; Guarino, M.J.; Conkling, P.R.; Cohen, S.J.; Bahary, N.; et al. 90Y-clivatuzumab tetraxetan with or without low-dose gemcitabine: A phase Ib study in patients with metastatic pancreatic cancer after two or more prior therapies. Eur. J. Cancer 2015, 51, 1857–1864. [Google Scholar] [CrossRef] [PubMed]
- Ocean, A.J.; Pennington, K.L.; Guarino, M.J.; Sheikh, A.; Bekaii-Saab, T.; Serafini, A.N.; Lee, D.; Sung, M.W.; Gulec, S.A.; Goldsmith, S.J.; et al. Fractionated radioimmunotherapy with 90Y-clivatuzumab tetraxetan and low-dose gemcitabine is active in advanced pancreatic cancer: A phase 1 trial. Cancer 2012, 118, 5497–5506. [Google Scholar] [CrossRef] [PubMed]
- Gulec, S.A.; Cohen, S.J.; Pennington, K.L.; Zuckier, L.S.; Hauke, R.J.; Horne, H.; Wegener, W.A.; Teoh, N.; Gold, D.V.; Sharkey, R.M.; et al. Treatment of advanced pancreatic carcinoma with 90Y-clivatuzumab tetraxetan: A phase I single-dose escalation trial. Clin. Cancer Res. 2011, 17, 4091–4100. [Google Scholar] [CrossRef] [PubMed][Green Version]
- ClinicalTrials.gov. Phase 3 Trial of 90Y-Clivatuzumab Tetraxetan & Gemcitabine vs. Placebo & Gemcitabine in Metastatic Pancreatic Cancer (PANCRIT®-1) (NCT01956812). Available online: https://clinicaltrials.gov/ct2/show/NCT01956812 (accessed on 7 July 2019).
- Oberstein, P.E.; Olive, K.P. Pancreatic cancer: Why is it so hard to treat? Therap. Adv. Gastroenterol. 2013, 6, 321–337. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.J.; Huang, C.-Y.; Clarke, R.A. Targeted alpha anticancer therapies: Update and future prospects. Biologics 2014, 8, 255–267. [Google Scholar] [CrossRef]
- Wang, Y.; Probin, V.; Zhou, D. Cancer therapy-induced residual bone marrow injury-mechanisms of induction and implication for therapy. Curr. Cancer Ther. Rev. 2006, 2, 271–279. [Google Scholar] [CrossRef]
- Mak, I.W.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Trans. Res. 2014, 6, 114–118. [Google Scholar]






| Half-Life | LET in Water (keV/µm) | Maximum Energy (MeV) | Maximum Range in Water (µm) | Number of PANC-1 Cells Traversed * | |
|---|---|---|---|---|---|
| A emitters | Maximum | ||||
| 225Ac | 9.92 d | 102 | 8.4 | 82.2 | 3.9 |
| 213Bi | 45.6 m | 102 | 8.4 | 82.2 | 3.9 |
| 212Pb | 10.6 h | 99.4 | 8.8 | 88.5 | 4.2 |
| Β emitters | Average | ||||
| 90Y | 2.67 d | 2.00 | 2.28 | 11400 | 570 |
| 177Lu | 6.65 d | 0.28 | 0.498 | 1800 | 125 |
| 131I | 8.02 d | 0.20 | 0.606 | 3032 | 202 |
| Target | Antibody | Expression in PDAC Cells or Tissues | Measurement Type | References |
|---|---|---|---|---|
| B7-H3 | Anti-B7-H3 (1027) | 88.2% (60/68) | IHC | Loos et al. [24] |
| CA19-9 | 5B1 CA19-9 | 77.7% (7/9) 90.7% (39/43) | IHC IHC | Viola-Villegas et al. [25] Shi et al. [26] |
| CD147 | MEM-M6/1 and HIM6 combination | 87.2% (34/39) | IHC | Riethdorf et al. [27] |
| CEA | Anti-CEA | 89.2% (223/250) | IHC | Yamaguchi et al. [28] |
| CENT 1 | Anti-CENT 1 | 50.0% (10/20) | IHC | Jiao et al. [29] |
| EGFR | Anti-EGFR (H11) | 88.5% (92/104) | IHC | Al-Ejeh et al. [6] |
| Ferritin | Bz-DTPA-AMB8LK | 52% (CAPAN-1 cells) | Immunoreactivity | Sabbah et al. [30] |
| HER 2 | Anti-HER 2 | 59.9% (124/207) | IHC | Harder et al. [31] |
| Integrin avß5 | 14C5 | 100 (4/4) | IHC | Vervoort et al. [32] |
| Integrin α6β4 | ITGA6B4 | 55% (range: 13.1–91.0% across four cell lines) | Flow cytometry | Aung et al. [33] |
| 439-9B | 92.0% (104/113) | IHC | Cruz-Monserrate et al. [34] | |
| MUC1 | C595 MA5 | 90.5% (48/53) 100% (43/43) | IHC IHC | Qu et al. [35] Shi et al. [26] |
| MUC1/MUC5AC | PAM4 | 79.1% (34/43) | IHC | Shi et al. [26] |
| PDGFR-β | Anti-PDGFR-β | 100% (5/5) | IHC | Hwang et al. [36] |
| TRFC | Anti-TFRC (Ber-T9) | 80.4% (41/51) | IHC | Ryschich et al. [13] |
| uPA/uPAR | PAI2 | 87% (26/30) | IHC | Qu et al. [37] |
| Study/Objective | Target | RIC | Dissociation Constant ± SEM (nmol/L) | Cell Binding (%) | Survival Using RIC | Survival Using Non-Specific Control RIC | Apoptotic Cells Or γ-H2AX Foci (% ± SEM Where Available) |
|---|---|---|---|---|---|---|---|
| A-RIT Studies | |||||||
| Kasten et al. [46] Cell binding | B7-H3 | 212Pb-376.96 | Adherent cells: 9.0 ± 1.1 CIC: 21.7 ± 0.7 | Internalisation: Adherent cells: 44 CIC: 40 | Measure: IC50 ± SEM Adherent cells: 41 ± 14 CIC: 26 ± 17 | Measure: IC50 ± SEM Adherent cells: 120 ± 12 CIC cells: 180 ± 150 | Not investigated |
| Qu et al. [37] In vitro cytotoxicity | uPA/uPAR | 213Bi-PAI2 | Not investigated | Not investigated | Measure: activity for 37% cell survival (kBq) CFPAC-1: 133 CAPAN-1 185 PANC-1: 170 At 370 kBq: 5%–10% cell survival | Measure: activity for 37% cell survival 2.2–2.7 MBq for all cell lines At 370 kBq: 90%–95% cell survival | Measure: apoptotic cells At 24 h: CFPAC-1: 92 CAPAN-1: 87 PANC-1: 90 Control RIC: <5% |
| Qu et al. [47] In vitro cytotoxicity | MUC1 | 213Bi-CHX.A”-C595 | Not investigated | Not investigated | Not investigated | Not investigated | Measure: apoptotic cells At 48 h: CAPAN: 73 ± 2.6 PANC-1: 78 ± 1.8 CFPAC-1: 81 ± 3.0 Control RIC: <12 ± 3.0 |
| Qu et al. [35] In vitro cytotoxicity | MUC1 | 213Bi-cDTPA-C595 | Not investigated | Not investigated | Measure: activity for 37% cell survival (kBq) CAPAN-1: 167 CFPAC-1: 141 PANC-1: 159 At 370 kBq: 5%–10% cell survival | Measure: activity for 37% cell survival (MBq) 2.2–2.6 for all cell lines At 370 kBq: 90–95% cell survival (control RIC) >95% cell survival (conjugated mAb or intact mAb only) | Measure: Combined cell counts 4 h: 11 8 h: 18 12 h: 42 24 h: 87 48 h: 92 72 h: 81 Control RIC: <15% at all time points |
| B-RIT Studies | |||||||
| Sugyo et al. [48] Cell binding | CD147 | 111In-059-053 (diagnostic agent to 90Y-059-053) | Intact 059-053: 0.35 CHX-A”-DTPA-059-053: 0.99 | 111In-059-053: 51 in BxPC-3 cells | Not investigated | Not investigated | Not investigated |
| Aghevlian et al. [49] In vitro cytotoxicity | EGFR | 177Lu-MCP-panitumumab | Not investigated | RIC: 4.16 ± 0.17 RIC with excess mAB: 0.35 ± 0.01 Control RIC with excess mAB: 1.21 ± 0.18 | Measure: clonogenic survival of treated cells compared to untreated controls 1.2 MBq RIC: 7.4-fold decrease 0.6 Mbq RIC: 9.0-fold decrease 0.3 MBq RIC: 1.9-fold decrease | Not investigated | Measure: γ-H2AX foci 1.2 MBq RIC: 3.8-fold increase in density vs. untreated controls |
| Al-Ejeh et al. [6] In vitro cytotoxicity | EGFR | 177Lu-anti-EGFR | Not investigated | Not investigated | Measure: clonogenic survival Triple combination therapy (RIT, Chk1i and gemcitabine) significantly reduced clonogenic survival vs. untreated controls | Quantitative results not presented | Not investigated at an in vitro level |
| Sabbah et al. [30] Cell binding | Ferritin | 111In- and 90Y-labelled Bz-DTPA-AMB8LK, Bz-CHX-AU-DTPA-AMB8LK and Bz-DOTA-AMB8LK. | Not investigated | 111In-DTPA-AMB8LK: 52 111In-CHX-A”-DTPA-AMB8LK: 43 111In-DOTA-AMB8LK: 24 | Not investigated | Not investigated | Not investigated |
| Vervoort et al. [32] Cell binding | Integrin avß5 | 125I-14C5, 111In-DOTA- 14C5 and 111In-DTPA-14C5 | 125I-14C5: 0.11 ± 0.01 111In-DTPA-14C5: 0.24 ± 0.02 111In-DOTA-14C5: 0.11 ± 0.03 Control RIC: no specific binding | Internalisation at 24 h 125I-14C5: 6.93 ± 0.88 111In-DOTA- 14C5: 49.44 ± 0.75 111In-DTPA-14C5: 36.66 ± 1.42 | Not investigated | Not investigated | Not investigated |
| Aung et al. [50] In vitro cytotoxicity | Integrin α6β4 | 90Y-ITGA6B4 | Not investigated | Not investigated | Measure: colony formation at 24 h RIC and BEZ235: 90.9% reduction of PE vs. control RIC only: 52.5% reduction of PE vs. control | Included in previous column | Measure: γ-H2AX positive cells (mean ± SD) at Day 3 RIC and BEZ235: 19.3 ± 7.0 RIC only: 12.7 ± 4.7 BEZ235 only: 4.0 ± 2.2 Control: 2.3 ± 1.2 |
| Sugyo et al. [51] Cell binding | Transferrin | 111In-labelled TSP-A01, DOTA-TSP-A01, TSP-A02 and DOTA-TSP-A02 (diagnostic agents for 90Y-TSP-A01) | 111In-TSP-A01: 0.22 111In-DOTA-TSP-A01: 0.28 111In-TSP-A02: 0.17 111In-DOTA-TSP-A02: 0.22 | Immunoreactive fraction: 111In-TSP-A01 and 111In-DOTA-TSP-A02: 1.0 | Not investigated | Not investigated | Not investigated |
| Study | Target | RIC | Therapies Assessed | Survival | Tumour Growth | Tumour Uptake (% ID/g ± SD) | Side Effects |
|---|---|---|---|---|---|---|---|
| A-RIT Studies | |||||||
| Kasten et al. [46] | B7-H3 | 212Pb-376.96 | RIT only | Not investigated | Significant inhibition of tumour growth at all RIT dose levels compared to untreated controls | At 24 h: 14.0 ± 2.1 (RIT) 6.5 ± 0.9 (212Pb-control) | Transient weight loss |
| Poty et al. [57] | CA19-9 | 225Ac-5B1 | PT-RIT | Median survival (orthotopic tumours): 67.5 days (37 kBq PT-RIT) 60.0 days (37 kBq RIT only) 32 days (18.5 kBq PT-RIT) 46 days (18.5 kBq RIT only) 28.5 days (vehicle-only control) | Not investigated | At 4 h: 4.6 ± 3.3 (PT-RIT) 15.4 ± 3.5 (conventional RIT) At 72 h: 29.6 ± 6.6 (PT-RIT) 31.1 ± 21.4 (conventional RIT) | All RIT groups: Transient weight loss, mild nephrotoxicity, transient haemotoxicity (more severe in conventional RIT group compared to PT-RIT group) Conventional RIT: disseminated intravascular coagulation (2/10) |
| Jiao et al. [29] | CENT1 | 213Bi-69-11 | Comparison of 213Bi-69-11 and 177Lu-69-11 | Not investigated | 3.7–7.4 MBq of 213Bi-69-11: Significant reduction in tumour growth rate compared to controls | Not investigated | Transient haemotoxicity |
| Milenic et al. [58] | HER2 | 213Bi-Herceptin | RIT only | Median survival: 15 days (untreated controls)2 2 days (213Bi-control) 26 days (18.5 MBq RIT) 28 days (37 MBq RIT) 26 days (74 MBq RIT) | Not investigated in PDAC xenografts | 111In-Herceptin 24 h: 19.47 ± 3.04 48 h: 31.00 ± 8.92 72 h: 34.00 ± 10.15 120 h: 29.89 ± 3.96 168 h: 15.34 ± 5.14 | Increasing weight loss with dose |
| Bryan et al. [59] | ssDNA and RNA | 213Bi-chTNT3 | RIT compared to gemcitabine and cisplatin | Survival: 100% at day 65 (RIT, cold chTNT3 and untreated) 40% at day 65 (gemcitabine) 0% at day 15 (cisplatin) | Significant reduction in tumour size for RIT and gemcitabine compared to controls | Ratio of sum of pixels in tumour region to sum of pixels in internal organs: 1 h: 0.18 2 h: 0.22 24 h: 0.72 48 h: 0.68 | No RIT-related side effects |
| Qu et al. [37] | uPA/uPAR | 213Bi-PAI2 | Comparing local and systemic RIT injections | Local injection Time to end point: 35 days (cold PAI2) >84 days (≥111 MBq/kg RIT) Systemic injection Time to end point: 35 days (cold PAI2) 50 days (111 MBq/kg RIT) 66 days (222 MBq/kg RIT) | Local injection Tumour growth in: 0/5 tumours (222 MBq/kg RIT) 3/5 tumours (111 MBq/kg RIT) 5/5 tumours (cold PAI2) Systemic injection Tumour growth in: 3/5 tumours (222 MBq/kg RIT) 5/5 tumours (111 MBq/kg RIT) 5/5 tumours (cold PAI2) | Not investigated | Not reported |
| Song et al. [60] | uPA/uPAR | 213Bi-PAI2 | RIT only | Time to end point: 175 days (470 MBq/kg RIT) 162 days (590 MBq/kg RIT) Did not reach end-point (350 MBq/kg RIT and control groups) | Not investigated | Not investigated | Body weight loss with increasing dose. Decline in renal function. |
| Qu et al. [47] | MUC1 | 213Bi-C595 | Comparing local and systemic RIT injections | Local injection Time to end point: 42 ± 7 days (cold C595) 74 ± 3 days (213Bi-control) 77 days (1.85 MBq RIT) >112 days (3.7–7.4 MBq RIT) Systemic injection 42 days (cold C595) 56 days (213Bi-control) >112 days (≥ 111 MBq/kg RIT) | Local injection Tumour growth in: 0/5 tumours (3.7–7.4 MBq RIT) 1/5 tumours (1.85 MBq RIT) 5/5 tumours (cold C595 and 213Bi-control) Systemic injection Tumour growth in: 0/5 tumours (≥222 MBq/kg RIT) 2/5 tumours (111 MBq/kg RIT) 5/5 tumours (cold C595 and 213Bi-control) | Not investigated | Transient weight loss |
| B-RIT Studies | |||||||
| Jiao et al. [29] | CENT1 | 177Lu-69-11 | Comparison of 213Bi-69-11 and 177Lu-69-11 | Not investigated | 177Lu-69-11: No significant reduction in tumour growth compared to control treatments | Clear localisation of RIC in tumour at 24 h | Transient haemotoxicity |
| Houghton et al. [61] | CA19-9 | 177Lu-DOTA-PEG7-Tz | PT-RIT using 5B1-TCO | Not investigated | Tumour doubling time was significantly increased in 44.4 MBq PT-RIT compared to controls and 14.8 MBq PT-RIT. Tumour volume was reduced in 44.4 and 29.6 MBq PT-RIT compared to controls. | At 120 h: 16.8 ± 3.87 (PT-RIT) | No side effects observed |
| Sharkey et al. [55] | MUC1 | 90Y-hPAM4 | Combined RIT and antibody-drug conjugate (ADC) (hRS7–SN-38) | Median time to progression: 4.3 weeks (untreated) 9.75 weeks (ADC only) 13 weeks (2.78 MBq RIT only) >21 weeks (Combined therapy and 4.8 MBq RIT only) | Tumour-free mice at 21 weeks: 0/9 (untreated control) 1/10 (ADC only) 1/10 (2.8 MBq RIT only) 5/10 (4.8 MBq RIT only) 4/10 (Combined therapy using 2.8 MBq RIT) 9/10 (Combined therapy using 4.8MBq RIT) | At 48 h: 48.4 ± 16.4 | Transient weight loss |
| Aung et al. [62] | Integrin | 90Y-ITGA6B4 | Single and double RIT cycles | Not calculated—all mice euthanised at day 27 | Growth rates significantly reduced in single and double RIT cycles compared to controls | Not investigated | Increasing haemotoxicity with RIT activity |
| Aung et al. [50] | Integrin | 90Y-ITGA6B4 | Combined RIT and PI3K/mTOR inhibitor (BEZ235) | Not investigated | Compared to controls, tumour growth significantly delayed for: 58 days (2.8 MBq RIT only) 23 days (BEZ235 only) Compared to RIT only, tumour growth significantly delayed for 27 days for combined therapy Compared to BEZ235 only, tumour growth significantly delayed for 41 days for combined therapy. | Not investigated | No side effects observed |
| Aghevlian et al. [63] | EGFR | 177Lu-panitumumab | RIT only | Not investigated | Not investigated | At 72 h: 6.9 ± 1.3 (111In-MCP-panitumumab) 6.6 ± 3.3 (111In-DOTA-panitumumab) 1.9 ± 0.3 (111In-DOTA-control) 5.4 ± 0.3 (111In-MCP-control) In 100-fold excess of panitumumab: 0.02 ± 0.00 (111In-MCP-panitumumab) 0.06 ± 0.02 (111In-DOTA-panitumumab) | Not investigated |
| Aghevlian et al. [49] | EGFR | 177Lu-panitumumab | RIT only | Not investigated | Tumour growth index at 33 days (mean ± SEM): 2.5 ± 0.3 (RIT) 4.0 ± 0.7 (Control RIC) 6.1 ± 1.1 (cold panitumumab) 5.8 ± 0.5 (untreated) | Absorbed tumour dose for 6 Mbq of RIC: 12.33 ± 0.86 Gy | No significant effects over 14 days |
| Al-Ejeh et al. [6] | EGFR | 177Lu-anti EGFR | Combined RIT, gemcitabine and Chk1 inhibition (triple therapy) | Not investigated | Tumour growth rate of all triple therapy dose combinations was significantly less than combined gemcitabine and Chk1 inhibition. Complete tumour regression in triple therapy. | Not investigated | Weight loss with high doses of gemcitabine or RIT |
| Sugyo et al. [48] | CD147 | 90Y-059-053 | Combined RIT and gemcitabine | Survival at day 42: 0% (untreated, cold CD147, 0.925 and 1.85 MBq RIT) 0% and 20% (3.7 MBq RIT in two experiments) 40% (Combined therapy) | Significant suppression of tumour growth in 3.7 MBq RIT and combined therapy groups compared to untreated and gemcitabine only groups | 111In-059-053 30 min: 1.04 ± 0.16 24 h: 9.23 ± 0.67 48 h: 16.13 ± 0.92 96 h: 16.78 ± 2.61 168 h: 14.98 ± 1.63 | Weight loss, diarrhea and decreasing activity with multiple cycles |
| Sabbah et al. [30] | Ferritin | 90Y and 111In-labelled Bz-DTPA-AMB8LK, Bz-CHX-AU-DTPA-AMB8LK and Bz-DOTA-AMB8LK | RIT only—comparing different conjugates | Not investigated | Not investigated | 90Y-DTPA-AMB8LK: 24 h: 14.0 ± 7.5 48 h: 18.6 ± 1.9 120 h: 16.2 ± 2.9 90Y-DOTA-AMB8LK: 24 h: 14.1 ± 1.2 48 h: 12.9 ± 2.3 120 h: 11.2 ± 4.5 | Not investigated |
| Vervoort et al. [32] | Integrin avß5 | 131I-14C5 | RIT only | Not investigated | Not investigated | 131I-14C5 1 h: 3.63 ± 0.50 24 h: 11.22 ± 3.31 48 h: 12.16 ± 1.03 72 h: 8.45 ± 0.57 168 h: 6.91 ± 1.84 | Not investigated |
| Al-Ejeh et al. [64] | Intracellular La ribonucleoprotein | 90Y-DOTA-DAB4 | Combination RIT, gemcitabine and cisplatin | Median survival 31 days (2.40 MBq RIT only) 47 days (Combined therapy) 24 days (untreated control) | Tumour doubling time (days ± SEM): 4.44 ± 0.02 (control) 5.87 ± 0.04 (RIT only) 4.88 ± 0.01 (chemotherapy only) 8.53 ± 0.02 (combined therapy) | Not investigated in PDAC model | Not investigated in PDAC model |
| Sugyo et al. [51] | Transferrin | 90Y-TSP-A01 | RIT only | Not investigated | BxPC-3 tumours: 1.85 and 3.7 MBq RIT significantly delayed tumour growth compared to unlabelled A01. No significant difference in tumour volume between 0.74 MBq RIT and unlabelled A01. MIAPaCa-2: Tumour volumes in 1.85 MBq and 3.7 MBq RIT groups were reduced to 20%. Complete resolution of tumours treated with 3.7 MBq RIT by 6 weeks. | Peak 111In-TSP-A01 uptake: 37.5 ± 5.3 at 24 h (MIAPaCa-2) 27.0 ± 10.7 at 96 h (BxPC-3) | Transient decrease in body weight |
| Baranowska-Kortylewicz et al. [65] | PDGFR | 131I-CC49 | Combined RIT and PDGFR inhibitor (imatinib) | Not investigated | Tumour doubling time (days): 12.86 ± 0.19 (RIT only) 26.06 ± 1.47 (combined therapy) 13.03 ± 0.27 (imatinib only) 9.05 ± 0.05 (untreated control) | At 120 h: 6.06 ± 1.76 (RIC only) 9.03 ± 1.59 (RIC and imatinib) | No side effects |
| Cardillo et al. [66] | MUC1 | 131I-PAM4 and 90Y-PAM4 | Comparing RICs as stand-alone treatments | Median survival: 6 weeks (untreated) 13 weeks (13 MBq 131I RIT) 12 weeks (19 MBq 131I RIT) 17.5 weeks (26 MBq 131I RIT) 16 weeks (4.8 MBq 90Y RIT) >26 weeks (≥6.5 MBq 90Y RIT) | Mean size of tumours at nadir (cm3): N/A (untreated and ≤19 MBq 131I RIT as tumours never regressed) 0.61 ± 0.24 (26 MBq 131I RIT at 7 weeks) 0.78 ± 0.61 (4.8 MBq 90Y RIT at 6 weeks) 0.33 ± 0.40 (6.5 MBq 90Y RIT at 7 weeks) 0.10 ± 0.07 (8.1 MBq 90Y RIT at 9 weeks) 0.19 ± 0.13 (9.6 MBq 90Y RIT at 10 weeks) | Radiation dose estimates to tumour (cGy): 8559 (26 MBq 131I RIT) 8068 (9.6 MBq 90Y RIT) | Weight loss |
| Gold et al. [67] | MUC1 | 90Y-PAM4 | Single RIT | Not investigated | Not investigated | 96 h: 39.5 ± 16.4 | Not investigated |
| Cardillo et al. [68] | MUC1 | 131I-PAM4 | Combined RIT and gemcitabine | Median survival: 6 weeks (3.7 MBq RIT only) 10 weeks (combined therapy using 3.7 MBq RIT) 9 weeks (combined therapy using 3.7 MBq 131I control) 13 weeks (7.4 MBq RIT only) 13 weeks (combined therapy using 7.4 MBq RIT) 10 weeks (combined therapy using 7.4 MBq control RIC) 6 weeks (untreated controls) 5 weeks (gemcitabine only) | Normalised tumour growth at week 4: 3.91 ± 2.54 (3.7 MBq RIT only) 1.69 ± 1.26 (combined therapy using 3.7 MBq RIT) 1.45 ± 1.05 (combined therapy using 3.7MBq 131I control) 1.58 ± 0.84 (7.4 MBq RIT only) 1.13 ± 0.50 (combined therapy using 7.4 MBq RIT) 1.92 ± 1.02 (combined therapy using 7.4 MBq control RIC) 4.16 ± 0.89 (untreated control) 4.35 ± 1.80 (gemcitabine only) | 131I-PAM4: 24 h: 12.08 ± 6.85 72 h: 11.08 ± 5.56 168 h: 8.04 ± 6.13 336 h: 4.00 ± 2.80 131I-PAM4 and gemcitabine: 24 h: 12.21 ± 5.73 72 h: 14.29 ± 7.31 168 h: 8.39 ± 6.50 336 h: 2.52 ± 2.30 | Weight loss |
| Gold et al. [69] | MUC1 | 90Y-PAM4 | Combined RIT and gemcitabine | Median survival 16 weeks (RIT only) 24 weeks (combined therapy) 11 weeks (combined therapy with 90Y control) 8 weeks (90Y control only) 10 weeks (gemcitabine only) 8.5 weeks (untreated controls) | Tumour response Week 10: 1/9 PR (RIT only) 1/9 CR, 3/9 PR, 2/9 SD (combined therapy) Disease progression in all other groups Week 26: 4/9 CR (combined therapy) | 111In-cPAM4: 24 h: 18.65 ± 2.93 96 h: 26.93 ± 11.81 168 h: 18.05 ± 11.02 111In-cPAM4 and gemcitabine: 24 h: 21.79 ± 4.55 96 h: 36.70 ± 9.58 168 h: 25.47 ± 10.35 | Weight loss and transient reduction in white blood cell counts |
| Gold et al. [70] | MUC1 | 90Y-PAM4 | Combined RIT and gemcitabine | Median survival 12 weeks (single cycle combined therapy) 9 weeks (single RIT only) 7 weeks (single cycle combined 90Y control) 4 weeks (gemcitabine only) 6 weeks (untreated controls) 21 weeks (double cycle combined therapy) 16 weeks (double RIT only) 10 weeks (double cycle combined 90Y control) | Tumour response: 1/13 PR, 8/13 SD (single cycle combined therapy) 7/12 SD (single RIT only) 1/8 PR (90Y control only) 1/10 CR (single cycle combined 90Y control therapy) 1/13 SD (double cycle 1.85 MBq 90Y control) 3/13 SD, 1/13 PR (double cycle combined 1.85 MBq 90Y control therapy) 3/12 SD, 4/12 PR (double cycle 3.7 MBq RIT) 4/12 SD, 7/12 PR (double cycle combined 3.7 MBq RIT) | Not investigated | Transient weight loss |
| Karacay et al. [56] | MUC1 | PT-RIT: 90Y-IMP-288 RIT only: 90Y-PAM4 | Combined TF10 PT-RIT and gemcitabine | Time to progression 16.3 weeks (9.25 MBq PT-RIT) 5.4 week (untreated controls) >30 weeks (18.5 MBq PT-RIT and 5.55 MBq 90Y RIT only) 4.8 weeks (9.25MBq PT-RIT) 18.1 weeks (combined PT-RIT and gemcitabine) | Tumour-free mice at week 19: 8/10 (18.5 MBq PT-RIT) 3/10 (9.25 MBq PT-RIT) 8/9 (5.55 MBq RIT only) 0/10 (untreated controls) | Not investigated | Transient decrease in white blood cell counts, diarrhea (1/11). No nephrotoxicity observed. |
| Study | Target | RIC | Study Type | Sample (n) | Patient Group | Median Overall Survival (Months) | Tumour Responses | Disease Control Rate | Adverse Events * (Grade ≥ 3) |
|---|---|---|---|---|---|---|---|---|---|
| Sultana et al. [77] | CEA | 131I-KAb201 | Single RIT comparing IV and IA administration | 18 | Locally advanced or metastatic PDAC, with at least one tumour site in head of pancreas. KPS ≥ 70, life expectancy < 3 months. Prior treatment allowed but not necessary for inclusion. | 5.2 No survival difference between IV and IA administrations | 1/18 (5.6%) partial responses 1/18 (5.6%) stable disease 16/18 (88.9%) progression | 11.2% | In total, 31 therapy related adverse events were observed. Haemotological toxicity, 18 events; sepsis and vomiting, two events each; alanine aminotransferase, anaemia, anorexia, aspartate aminotransferase, blood alkaline phosphatase, febrile neutropenia, haematemesis, neutrophilia and thrombosis, one event each. |
| Picozzi et al. [78] | MUC1/MUC5ac | 90Y-hPAM4 | Combination with gemcitabine | 58 | Metastatic PDAC, ≥ 2 prior chemotherapy regimens with measurable disease by CT. No CNS metastases of single masses ≥10cm. KPS ≥ 70. Adequate haematologic parameters. | 2.7 (overall survival for all patients) 7.9 (multiple cycles of combined therapy) 3.4 (multiple cycles of RIT only) | 2/29 (6.9%) partial responses (combined therapy group) 10/29 (34.5%) stable disease (combined therapy group) 12/29 (41.4%) stable disease (RIT only group) | 41.4% | Thrombocytopenia, 19% of patients (11/58); anaemia, leukopenia and neutropenia, 7% each (4/58), unspecified, 2% (1/58). |
| Ocean et al. [79] | MUC1 | 90Y-hPAM4 | Fractionated RIT combined with gemcitabine | 38 | Untreated adults with locally advanced or metastatic PDAC. KPS ≥ 70. Life expectancy > 3 months, no CNS tumours or single tumour mass > 10cm. Adequate haematologic parameters. | 7.7 | 6/38 (15.8%) partial response 16/38 (42.1%) stable disease 16/38 (42.1%) disease progression | 57.9% | No significant therapy related adverse events occurred. |
| Gulec et al. [80] | MUC1 | 90Y-hPAM4 | Single RIT | 20 | Stage III or IV PDAC. If stage III, must have progressed after therapy. Stage IV patients must have had no more than one prior chemotherapy regimen. No CNS tumours or single mass > 10cm. KPS ≥ 70 or ECOG ≤1. Adequate haematological parameters. | 4.3 | At 4 weeks: 3/20 (15.0%) partial response 4/20 (20.0%) stable disease | At 4 weeks: 35.0% Follow up: 0% | Eight therapy related adverse events occurred consisting of seven cytopenia events and a single vomiting event. |
| ClinicalTrials.gov [81] | MUC1 | 90Y-hPAM4 | RIT with gemcitabine | Data not available | Metastatic PDAC, completed at least one prior treatment cycle, progressed following gemcitabine regimen, KPS ≥ 70. No CNS tumours or single mass > 10 cm. | No significant improvements in survival in combined therapy group compared to gemcitabine only group | Data not available | Data not available | Data not available. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hull, A.; Li, Y.; Bartholomeusz, D.; Hsieh, W.; Allen, B.; Bezak, E. Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma: A Review of the Current Status of Literature. Cancers 2020, 12, 481. https://doi.org/10.3390/cancers12020481
Hull A, Li Y, Bartholomeusz D, Hsieh W, Allen B, Bezak E. Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma: A Review of the Current Status of Literature. Cancers. 2020; 12(2):481. https://doi.org/10.3390/cancers12020481
Chicago/Turabian StyleHull, Ashleigh, Yanrui Li, Dylan Bartholomeusz, William Hsieh, Barry Allen, and Eva Bezak. 2020. "Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma: A Review of the Current Status of Literature" Cancers 12, no. 2: 481. https://doi.org/10.3390/cancers12020481
APA StyleHull, A., Li, Y., Bartholomeusz, D., Hsieh, W., Allen, B., & Bezak, E. (2020). Radioimmunotherapy of Pancreatic Ductal Adenocarcinoma: A Review of the Current Status of Literature. Cancers, 12(2), 481. https://doi.org/10.3390/cancers12020481