1. Introduction
Ovarian cancer remains a significant cause of female cancer-related deaths, with approximately 21,750 new cases and 13,940 deaths for 2020 in the US [
1]. Its high death rate is reflective of the fact that most ovarian cancer patients are diagnosed with advanced-stage disease [
2]. Epithelial ovarian cancer (EOC)—the most common type of ovarian malignancies—is a complex group of diseases that, based on histopathology and molecular genetic alterations, could be divided into five main types including high-grade serous, endometrioid, clear-cell, mucinous, and low-grade serous carcinomas [
3,
4,
5]. High-grade serous ovarian cancer (HGSOC) represents 70% of all EOCs [
5]. Despite initial response to first-line treatment with platinum-based combination chemotherapy, most HGSOC patients will relapse and eventually develop platinum-resistant disease with a poor overall prognosis [
6]. Therefore, novel therapies targeting key survival pathways are urgently needed to improve clinical outcomes for women with this deadly disease.
Integrin-linked kinase (ILK) is a highly-conserved serine/threonine kinase and adaptor protein connecting the extracellular matrix (ECM), cell membrane-associated proteins (i.e., integrins), and cell signaling pathways [
7,
8]. ILK activity is stimulated by integrins and growth factors in a phosphatidylinositol 3-kinase (PI3K)-dependent manner [
9]. On the other hand, ILK is negatively regulated by phosphatase and tensin homolog (PTEN) and ILK-associated serine/threonine phosphatase 2C (ILKAP) [
9]. In addition, ILK-mediated signaling and intracellular translocation is regulated by p21-activated kinase 1 (PAK1) through ILK phosphorylation at threonine 173 (T173) and serine 246 (S246) residues [
10]. Once activated, ILK can mediate a variety of cellular functions through regulation of its downstream targets including protein kinase B (AKT), glycogen synthase kinase 3β (GSK3β), and nuclear factor kappa B (NFκB) [
11].
Increased levels of ILK have been associated with cancer progression, epithelial to mesenchymal transition (EMT), angiogenesis, and metastasis [
7,
8,
9,
12,
13]. In ovarian cancer, ILK expression has been previously associated with tumor progression [
14]. Early reports show that subcutaneous injection of ovarian cancer cells transfected with either ILK-short hairpin RNA (shRNA) or ILK-antisense oligonucleotides (ASOs) suppresses tumor formation [
15] and growth [
16] in vivo. Other studies have shown that ILK activity modulates the pro-metastatic behavior of ovarian cancer [
17] by stimulating cell invasion and migration [
18]. Moreover, increased ILK activation has been shown to protect platinum-resistant ovarian cancer cells from cisplatin-induced apoptosis [
19]. Targeting ILK abrogates the invasive potential of ovarian cancer cells [
15], induces apoptosis [
20,
21], and decreases cell viability [
22].
The molecular and biological effects of siRNA-mediated ILK targeting in cisplatin-resistant ovarian cancer and the identification of associated downstream effectors have not been fully explored. In this study, we assessed the phosphorylation levels of ILK in ovarian cancer cell lines and tissue samples and investigated the effects of targeting ILK in cisplatin-resistant ovarian cancer. We report that targeting ILK with siRNA induces significant changes in the expression of several cancer-associated protein-coding genes and non-coding RNAs involved in cell growth, survival, and metastasis. Expression-based survival analysis using the Kaplan–Meier (KM) plotter database revealed significant associations between potential ILK target genes and ovarian cancer prognosis. Results from this study suggest that ILK-regulated genes may serve as potential prognostic markers and therapeutic targets in ovarian cancer.
3. Discussion
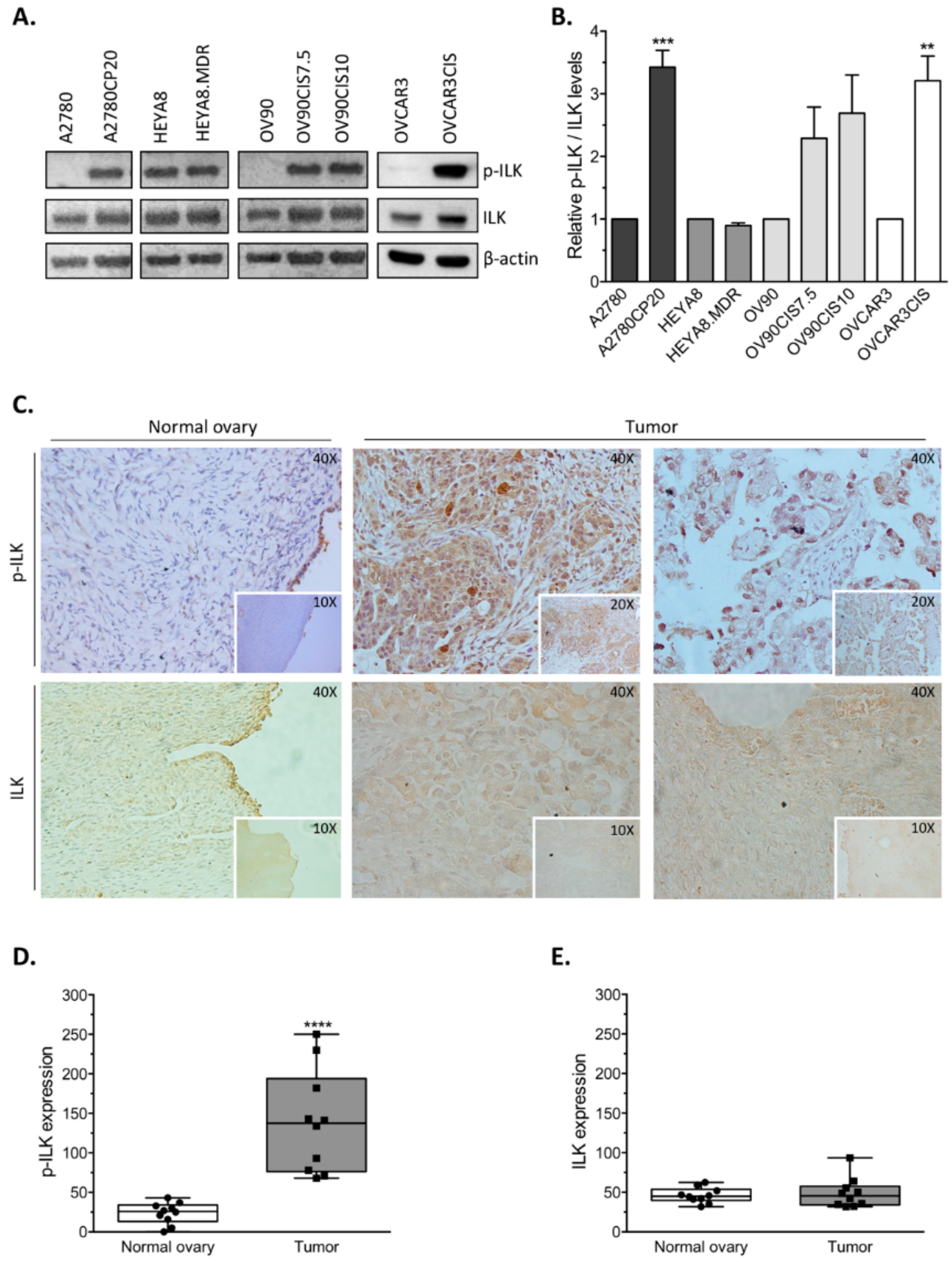
Increased levels of ILK, a serine/threonine kinase and adaptor protein, have been well documented in different tumor types, including ovarian cancer [
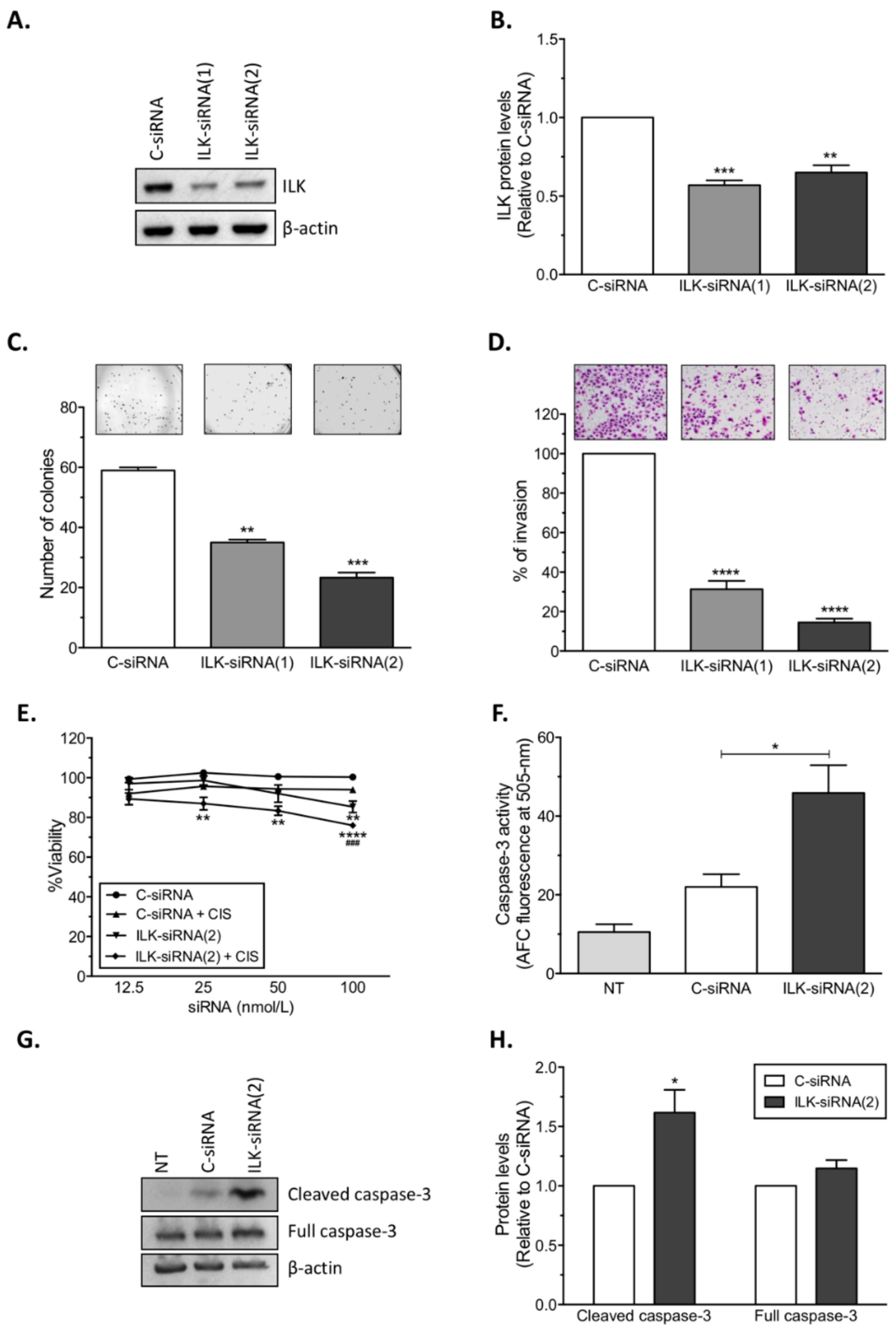
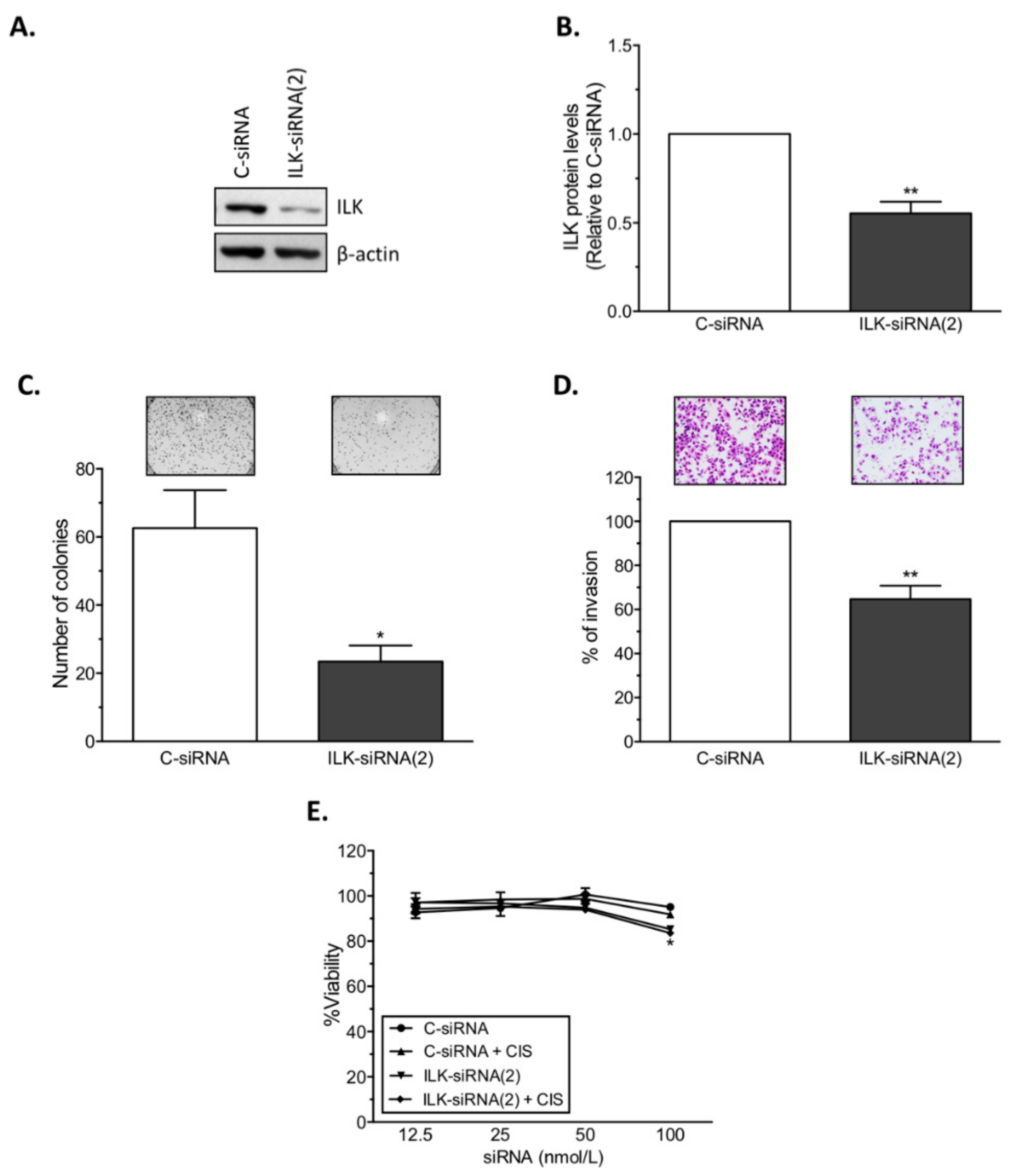
14]. Here, we present further evidence that phosphorylated ILK (p-ILK) levels are higher in human ovarian cancer tissues compared with normal ovary samples, and that p-ILK levels relative to total ILK protein (p-ILK/ILK) are higher in cisplatin-resistant compared with cisplatin-sensitive ovarian cancer cells. Transfection of ILK-siRNA into cisplatin-resistant ovarian cancer cells induced long-term effects on cell growth, short-term effects on cell viability in combination with cisplatin treatment, and increased caspase-3 activity. These results are in accordance with previous reports showing that targeting ILK decreases cell viability [
22] and induces apoptosis in ovarian cancer [
20,
21], and increases the sensitivity of lung cancer [
26], gastric carcinoma [
27], and colon cancer [
28] cells to cisplatin or oxaliplatin treatment. In fact, studies in platinum-resistant ovarian cancer have shown that ILKAP, a phosphatase which inactivates ILK, is downregulated by cisplatin treatment, and that silencing
ILKAP increases ILK activation and protects cells from cisplatin-induced apoptosis [
19]. We also observed that transfection of ILK-siRNA reduced the invasive potential of cisplatin-resistant ovarian cancer cells, which is in agreement with previous studies showing that ILK activity modulates the pro-metastatic behavior of ovarian cancer [
17]. Similar to our results, ILK activity stimulates invasion and migration of the SKOV3 human ovarian cancer cell line [
18], whereas ILK depletion by shRNA (short hairpin RNA) abrogates the invasive potential of SKOV3 cells [
15].
Inhibition of ILK activity by Cpd22 treatment significantly reduced the viability of all cancer cell lines tested. These results are in accordance with a previous study showing that Cpd22 treatment suppresses the viability of prostate and breast cancer cells compared with normal prostate and mammary epithelial cells [
23]. Cpd22 inhibits ILK kinase activity, facilitating dephosphorylation of ILK targets, suppressing the expression of YB-1 (Y-box binding protein) transcription factor and its targets, and inducing autophagy and apoptosis [
23]. In addition, ILK expression levels positively correlate with the efficacy of Cpd22 in leukemia [
29]. While siRNAs have high specificity in a sequence specific manner, small-molecule inhibitors can act over multiple targets [
30]. These observations could explain in part the fact that the viability of both cisplatin-resistant and cisplatin-sensitive cells was reduced upon Cpd22 treatment.
Results from RNA-Seq analysis of ILK-siRNA-transfected ovarian cancer cells showed significant changes in the expression of cancer-associated molecules including protein-coding genes and lncRNAs. Repression of glutamate receptor
GRIA4, one of the top upregulated genes in our study, increases cell viability, proliferation, and migratory potential in rhabdomyosarcoma/medulloblastoma and multiple myeloma cells [
31]. Another
GRIA family member,
GRIA2, is downregulated in chemoresistant advanced serous papillary ovarian carcinomas and upregulated in chemosensitive tumors [
32].
TOMM40L, also upregulated in our study, is upregulated in epithelial ovarian cancer cell lines overexpressing DOK1, a candidate tumor suppressor associated with cisplatin sensitivity [
33]. Similarly,
TOMM40L is downregulated in gemcitabine-resistant pancreatic cancer cells compared with their parental sensitive counterparts [
34]. Inhibition of NDCBE in cholangiocarcinoma cells, a product of the
SLC4A8 gene downregulated in our study, decreases proliferation and induces apoptosis [
35].
TEX41, also downregulated in our study, is upregulated in portal vein tumor thrombosis (PVTT), the main route for intrahepatic metastasis in hepatocellular carcinoma [
36]. Therefore, changes in the expression of these genes following siRNA-mediated ILK depletion in cisplatin-resistant ovarian cancer cells could explain in part the reduction in cell growth, invasion, and viability, and the increase in caspase-3 activity.
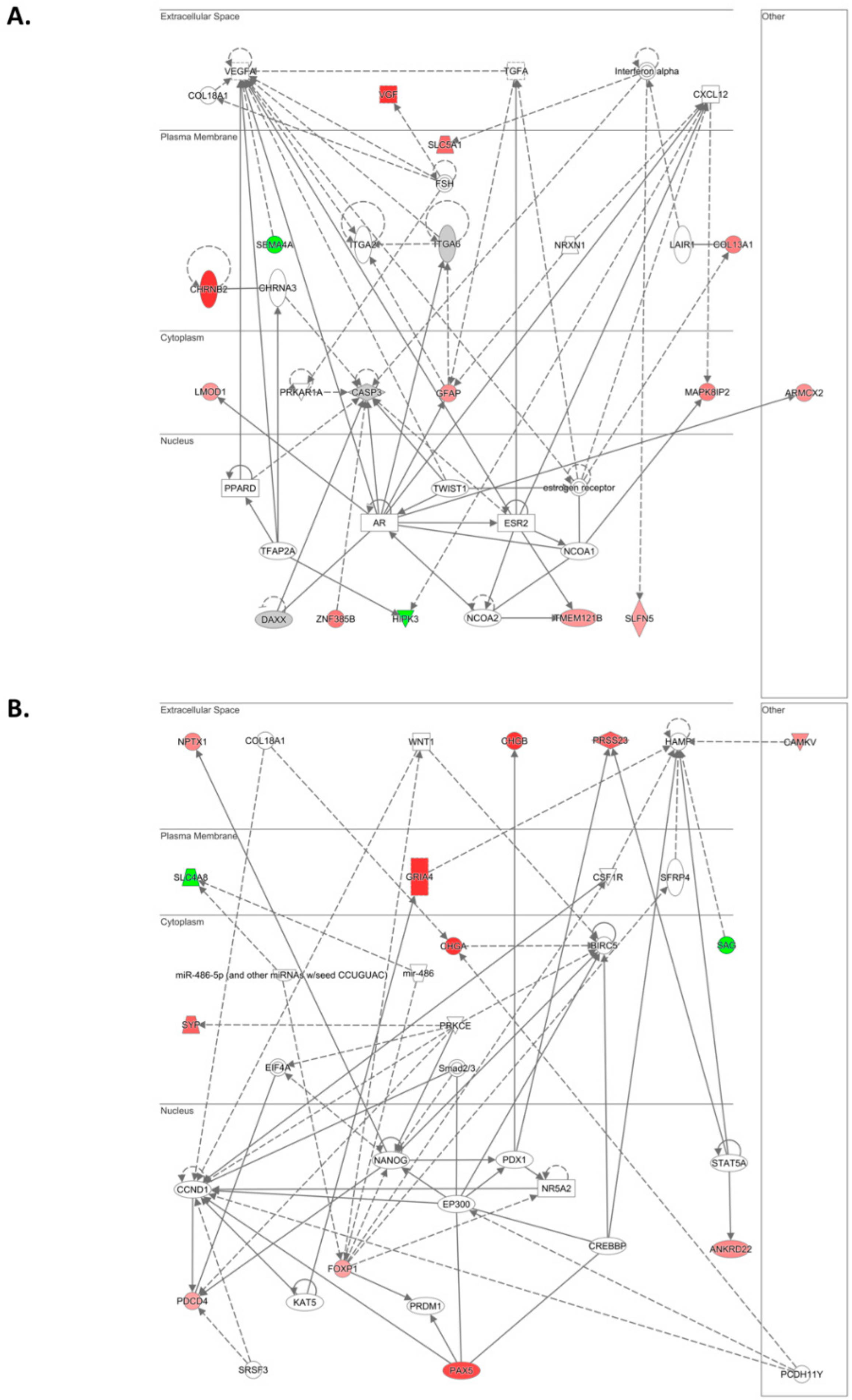
In agreement with these observations, within the list of differentially abundant transcripts, we identified additional genes involved in the regulation of cell proliferation, survival, migration, invasion, metastasis, drug resistance, and apoptosis. Interestingly,
DUSP8,
MARVELD3,
PDCD4,
MAPK8IP1,
MAPK8IP2, and
HIPK3 converge on JNK signaling, one of the top canonical pathways (SAPK/JNK) identified in our IPA analysis. In epithelial ovarian cancer, activated JNK is associated with decreased PFS [
37]. In addition, we have previously shown that the JNK-1/c-JUN/
miR-21 pathway contributes to cisplatin resistance in ovarian cancer cells [
38]. ILK negatively regulates the expression of
DUSP8 [
39], a MAPK phosphatase upregulated in our study, and DUSP8 dephosphorylates and inactivates JNK [
40,
41]. Inhibition of ILK/AKT decreases
miR-21 in vestibular schwannoma and meningioma cells [
42], and targeting
miR-21 with antisense oligonucleotides (ASOs) inhibits growth and metastasis via upregulation of DUSP8 in colorectal carcinoma [
43]. MARVELD3, a transmembrane protein of tight junctions [
44,
45] upregulated in our study, inhibits JNK activity via recruitment of MEKK1 to cell junctions [
46]. MARVELD3 is downregulated in SNAIL-induced EMT during pancreatic cancer progression [
47]. Similarly,
MARVELD3 silencing decreases
CDH1 and increases
SNAI2 expression in lung cancer [
48]. Downregulation of MARVELD3 in colorectal cancer cells increases migration and proliferation, whereas upregulation inhibits migration, proliferation, and in vivo tumor formation [
46]. PDCD4, a well-known tumor suppressor [
49] upregulated in our study, has been suggested to interact with c-JUN, blocking c-JUN phosphorylation by JNK [
50]. In ovarian cancer, lower PDCD4 expression correlates with shorter disease-free survival (DFS) [
51]. Moreover, PDCD4 overexpression enhances the sensitivity of ovarian cancer cells to cisplatin by activating the death receptor pathway [
52]. In bladder cancer cells, PDCD4 overexpression enhances sensitivity to cisplatin via regulation of the JNK/c-JUN pathway [
53]. The JIP family of MAPK scaffold proteins, including MAPK8IP1 and MAPK8IP2 which were upregulated in our study, have been previously identified as regulators of JNK signaling [
54]. MAPK8IP1, a negative regulator of MAPK activity [
55], inhibits c-JUN phosphorylation by JNK [
56]. In gastric cancer,
miR-10a promotes cell migration and invasion by downregulating
MAPK8IP1 [
55]. HIPK3, a FAS/FADD-interacting kinase [
57] downregulated in our study, has been previously implicated in multidrug resistance in cancer [
58,
59]. In fact, overexpression of
HIPK3 protects osteosarcoma cells from cisplatin-induced death in vitro and in vivo [
60]. In prostate cancer, JNK activity increases the expression of HIPK3 and promotes resistance to FAS receptor-mediated apoptosis [
61]. Overall, these observations suggest that ILK regulates the JNK/c-JUN pathway in cisplatin-resistant ovarian cancer via modulation of
DUSP8,
MARVELD3,
PDCD4,
MAPK8IP1,
MAPK8IP2, and
HIPK3.
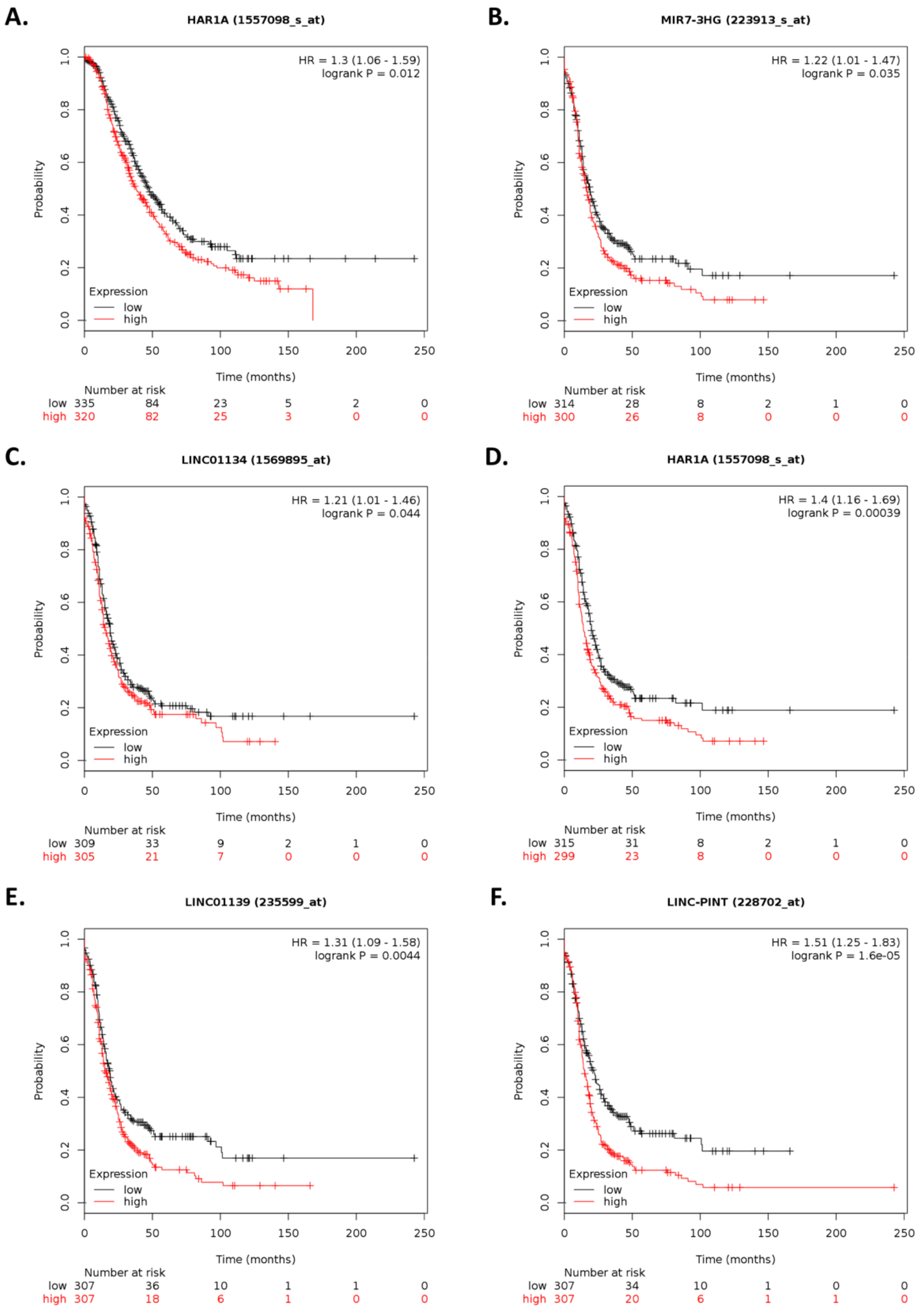
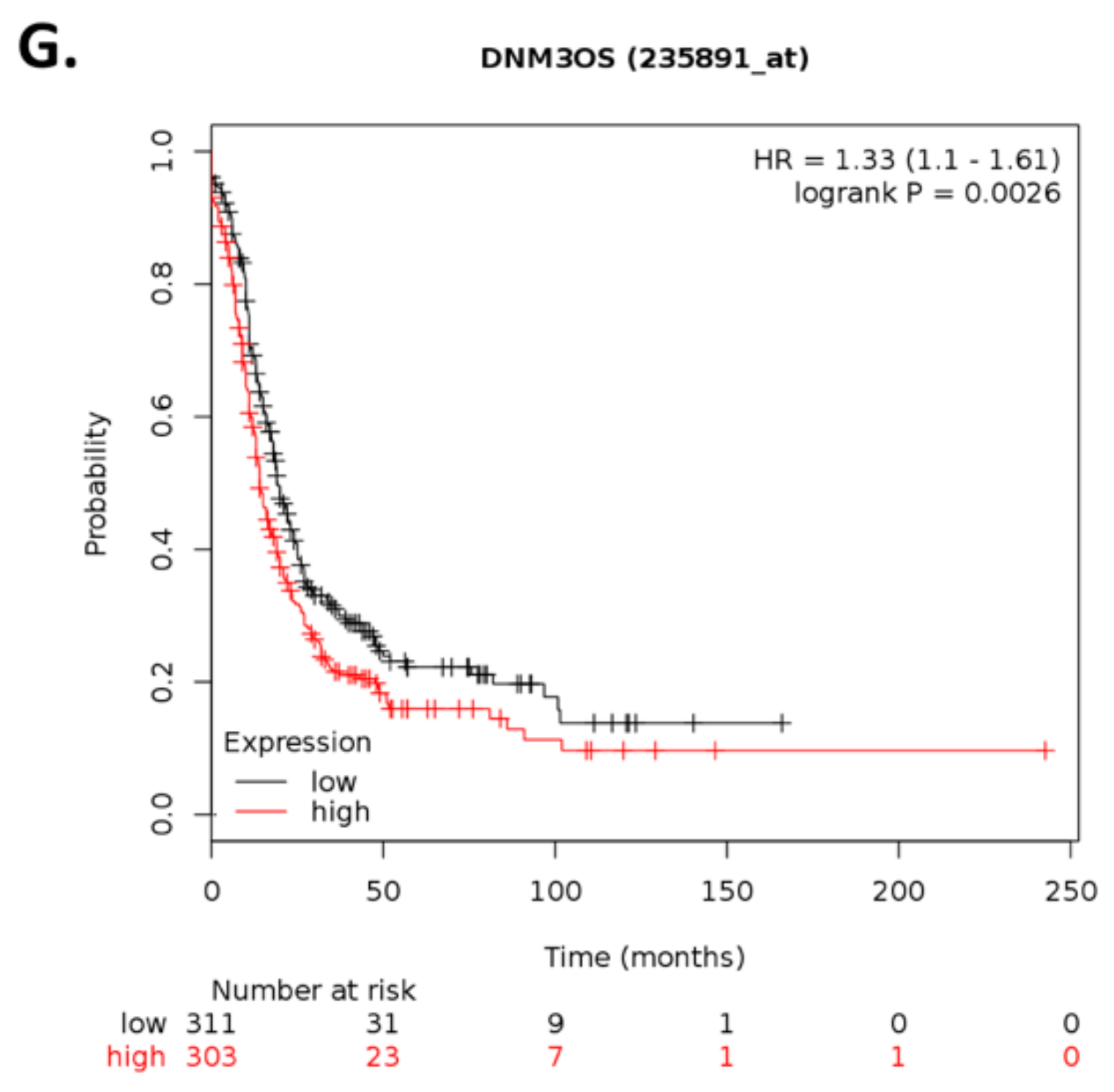
Within the top 10 differentially expressed lncRNAs,
ARHGEF26-AS1 and
MIR7-3HG have been previously reported in cancer.
ARHGEF26-AS1 is downregulated in cancer-associated fibroblasts (CAFs) of HGSOC patients compared with normal ovarian fibroblasts (NOFs) [
62]. As CAFs promote cancer metastasis [
62], our results showing that ILK depletion leads to
ARHGEF26-AS1 downregulation suggest additional roles for
ARHGEF26-AS1 in cisplatin-resistant ovarian cancer. Similarly,
MIR7-3HG upregulation in our analysis contrasts a report showing that
MIR7-3HG downregulates the tumor suppressor AMBRA1 and prevents MYC dephosphorylation in lung cancer [
63]. Nevertheless, our results that
LINC-PINT was upregulated upon ILK-siRNA transfection is consistent with reports that show that this lncRNA inhibits cell invasion [
24]. Interestingly, in osteosarcoma and gastric cancer,
LINC-PINT inhibits cell invasion, migration, and proliferation by downregulating
miR-21 [
64,
65]. In ovarian cancer,
LINC-PINT is downregulated relative to normal ovary cells and tissue samples [
66], whereas
miR-21 is upregulated [
67]. These observations warrant further investigation to elucidate the role of these lncRNAs in cisplatin-resistant ovarian cancer.
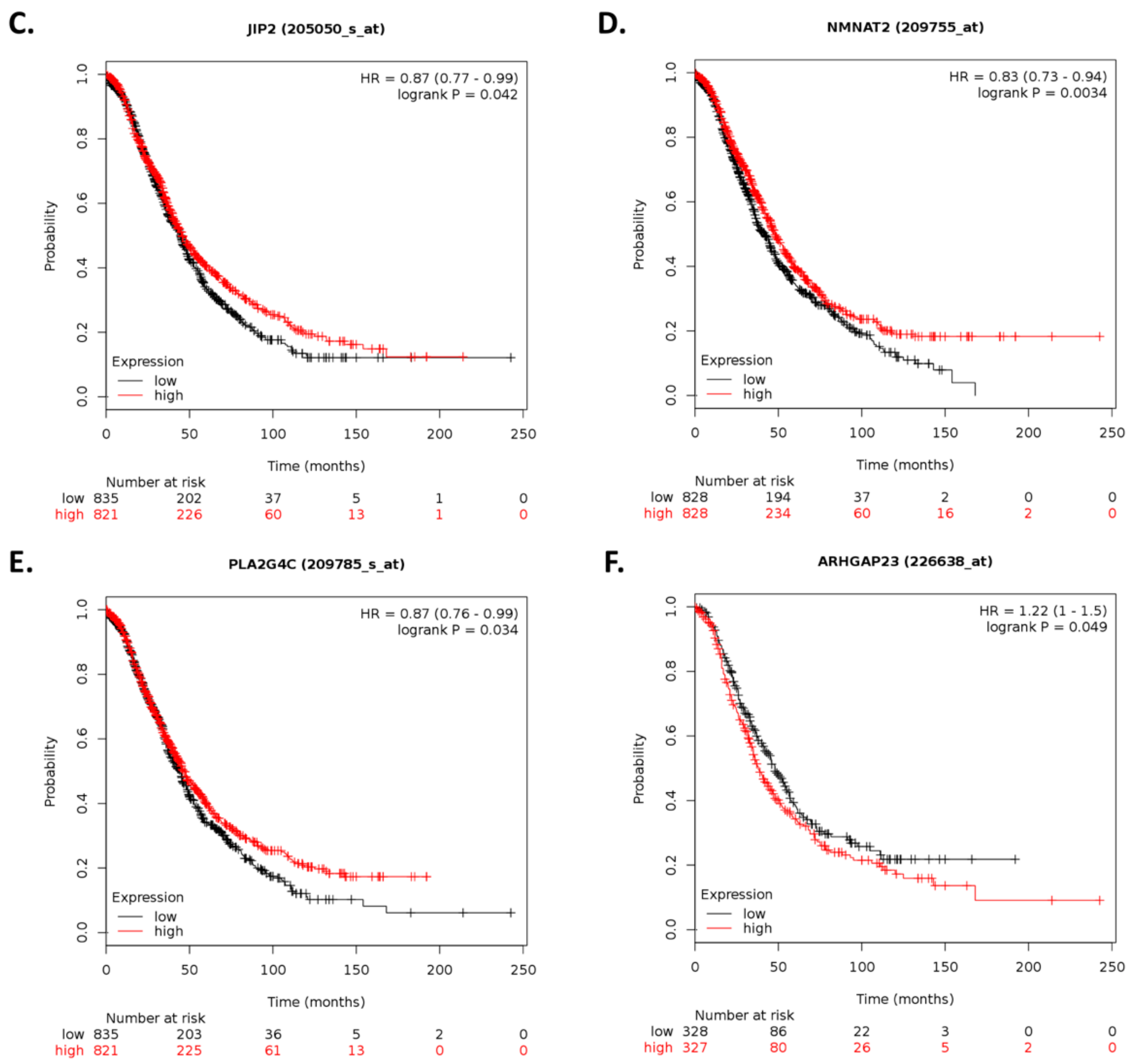
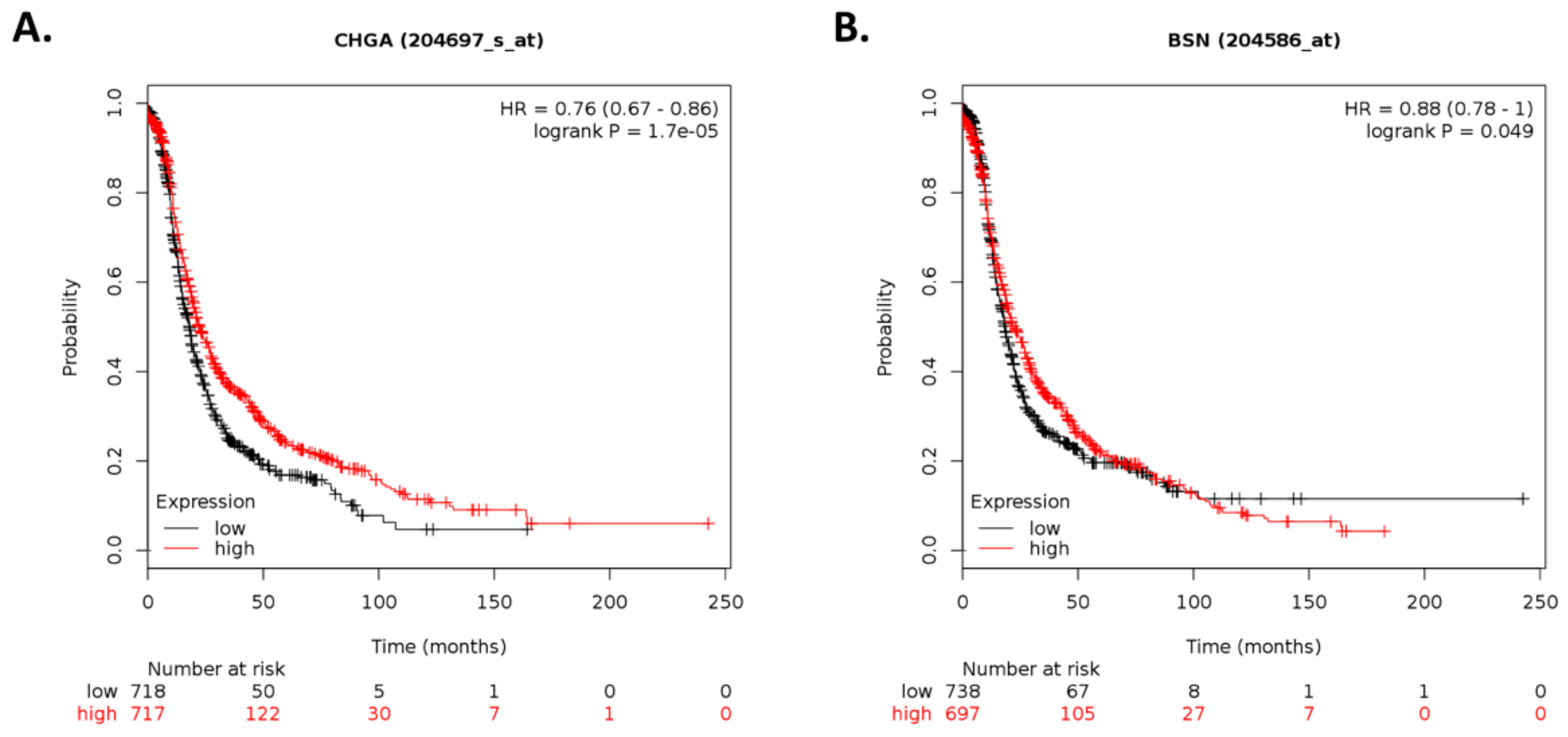
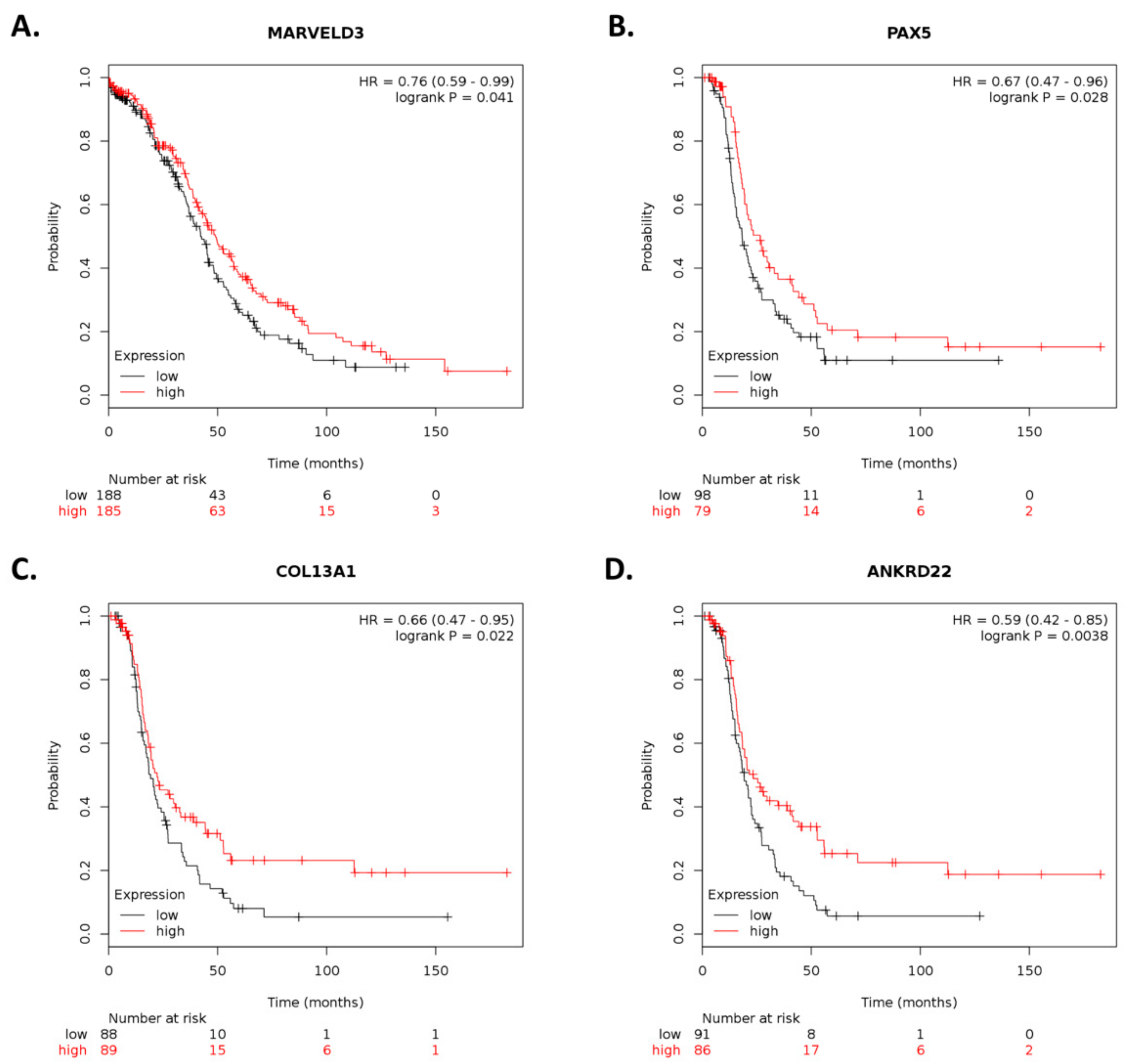
By using Kaplan–Meier analysis of publicly available mRNA expression (gene chip and RNA-Seq data) we further show that several genes differentially altered upon ILK depletion are significantly associated with survival outcomes in ovarian cancer. Based on expression levels of survival-relevant genes, we identified
CHGA,
BSN,
SLC5A1,
MAPK8IP2,
NMNAT2,
SEMA3G,
ARHGAP23,
SAG,
SLC4A8,
MARVELD3,
PAX5,
COL13A1,
ANKRD22,
VGF,
ACTL6B,
NKAIN1,
CAMKV,
LTF, and
ILK as potential prognostic markers. Of these genes, we were able to validate
CHGA,
BSN,
SLC5A1,
MAPK8IP2,
NMNAT2,
SEMA3G,
SLC4A8,
MARVELD3,
PAX5,
ANKRD22,
VGF,
ACTL6B,
CAMKV, and
ILK by qRT-PCR analysis. In accordance with previous reports, CHGA can exert antiangiogenic effects and inhibit tumor growth in vivo [
68]. Overexpression of PAX5 induces apoptosis in multiple myeloma [
69], whereas knockdown of
PAX5 increases cell proliferation and cisplatin resistance in esophageal squamous cell carcinoma [
70]. Interestingly,
PAX5 promoter hypermethylation has been observed in ovarian cancer [
71]. Overexpression of
VGF inhibits colony formation of ovarian cancer cells, however,
VGF promoter hypermethylation correlates with better patient survival [
72]. In contrast to our results, SEMA3G inhibits cell migration and invasion in glioma [
73].
SLC5A1 promotes growth and proliferation of pancreatic cancer [
74]. High expression of SGLT1, which is encoded by
SLC5A1, is associated with tumor development and poor prognosis in ovarian cancer [
75]. In addition,
ANKRD22 promotes progression of non-small-cell lung cancer [
76]. However, higher
ANKRD22 expression levels in prostate cancer are associated with longer DFS following radical prostatectomy [
77]. Therefore, further studies are required to elucidate the role of these genes and their therapeutic potential in cisplatin-resistant ovarian cancer.
4. Materials and Methods
4.1. Cells and Culture Conditions
The human ovarian epithelial cancer cells, A2780CP20, HEYA8, and HEYA8.MDR, were provided by Dr. Anil K. Sood (MD Anderson Cancer Center, Houston, TX, USA) and have been described elsewhere [
78]. A2780 was purchased from the European Collection of Cell Cultures (ECACC, Porton Down, Salisbury, UK), and OV90 and OVCAR3 from the American Type Culture Collection (ATCC, Manassas, VA, USA)—both of which provide authenticated cell lines. OV90CIS and OVCAR3CIS cells were generated by exposing OV90 and OVCAR3 to increasing concentrations of cisplatin (CIS; Sigma-Aldrich, St. Louis, MO, USA). Cells were maintained in RPMI1640 (A2780, A2780CP20, HEYA8, and HEYA8.MDR; HyClone, GE Healthcare Life Sciences, Logan, UT, USA), RPMI1640 + insulin (0.01 mg/mL, OVCAR3 and OVCAR3CIS; Sigma) or M199/MCDB-105 (OV90 and OV90CIS; Gibco, Thermo Fisher Scientific, Grand Island, NY/Sigma) medium supplemented with 10% fetal bovine serum (FBS; HyClone) and 0.1% antibiotic/antimycotic solution (HyClone) at 37 °C in 5% CO
2 and 95% air. All cell lines were screened for mycoplasma using the LookOut
® Mycoplasma PCR detection kit (Sigma), and authenticated by Promega (Madison, WI, USA) and ATCC using Short Tandem Repeat (STR) analysis. In vitro assays were performed at 70%–85% cell density.
4.2. Western Blot Analysis
Cell pellets were lysed with ice-cold lysis buffer (1% Triton X, 150 mmol/L NaCl, 25 mmol/L Tris HCl, 0.4 mmol/L NaVO
4, 0.4 mmol/L NaF, and protease inhibitor cocktail from Sigma) and total protein concentration was determined using Bio-Rad DC Protein Assay reagents (Bio-Rad, Hercules, CA). Protein samples were separated by SDS-PAGE and blotted onto nitrocellulose membranes. The membranes were blocked in either 5% non-fat dry milk (Bio-Rad) or 5% BSA (HyClone) and probed with phospho-ILK (Ser 246; Millipore, Burlington, MA, USA), ILK (Cell Signaling, Danvers, MA, USA), phospho-AKT (Ser 473; Cell Signaling), AKT (Cell Signaling), full caspase-3 (Cell Signaling), or cleaved caspase-3 (Cell Signaling) primary antibodies. Membranes were then incubated with mouse or rabbit IgG horseradish peroxidase (HRP)-linked secondary antibodies (Cell Signaling) followed by enhanced chemiluminescence and autoradiography. Bands were imaged with a FluorChem system (Alpha Innotech Corporation, San Leandro, CA, USA) and the signal intensity of each band was quantified using AlphaEaseFC software. All membranes were reprobed with β-actin monoclonal antibody (Sigma) as a normalizing control. Western blot images with molecular weight markers are shown in
Figure S15.
4.3. Immunohistochemistry
Immunohistochemical (IHC) analysis of p-ILK and ILK was conducted on formalin-fixed paraffin-embedded (FFPE) specimens from chemonaïve serous papillary ovarian cancer (n = 10; ages 40–85, mean 64.70 ± 13.320) and normal ovary samples (n = 10; ages 43–73, mean 51.70 ± 9.673) provided by the Department of Pathology at UPR-MSC. The study was carried out with approval from the UPR-MSC Institutional Review Board (IRB protocol number A9180115; category 4 exemption; informed consent is not required). A pathologist manually delineated tumor tissue on a representative hematoxylin and eosin (H&E)-stained slide from each paraffin block. Briefly, sections were deparaffinized and rehydrated. After antigen retrieval (Vector Laboratories, Burlingame, CA, USA), endogenous peroxidase was blocked with 3% H2O2, followed by non-specific protein blocking (Dako, Carpinteria, CA, USA). Human sections were incubated with phospho-ILK (Ser 246; Abcam, Cambridge, MA, USA) or ILK (Cell Signaling) primary antibodies overnight at 4 °C. After washing with PBS, anti-rabbit EnVision+ System-HRP (Dako) was used as a secondary antibody. HRP was detected with 3,3′ diaminobenzidine (DAB; Dako), counterstained with Gill’s No.3 hematoxylin (American MasterTech Scientific, Lodi, CA, USA), and cover slipped with Permount (Fisher Scientific, Fair Lawn, NJ, USA). Scoring of p-ILK and ILK expression was based on the number of positive cells (DAB stained) in each group and quantified in 5 random fields at 20×. One slide per specimen and 10 specimens per group were examined.
4.4. Small-Interfering RNA (siRNA) and In Vitro siRNA Transfection
To target human ILK (NM_001014794.2), two siRNAs [5′-GTCAAGTTCTCTTTCCAAT-3′ (ILK-siRNA(1)), and 5′-CTCAATAGCCGTAGTGTAA-3′ (ILK-siRNA(2))] targeting ILK mRNA, and a non-silencing negative control siRNA (C-siRNA) were used (Sigma). For in vitro siRNA transfections, siRNAs were mixed with HiPerfect transfection reagent (Qiagen, Valencia, CA) at 1:2 ratio (siRNA: transfection reagent) in serum and antibiotic-free Opti-MEM medium (Gibco). Transfected cells were collected 24 h after treatment for assessment of ILK protein levels by Western blot analysis.
4.5. In Vitro Inhibitor Treatment
ILK inhibitor (Cpd22) was purchased from Millipore and dissolved in DMSO (Sigma). For in vitro inhibitor treatment, cells were treated with Cpd22 for 24 h and collected for assessment of p-ILK and ILK protein levels by Western blot analysis.
4.6. In Vitro Cell Viability and Cell Growth
For cell viability assays using siRNAs, A2780, A2780CP20, OVCAR3, OVCAR3CIS, and HEYA8 cells (2 × 104 cells/mL, 3 × 104 cells/mL, or 10 × 104 cells/mL) were seeded into 96-well plates. Twenty-four hours later, siRNA transfection was performed as described above. The transfection mix was replaced with CIS after 24 h of siRNA treatment. Forty-eight hours after CIS treatment, the medium was removed and Alamar blue dye (Invitrogen, Thermo Fisher Scientific, Eugene, OR, USA) was added following the manufacturer’s instructions. Optical density (OD) values were obtained using a plate reader (Bio-Rad) and percentages of cell viability were calculated after blank OD subtraction, taking the untreated cell values as 100% cell viability. For cell viability assays using ILK inhibitor (Cpd22), A2780, A2780CP20, OV90, OV90CIS, OVCAR3, OVCAR3CIS, and HEYA8 cells (2 × 104 cells/mL or 3 × 104 cells/mL) were seeded into 96-well plates. Twenty-four hours later, inhibitor treatment was performed as described above. Seventy-two hours after treatment, cell viability was assessed using Alamar blue dye.
For assessment of cell growth, colony formation assays were performed using Crystal violet dye (Sigma). Briefly, A2780, A2780CP20, OVCAR3, OVCAR3CIS, OV90, and OV90CIS cells (3 × 104 cells/mL or 4.5 × 104 cells/mL) were seeded into 6-well plates. Twenty-four hours later, siRNAs were added to the cells. After treatment, 1000 (A2780CP20) or 2500 (A2780, OVCAR3, OVCAR3CIS, OV90, and OV90CIS) cells were seeded into 10 cm Petri dishes. Seven (A2780, A2780CP20, OVCAR3, and OVCAR3CIS) or ten (OV90 and OV90CIS) days later, colonies were fixed and stained with 0.5% Crystal violet solution in methanol. Colonies of at least 50 cells were scored in five random fields using a light microscope (CKX41; Olympus, Center Valley, PA, USA) with a total magnification of 40×.
4.7. In Vitro Cell Invasion
Cell invasion was analyzed using the matrigel transwell method as previously described [
38,
79]. A2780CP20, OV90CIS, and OVCAR3CIS cells (3.5 × 10
4 cells/mL) were seeded into 10 cm Petri dishes and transfected with siRNAs. The next day, matrigel (BD Biosciences, San Jose, CA, USA) in serum-free medium was added onto the upper chambers of 24-well transwell plates and incubated at 37 °C for polymerization. Transfected cells were collected and resuspended in serum-free medium and re-seeded onto the matrigel-coated chamber. Medium containing 10% FBS was added to the lower wells. After 48 h at 37 °C, medium was removed and cells that invaded through the matrigel were fixed and stained using the Protocol Hema 3 Stain Set (Fisher Scientific, Kalamazoo, MI, USA). The invading cells were counted at 20× resolution on an Olympus IX71 microscope equipped with a digital camera (Olympus DP26). Percentages of cell invasion were calculated taking the C-siRNA-transfected cell values as 100% cell invasion.
4.8. Caspase-3 Activity
Caspase-3 activity was quantified using the Caspase-3/CPP32 Fluorometric Assay Kit (BioVision, Milpitas, CA, USA) following the manufacturer’s instructions. A2780CP20 cells (2.0 × 105 cells/mL) were seeded into 10 cm Petri dishes and transfected with C-siRNA or ILK-siRNA(2) for 24 h. Seventy-two hours after transfection, floating and attached cells were collected, pellets were lysed, and total protein concentration was determined. Equal amounts of protein were mixed with 2× Reaction Buffer and 1 mM DEVD-AFC substrate in a 96-well plate and incubated at 37 °C for 1 h. Fluorescence intensity at 400 nm excitation and 505 nm emission was measured on a Varioskan Flash reader from Thermo Scientific (Waltham, MA, USA).
4.9. RNA-Seq Analysis
For RNA-Seq analysis, total RNA was isolated from C-siRNA or ILK-siRNA(2)-transfected A2780CP20 cells using the mirVanaTM miRNA Isolation Kit (Invitrogen) following the manufacturer’s instructions. RNA quality was verified on all samples with High Sensitivity RNA system on an Agilent 2200 TapeStation instrument (Agilent Technologies, Santa Clara, CA, USA). mRNA was enriched and library was prepared using NEBNext® Poly(A) mRNA Magnetic Isolation and NEBNext® Ultra™ RNA Library Prep (New England Biolabs, Ipswich, MA, USA). Once library construction was complete, qPCR was performed with KAPA SYBR® FAST qPCR in a HiSeq 4000 system (Illumina, San Diego, CA, USA) with a 2 × 150 bp configuration.
4.10. KEGG Pathway Enrichment, Gene Ontology, and Network Analysis
Differentially expressed genes with a log2 fold change ≥1.5 and ≤−1.5 were enriched for their involvement in various biological pathways using KEGG (Kyoto Encyclopedia of Genes and Genomes) Pathway Enrichment. Ontological signatures, mainly biological processes, molecular functions, and cellular components were enriched based on STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) integration of interaction data.
Ingenuity Pathway Analysis (IPA; Ingenuity Systems, Qiagen, Redwood City, CA, USA) software was used to determine the functional networks and pathways associated with differentially expressed genes using a p-value cutoff ≤0.001 and a log2 fold change >1.5 or <−1.5. Gene networks and canonical pathways enrichment analysis were performed filtering for all tissues, all cell lines and human species. For the identification of differentially expressed lncRNAs, a p-value cutoff ≤0.001 and a log2 fold change >1.0 or <−1.0 were used.
4.11. Survival Analysis
Kaplan–Meier survival analysis was performed using publicly available gene chip and RNA-Seq datasets in Kaplan–Meier (KM) plotter (
www.kmplot.com) [
25]. For each gene symbol, the probe ID was selected, and ovarian cancer patients were split into high and low expression groups by the median values of mRNA expression. For genes with multiple probes, the best probe was selected. All available datasets were used for survival analysis. Data from ovarian cancer patients, ovarian cancer patients treated with platin, serous ovarian cancer patients, and serous ovarian cancer patients treated with platin were evaluated. KM survival plots for overall survival (OS), progression-free survival (PFS), and relapse-free survival (RFS) were generated with their respective hazard ratios (HR), confidence intervals (CI), and
p-values (log-rank).
p-values < 0.05 were considered to be statistically significant.
4.12. SYBR-Green Based qRT-PCR Analysis
For validation of RNA-Seq data, qPCR was performed to assess expression levels of specific genes upon ILK targeting. A custom 96-well plate containing pre-designed primers was purchased from Bio-Rad. Total RNA was isolated from C-siRNA or ILK-siRNA(2)-transfected A2780CP20 cells using the GenElute Mammalian Total RNA Mini Kit (Sigma) following the manufacturer’s instructions, and reverse transcribed using the iScript™ cDNA Synthesis Kit (Bio-Rad). SYBR Green-based qPCR was conducted using SsoAdvanced™ Universal SYBR® Green Supermix (Bio-Rad) and StepOne Plus Real-Time PCR system with the suggested PrimePCR cycling protocol (activation at 95 °C for 2 min, and 40 cycles of 95 °C for 5 s and 60 °C for 30 s).
4.13. Statistical Analysis
Graphing and statistical analysis were performed using Student’s t test or ANOVA in GraphPad Prism (San Diego, CA, USA) software. p-values < 0.05 were considered to be statistically significant. Experiments were performed at least in triplicate.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}