The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
1. Identification of the Tumor Susceptibility Gene 101 (Tsg101)
2. TSG101 is a Housekeeping Gene
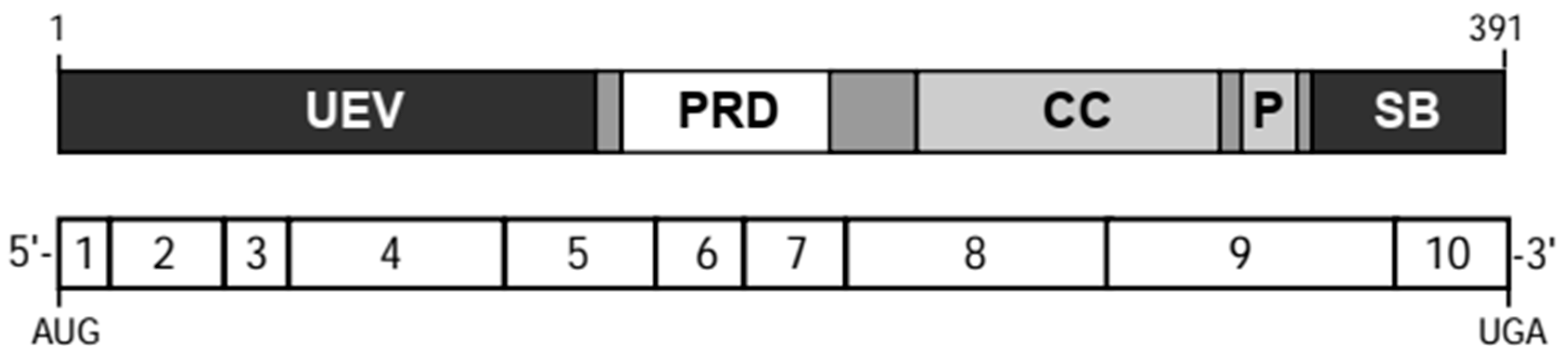
3. TSG101 Encodes a Multidomain Protein
4. The TSG101 Protein Mediates Diverse Intracellular Processes
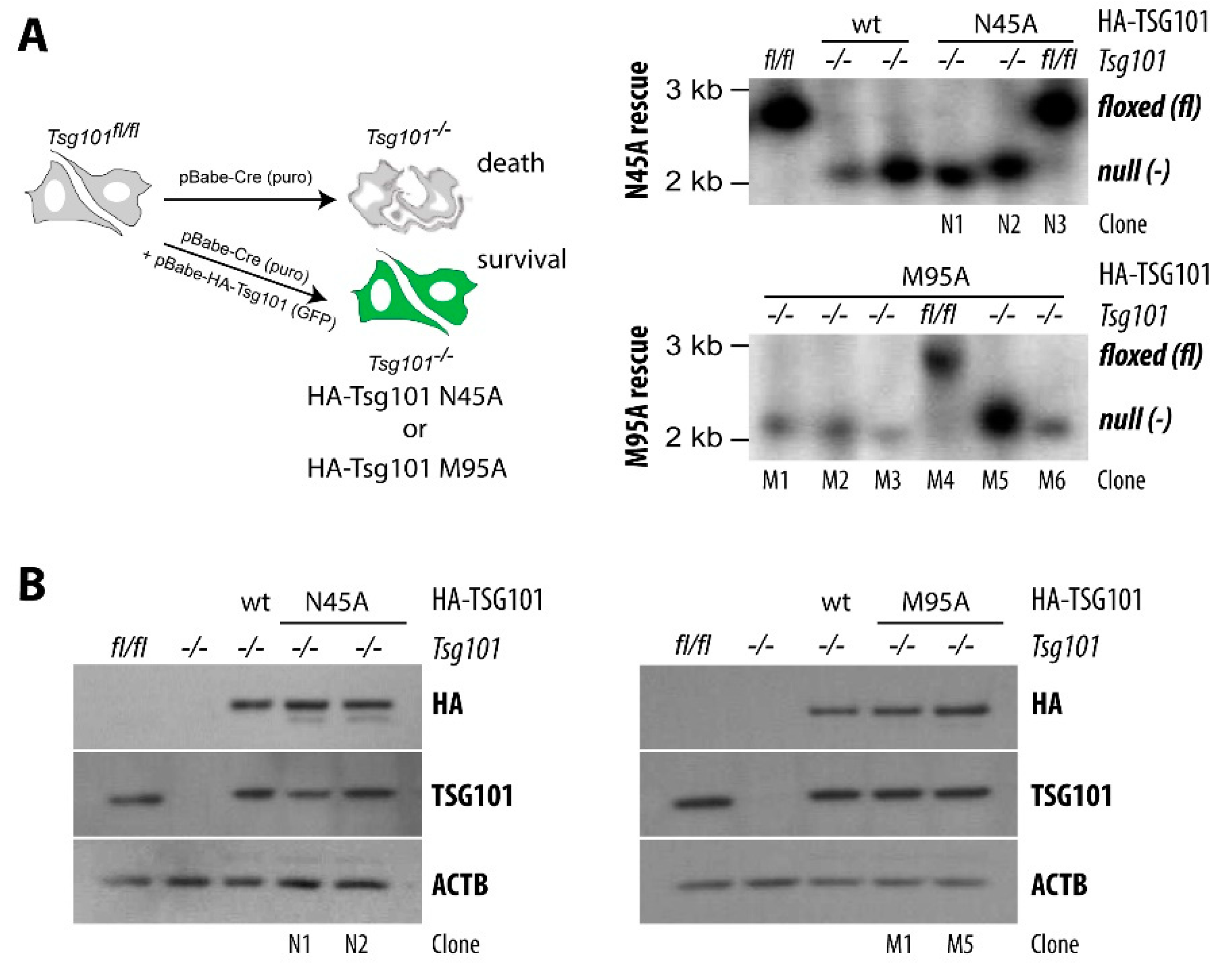
5. A Knockout of TSG101 Causes Cell Cycle Arrest and Cell Death
6. TSG101 Knockout Cells Are Stressed and Undergo Autophagy Prior to Cell Death
7. Lack of Clear Evidence for a Tumor-Suppressive Role of TSG101 in Mammalian in vivo Models
8. TSG101 Might Function as an Oncoprotein
9. Post-Translational Control of TSG101 Expression and Its Potential Role in Cancer
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, L.; Cohen, S.N. Tsg101: A novel tumor susceptibility gene isolated by controlled homozygous functional knockout of allelic loci in mammalian cells. Cell 1996, 85, 319–329. [Google Scholar] [CrossRef]
- Li, L.; Li, X.; Francke, U.; Cohen, S.N. The TSG101 tumor susceptibility gene is located in chromosome 11 band p15 and is mutated in human breast cancer. Cell 1997, 88, 143–154. [Google Scholar] [CrossRef]
- Steiner, P.; Barnes, D.M.; Harris, W.H.; Weinberg, R.A. Absence of rearrangements in the tumour susceptibility gene TSG101 in human breast cancer. Nat. Genet. 1997, 16, 332–333. [Google Scholar] [CrossRef]
- Li, L.; Francke, U.; Cohen, S.N. Retraction. The TSG101 tumor susceptibility gene is located in chromosome 11 band p15 and is mutated in human breast cancer. Cell 1998, 93, 660. [Google Scholar] [CrossRef]
- Zhong, Q.; Chen, C.F.; Chen, Y.; Chen, P.L.; Lee, W.H. Identification of cellular TSG101 protein in multiple human breast cancer cell lines. Cancer Res. 1997, 57, 4225–4228. [Google Scholar] [PubMed]
- Wang, Q.; Driouch, K.; Courtois, S.; Champeme, M.H.; Bieche, I.; Treilleux, I.; Briffod, M.; Rimokh, R.; Magaud, J.P.; Curmi, P.; et al. Low frequency of TSG101/CC2 gene alterations in invasive human breast cancers. Oncogene 1998, 16, 677–679. [Google Scholar] [CrossRef][Green Version]
- Krempler, A.; Henry, M.D.; Triplett, A.A.; Wagner, K.U. Targeted deletion of the Tsg101 gene results in cell cycle arrest at G1/S and p53-independent cell death. J. Biol. Chem. 2002, 277, 43216–43223. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.U.; Dierisseau, P.; Rucker, E.B.; Robinson, G.W.; Hennighausen, L. Genomic architecture and transcriptional activation of the mouse and human tumor susceptibility gene TSG101: Common types of shorter transcripts are true alternative splice variants. Oncogene 1998, 17, 2761–2770. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; Barski, P.; Batley, S.J.; Li, L.; de Foy, K.A.; Cohen, S.N.; Ponder, B.A.; Caldas, C. Aberrant splicing of the TSG101 and FHIT genes occurs frequently in multiple malignancies and in normal tissues and mimics alterations previously described in tumours. Oncogene 1997, 15, 2119–2126. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.P.; Feinberg, A.P. Aberrant splicing but not mutations of TSG101 in human breast cancer. Cancer Res. 1997, 57, 3131–3134. [Google Scholar]
- Sun, Z.; Pan, J.; Hope, W.X.; Cohen, S.N.; Balk, S.P. Tumor susceptibility gene 101 protein represses androgen receptor transactivation and interacts with p300. Cancer 1999, 86, 689–696. [Google Scholar] [CrossRef]
- Kozak, M. Recognition of AUG and alternative initiator codons is augmented by G in position +4 but is not generally affected by the nucleotides in positions +5 and +6. EMBO J. 1997, 16, 2482–2492. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.U.; Dierisseau, P.; Hennighausen, L. Assignment of the murine tumor susceptibility gene 101 (tsg101) and a processed tsg101 pseudogene (tsg101-ps1) to mouse chromosome 7 band B5 and chromosome 15 band D1 by in situ hybridization. Cytogenet. Cell Genet. 1999, 84, 87–88. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J.; Sirard, C.; Elia, A.; MacPherson, D.; Wakeham, A.; Li, L.; Luis, D.L.P.; Cohen, S.N.; Mak, T.W. p53 Accumulation, defective cell proliferation, and early embryonic lethality in mice lacking tsg101. Proc. Natl. Acad. Sci. USA 2001, 98, 1859–1864. [Google Scholar] [CrossRef]
- Wagner, K.U.; Krempler, A.; Qi, Y.; Park, K.; Henry, M.D.; Triplett, A.A.; Riedlinger, G.; Rucker III, E.B.; Hennighausen, L. Tsg101 Is Essential for Cell Growth, Proliferation, and Cell Survival of Embryonic and Adult Tissues. Mol. Cell Biol. 2003, 23, 150–162. [Google Scholar] [CrossRef]
- Koonin, E.V.; Abagyan, R.A. TSG101 may be the prototype of a class of dominant negative ubiquitin regulators. Nat. Genet. 1997, 16, 330–331. [Google Scholar] [CrossRef]
- Ponting, C.P.; Cai, Y.D.; Bork, P. The breast cancer gene product TSG101: A regulator of ubiquitination? J. Mol. Med. 1997, 75, 467–469. [Google Scholar]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for hiv-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Katzmann, D.J.; Babst, M.; Emr, S.D. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell 2001, 106, 145–155. [Google Scholar] [CrossRef]
- Pornillos, O.; Alam, S.L.; Rich, R.L.; Myszka, D.G.; Davis, D.R.; Sundquist, W.I. Structure and functional interactions of the Tsg101 UEV domain. EMBO J. 2002, 21, 2397–2406. [Google Scholar] [CrossRef]
- Pornillos, O.; Alam, S.L.; Davis, D.R.; Sundquist, W.I. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat. Struct. Biol. 2002, 9, 812–817. [Google Scholar] [CrossRef]
- Pornillos, O.; Higginson, D.S.; Stray, K.M.; Fisher, R.D.; Garrus, J.E.; Payne, M.; He, G.P.; Wang, H.E.; Morham, S.G.; Sundquist, W.I. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 2003, 162, 425–434. [Google Scholar] [CrossRef]
- Sundquist, W.I.; Schubert, H.L.; Kelly, B.N.; Hill, G.C.; Holton, J.M.; Hill, C.P. Ubiquitin recognition by the human TSG101 protein. Mol. Cell 2004, 13, 783–789. [Google Scholar] [CrossRef]
- Maucuer, A.; Camonis, J.H.; Sobel, A. Stathmin interaction with a putative kinase and coiled-coil-forming protein domains. Proc. Natl. Acad. Sci. USA 1995, 92, 3100–3104. [Google Scholar] [CrossRef]
- Feng, G.H.; Lih, C.J.; Cohen, S.N. TSG101 protein steady-state level is regulated post-translationally by an evolutionarily conserved COOH-terminal sequence. Cancer Res. 2000, 60, 1736–1741. [Google Scholar]
- Lu, Q.; Hope, L.W.; Brasch, M.; Reinhard, C.; Cohen, S.N. TSG101 interaction with HRS mediates endosomal trafficking and receptor down-regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 7626–7631. [Google Scholar] [CrossRef]
- Zhong, Q.; Chen, Y.; Jones, D.; Lee, W.H. Perturbation of TSG101 protein affects cell cycle progression. Cancer Res. 1998, 58, 2699–2702. [Google Scholar]
- Xie, W.; Li, L.; Cohen, S.N. Cell cycle-dependent subcellular localization of the TSG101 protein and mitotic and nuclear abnormalities associated with TSG101 deficiency. Proc. Natl. Acad. Sci. USA 1998, 95, 1595–1600. [Google Scholar] [CrossRef]
- Hittelman, A.B.; Burakov, D.; Iniguez-Lluhi, J.A.; Freedman, L.P.; Garabedian, M.J. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 1999, 18, 5380–5388. [Google Scholar] [CrossRef]
- Lin, Y.S.; Chen, Y.J.; Cohen, S.N.; Cheng, T.H. Identification of TSG101 functional domains and p21 loci required for TSG101-mediated p21 gene regulation. PloS ONE 2013, 8, e79674. [Google Scholar] [CrossRef]
- Oh, H.; Mammucari, C.; Nenci, A.; Cabodi, S.; Cohen, S.N.; Dotto, G.P. Negative regulation of cell growth and differentiation by TSG101 through association with p21Cip1/WAF1. Proc. Natl. Acad. Sci. USA 2002, 99, 5430–5435. [Google Scholar] [CrossRef]
- Lee, H.H.; Elia, N.; Ghirlando, R.; Lippincott-Schwartz, J.; Hurley, J.H. Midbody targeting of the ESCRT machinery by a noncanonical coiled coil in CEP55. Science 2008, 322, 576–580. [Google Scholar] [CrossRef]
- Li, Y.; Kane, T.; Tipper, C.; Spatrick, P.; Jenness, D.D. Yeast mutants affecting possible quality control of plasma membrane proteins. Mol. Cell Biol. 1999, 19, 3588–3599. [Google Scholar] [CrossRef]
- Babst, M.; Odorizzi, G.; Estepa, E.J.; Emr, S.D. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic 2000, 1, 248–258. [Google Scholar] [CrossRef]
- Doyotte, A.; Russell, M.R.; Hopkins, C.R.; Woodman, P.G. Depletion of TSG101 forms a mammalian “Class E” compartment: a multicisternal early endosome with multiple sorting defects. J. Cell Sci. 2005, 118, 3003–3017. [Google Scholar] [CrossRef]
- Bishop, N.; Horman, A.; Woodman, P. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 2002, 157, 91–101. [Google Scholar] [CrossRef]
- Raiborg, C.; Malerod, L.; Pedersen, N.M.; Stenmark, H. Differential functions of Hrs and ESCRT proteins in endocytic membrane trafficking. Exp. Cell Res. 2008, 314, 801–813. [Google Scholar] [CrossRef]
- Morris, C.R.; Stanton, M.J.; Manthey, K.C.; Oh, K.B.; Wagner, K.U. A knockout of the Tsg101 gene leads to decreased expression of ErbB receptor tyrosine kinases and induction of autophagy prior to cell death. PLoS ONE 2012, 7, e34308. [Google Scholar] [CrossRef]
- Carstens, M.J.; Krempler, A.; Triplett, A.A.; van Lohuizen, M.; Wagner, K.U. Cell cycle arrest and cell death are controlled by p53-dependent and p53-independent mechanisms in Tsg101-deficient cells. J. Biol. Chem. 2004, 279, 35984–35994. [Google Scholar] [CrossRef]
- Von Schwedler, U.K.; Stuchell, M.; Muller, B.; Ward, D.M.; Chung, H.Y.; Morita, E.; Wang, H.E.; Davis, T.; He, G.P.; Cimbora, D.M.; et al. The protein network of HIV budding. Cell 2003, 114, 701–713. [Google Scholar] [CrossRef]
- VerPlank, L.; Bouamr, F.; LaGrassa, T.J.; Agresta, B.; Kikonyogo, A.; Leis, J.; Carter, C.A. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag). Proc. Natl. Acad. Sci. USA 2001, 98, 7724–7729. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Strickland M, E.L.; Watanabe, S.; Khan, M.; Strub, M.P.; Luan, C.H.; Powell, M.D.; Leis, J.; Tjandra, N.; Carter, C.A. Tsg101 chaperone function revealed by HIV-1 assembly inhibitors. Nat. Comm. 2017, 8. [Google Scholar] [CrossRef]
- Thery, C.; Boussac, M.; Veron, P.; Ricciardi-Castagnoli, P.; Raposo, G.; Garin, J.; Amigorena, S. Proteomic analysis of dendritic cell-derived exosomes: A secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 2001, 166, 7309–7318. [Google Scholar] [CrossRef]
- Thery, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Thery, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Stanton, M.J.; Manthey, K.C.; Triplett, A.A.; Wagner, K.U. Novel Roles of Tsg101 in Endocytic Recycling Revealed by Analyses in Cre-Inducible Tsg101 Knockout Cells; University of Nebraska Medical Center: Omaha, NE, USA, 2011; unpublished. [Google Scholar]
- Oh, K.B.; Stanton, M.J.; West, W.W.; Todd, G.L.; Wagner, K.U. Tsg101 is upregulated in a subset of invasive human breast cancers and its targeted overexpression in transgenic mice reveals weak oncogenic properties for mammary cancer initiation. Oncogene 2007, 26, 5950–5959. [Google Scholar] [CrossRef]
- Essandoh, K.; Deng, S.; Wang, X.; Jiang, M.; Mu, X.; Peng, J.; Li, Y.; Peng, T.; Wagner, K.U.; Rubinstein, J.; et al. Tsg101 positively regulates physiologic-like cardiac hypertrophy through FIP3-mediated endosomal recycling of IGF-1R. FASEB J. 2019, 33, 7451–7466. [Google Scholar] [CrossRef]
- Essandoh, K.; Wang, X.; Huang, W.; Deng, S.; Gardner, G.; Mu, X.; Li, Y.; Kranias, E.G.; Wang, Y.; Fan, G.C. Tumor susceptibility gene 101 ameliorates endotoxin-induced cardiac dysfunction by enhancing Parkin-mediated mitophagy. J. Biol. Chem. 2019, 294, 18057–18068. [Google Scholar] [CrossRef]
- Kim, J.; Wende, A.R.; Sena, S.; Theobald, H.A.; Soto, J.; Sloan, C.; Wayment, B.E.; Litwin, S.E.; Holzenberger, M.; LeRoith, D.; et al. Insulin-like growth factor I receptor signaling is required for exercise-induced cardiac hypertrophy. Mol. Endocrinol. 2008, 22, 2531–2543. [Google Scholar] [CrossRef]
- Walker, W.P.; Oehler, A.; Edinger, A.L.; Wagner, K.U.; Gunn, T.M. Oligodendroglial deletion of ESCRT-I component TSG101 causes spongiform encephalopathy. Biol. Cell 2016, 108, 324–337. [Google Scholar] [CrossRef]
- Langheinrich, U.; Hennen, E.; Stott, G.; Vacun, G. Zebrafish as a model organism for the identification and characterization of drugs and genes affecting p53 signaling. Curr. Biol. 2002, 12, 2023–2028. [Google Scholar] [CrossRef]
- Li, L.; Liao, J.; Ruland, J.; Mak, T.W.; Cohen, S.N. A TSG101/MDM2 regulatory loop modulates MDM2 degradation and MDM2/p53 feedback control. Proc. Natl. Acad. Sci. USA 2001, 98, 1619–1624. [Google Scholar] [CrossRef]
- Razi, M.; Futter, C.E. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol. Biol. Cell 2006, 17, 3469–3483. [Google Scholar] [CrossRef]
- Horgan, C.P.; Hanscom, S.R.; Kelly, E.E.; McCaffrey, M.W. Tumor Susceptibility Gene 101 (TSG101) Is a Novel Binding-Partner for the Class II Rab11-FIPs. PloS ONE 2012, 7, e32030. [Google Scholar] [CrossRef]
- Krempler, A.; Qi, Y.; Triplett, A.A.; Zhu, J.; Rui, H.; Wagner, K.U. Generation of a conditional knockout allele for the Janus kinase 2 (Jak2) gene in mice. Genesis 2004, 40, 52–57. [Google Scholar] [CrossRef]
- Henry, M.D.; Triplett, A.A.; Oh, K.B.; Smith, G.H.; Wagner, K.U. Parity-induced mammary epithelial cells facilitate tumorigenesis in MMTV-neu transgenic mice. Oncogene 2004, 23, 6980–6985. [Google Scholar] [CrossRef]
- Triplett, A.A.; Oh, K.B.; Wagner, K.U. Deletion of both TSG101 alleles in Erbb2-induced mammary tumors results in cell death; University of Nebraska Medical Center: Omaha, NE, USA, 2007; unpublished. [Google Scholar]
- Zhu, G.; Gilchrist, R.; Borley, N.; Chng, H.W.; Morgan, M.; Marshall, J.F.; Camplejohn, R.S.; Muir, G.H.; Hart, I.R. Reduction of TSG101 protein has a negative impact on tumor cell growth. Int. J. Cancer 2004, 109, 541–547. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, M.; Cui, Z.S.; Li, C.Y.; Xue, X.X.; Yu, M.; Lu, Y.; Zhang, S.Y.; Wang, E.H.; Wen, Y.Y. Down-regulation of TSG101 by small interfering RNA inhibits the proliferation of breast cancer cells through the MAPK/ERK signal pathway. Histol. Histopathol. 2011, 26, 87–94. [Google Scholar] [CrossRef]
- Xu, C.; Zheng, J. siRNA against TSG101 reduces proliferation and induces G0/G1 arrest in renal cell carcinoma - involvement of c-myc, cyclin E1, and CDK2. Cell Mol. Biol. Lett. 2019, 24, 7. [Google Scholar] [CrossRef]
- Young, T.W.; Mei, F.C.; Rosen, D.G.; Yang, G.; Li, N.; Liu, J.; Cheng, X. Up-regulation of tumor susceptibility gene 101 protein in ovarian carcinomas revealed by proteomics analyses. Mol. Cell Proteom. 2007, 6, 294–304. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Ji, W.; Liu, A.; Qin, A.; Shen, L.; Li, G.; Zhou, Y.; Hu, X.; Yu, E.; Jin, G. TSG101 Silencing Suppresses Hepatocellular Carcinoma Cell Growth by Inducing Cell Cycle Arrest and Autophagic Cell Death. Med. Sci. Monit. 2015, 21, 3371–3379. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Ortega-Cava, C.F.; Winograd, P.; Stanton, M.J.; Reddi, A.L.; Dodge, I.; Arya, R.; Dimri, M.; Clubb, R.J.; Naramura, M.; et al. Endosomal-sorting complexes required for transport (ESCRT) pathway-dependent endosomal traffic regulates the localization of active Src at focal adhesions. Proc. Natl. Acad. Sci. USA 2010, 107, 16107–16112. [Google Scholar] [CrossRef]
- Moberg, K.H.; Schelble, S.; Burdick, S.K.; Hariharan, I.K. Mutations in erupted, the Drosophila ortholog of mammalian tumor susceptibility gene 101, elicit non-cell-autonomous overgrowth. Dev. Cell 2005, 9, 699–710. [Google Scholar] [CrossRef]
- Sevrioukov, E.A.; Moghrabi, N.; Kuhn, M.; Kramer, H. A mutation in dVps28 reveals a link between a subunit of the endosomal sorting complex required for transport-I complex and the actin cytoskeleton in Drosophila. Mol. Biol. Cell 2005, 16, 2301–2312. [Google Scholar] [CrossRef]
- Bishop, N.; Woodman, P. TSG101/mammalian VPS23 and mammalian VPS28 interact directly and are recruited to VPS4-induced endosomes. J. Biol. Chem. 2001, 276, 11735–11742. [Google Scholar] [CrossRef]
- Bache, K.G.; Slagsvold, T.; Cabezas, A.; Rosendal, K.R.; Raiborg, C.; Stenmark, H. The growth-regulatory protein HCRP1/hVps37A is a subunit of mammalian ESCRT-I and mediates receptor down-regulation. Mol. Biol. Cell 2004, 15, 4337–4346. [Google Scholar] [CrossRef]
- Liu, R.T.; Huang, C.C.; You, H.L.; Chou, F.F.; Hu, C.C.; Chao, F.P.; Chen, C.M.; Cheng, J.T. Overexpression of tumor susceptibility gene TSG101 in human papillary thyroid carcinomas. Oncogene 2002, 21, 4830–4837. [Google Scholar] [CrossRef]
- Liu, F.; Yu, Y.; Jin, Y.; Fu, S. TSG101, identified by screening a cancer cDNA library and soft agar assay, promotes cell proliferation in human lung cancer. Mol. Biol. Rep. 2010, 37, 2829–2838. [Google Scholar] [CrossRef]
- Young, T.W.; Rosen, D.G.; Mei, F.C.; Li, N.; Liu, J.; Wang, X.F.; Cheng, X. Up-regulation of tumor susceptibility gene 101 conveys poor prognosis through suppression of p21 expression in ovarian cancer. Clin. Cancer Res. 2007, 13, 3848–3854. [Google Scholar] [CrossRef]
- Ma, X.R.; Edmund Sim, U.H.; Pauline, B.; Patricia, L.; Rahman, J. Overexpression of WNT2 and TSG101 genes in colorectal carcinoma. Trop. Biomed. 2008, 25, 46–57. [Google Scholar] [PubMed]
- Cheng, X.; Young, T.W.; Mei, F.C. Proteomics analyses of ovarian cancer using genetically defined human ovarian cancer models. Front Biosci. 2007, 12, 5166–5174. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Creamer, B.A.; Sakamoto, K.; Schmidt, J.W.; Triplett, A.A.; Moriggl, R.; Wagner, K.U. Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol. Cell Biol. 2010, 30, 2957–2970. [Google Scholar] [CrossRef] [PubMed]
- Amit, I.; Yakir, L.; Katz, M.; Zwang, Y.; Marmor, M.D.; Citri, A.; Shtiegman, K.; Alroy, I.; Tuvia, S.; Reiss, Y.; et al. Tal, a Tsg101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004, 18, 1737–1752. [Google Scholar] [CrossRef]
- Nicolaou, P.; Cianchetti, C.; Minaidou, A.; Marrosu, G.; Zamba-Papanicolaou, E.; Middleton, L.; Christodoulou, K. A novel LRSAM1 mutation is associated with autosomal dominant axonal Charcot-Marie-Tooth disease. Eur. J. Hum. Genet. 2013, 21, 190–194. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Weterman, M.A.; Sorrentino, V.; Kasher, P.R.; Jakobs, M.E.; van Engelen, B.G.; Fluiter, K.; de Wissel, M.B.; Sizarov, A.; Nurnberg, G.; Nurnberg, P.; et al. A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum. Mol. Genet. 2012, 21, 358–370. [Google Scholar] [CrossRef]
- Guernsey, D.L.; Jiang, H.; Bedard, K.; Evans, S.C.; Ferguson, M.; Matsuoka, M.; Macgillivray, C.; Nightingale, M.; Perry, S.; Rideout, A.L.; et al. Mutation in the gene encoding ubiquitin ligase LRSAM1 in patients with Charcot-Marie-Tooth disease. Plos Genet. 2010, 6. [Google Scholar] [CrossRef]
- McDonald, B.; Martin-Serrano, J. Regulation of Tsg101 Expression by the Steadiness Box: A Role of Tsg101-associated Ligase. Mol. Biol. Cell 2008, 19, 754–763. [Google Scholar] [CrossRef]
- Bogdanik, L.P.; Sleigh, J.N.; Tian, C.; Samuels, M.E.; Bedard, K.; Seburn, K.L.; Burgess, R.W. Loss of the E3 ubiquitin ligase LRSAM1 sensitizes peripheral axons to degeneration in a mouse model of Charcot-Marie-Tooth disease. Dis. Models Mech. 2013, 6, 780–792. [Google Scholar] [CrossRef]
- Kim, B.Y.; Olzmann, J.A.; Barsh, G.S.; Chin, L.S.; Li, L. Spongiform neurodegeneration-associated E3 ligase Mahogunin ubiquitylates TSG101 and regulates endosomal trafficking. Mol. Biol. Cell 2007, 18, 1129–1142. [Google Scholar] [CrossRef]
- Jiao, J.; Sun, K.; Walker, W.P.; Bagher, P.; Cota, C.D.; Gunn, T.M. Abnormal regulation of TSG101 in mice with spongiform neurodegeneration. Biochim. Biophys. Acta 2009, 1792, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Man, T.K.; Lu, X.Y.; Jaeweon, K.; Perlaky, L.; Harris, C.P.; Shah, S.; Ladanyi, M.; Gorlick, R.; Lau, C.C.; Rao, P.H. Genome-wide array comparative genomic hybridization analysis reveals distinct amplifications in osteosarcoma. BMC Cancer 2004, 4, 45. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferraiuolo, R.-M.; Manthey, K.C.; Stanton, M.J.; Triplett, A.A.; Wagner, K.-U. The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease. Cancers 2020, 12, 450. https://doi.org/10.3390/cancers12020450
Ferraiuolo R-M, Manthey KC, Stanton MJ, Triplett AA, Wagner K-U. The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease. Cancers. 2020; 12(2):450. https://doi.org/10.3390/cancers12020450
Chicago/Turabian StyleFerraiuolo, Rosa-Maria, Karoline C. Manthey, Marissa J. Stanton, Aleata A. Triplett, and Kay-Uwe Wagner. 2020. "The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease" Cancers 12, no. 2: 450. https://doi.org/10.3390/cancers12020450
APA StyleFerraiuolo, R.-M., Manthey, K. C., Stanton, M. J., Triplett, A. A., & Wagner, K.-U. (2020). The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease. Cancers, 12(2), 450. https://doi.org/10.3390/cancers12020450