Patient Derived Models to Study Head and Neck Cancer Radiation Response

 ,
,
Abstract
1. Introduction
2. Patient-Derived Models of Head and Neck Cancer
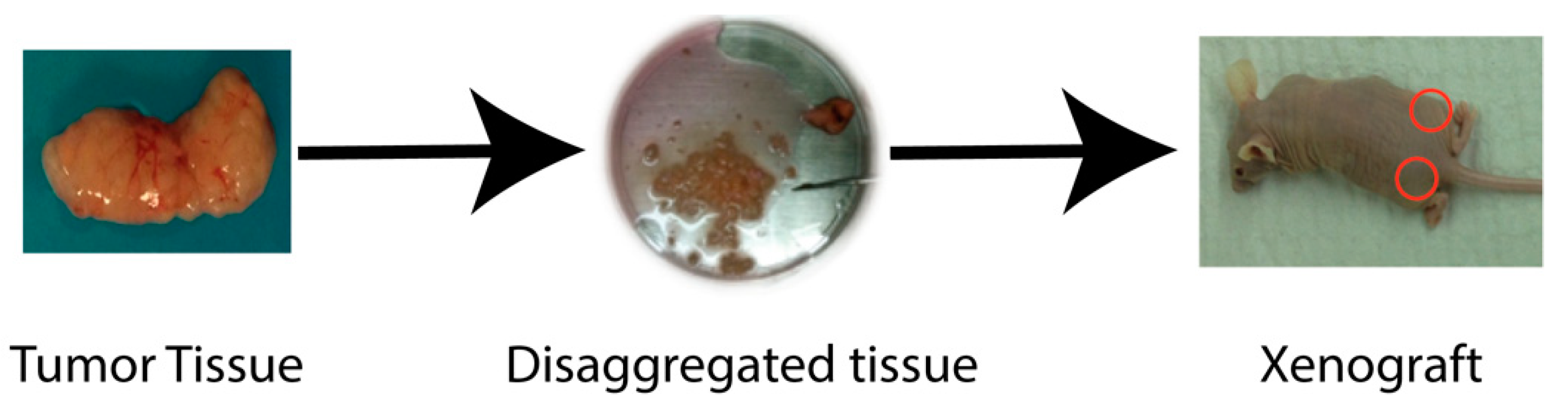
2.1. Xenograft Models
2.2. Organoid Models
2.3. Zebrafish Models
2.4. Establishing Patient-Derived Models
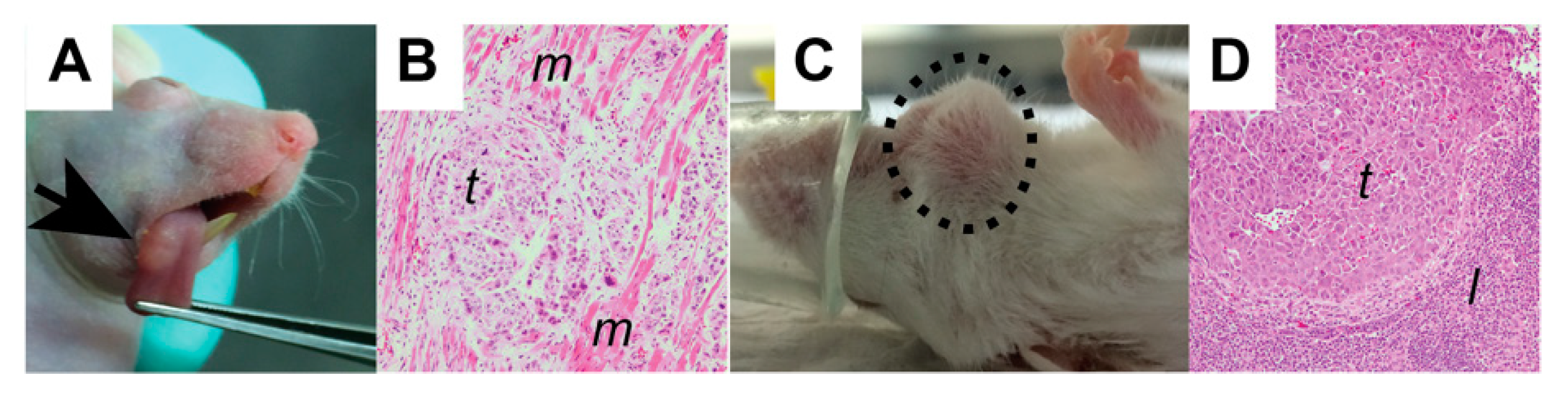
2.5. Flank Models
2.6. Orthotopic Models
2.7. Humanized Models
3. Radiation Delivery
Challenges in Drug and Radiation Delivery
4. Experimental Design
5. Statistical Analysis
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Greene, H.S. The significance of the heterologous transplantability of human cancer. Cancer 1952, 5, 24–44. [Google Scholar] [CrossRef]
- Goldenberg, D.M.; Muller, E.; Witte, S. In vivo proliferation of heterotransplanted human cancer cells. Eur. J. Cancer 1967, 3, 315–319. [Google Scholar] [CrossRef]
- Cobb, L.M.; Mitchley, B.C.; Wood, J.M. Proceedings: Factors influencing the establishment of human tumour cells as a xenograft. Br. J. Cancer 1974, 29, 97. [Google Scholar] [CrossRef][Green Version]
- Ishikawa, F.; Yasukawa, M.; Lyons, B.; Yoshida, S.; Miyamoto, T.; Yoshimoto, G.; Watanabe, T.; Akashi, K.; Shultz, L.D.; Harada, M. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood 2005, 106, 1565–1573. [Google Scholar] [CrossRef]
- Ito, M.; Hiramatsu, H.; Kobayashi, K.; Suzue, K.; Kawahata, M.; Hioki, K.; Ueyama, Y.; Koyanagi, Y.; Sugamura, K.; Tsuji, K.; et al. NOD/SCID/gamma(c)(null) mouse: An excellent recipient mouse model for engraftment of human cells. Blood 2002, 100, 3175–3182. [Google Scholar] [CrossRef]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef]
- Prabakaran, P.J.; Javaid, A.M.; Swick, A.D.; Werner, L.R.; Nickel, K.P.; Sampene, E.; Hu, R.; Ong, I.M.; Bruce, J.Y.; Hartig, G.K.; et al. Radiosensitization of adenoid cystic carcinoma with mdm2 inhibition. Clin. Cancer Res. 2017, 23, 6044–6053. [Google Scholar] [CrossRef]
- Kimple, R.J.; Harari, P.M.; Torres, A.D.; Yang, R.Z.; Soriano, B.J.; Yu, M.; Armstrong, E.A.; Blitzer, G.C.; Smith, M.A.; Lorenz, L.D.; et al. Development and characterization of HPV-positive and HPV-negative head and neck squamous cell carcinoma tumorgrafts. Clin. Cancer Res. 2013, 19, 855–864. [Google Scholar] [CrossRef]
- Stein, A.P.; Saha, S.; Liu, C.Z.; Hartig, G.K.; Lambert, P.F.; Kimple, R.J. Influence of handling conditions on the establishment and propagation of head and neck cancer patient derived xenografts. PLoS ONE 2014, 9, e100995. [Google Scholar] [CrossRef] [PubMed]
- Swick, A.D.; Stein, A.P.; McCulloch, T.M.; Hartig, G.K.; Ong, I.M.; Sampene, E.; Prabakaran, P.J.; Liu, C.Z.; Kimple, R.J. Defining the boundaries and expanding the utility of head and neck cancer patient derived xenografts. Oral Oncol. 2017, 64, 65–72. [Google Scholar] [CrossRef]
- Izumchenko, E.; Paz, K.; Ciznadija, D.; Sloma, I.; Katz, A.; Vasquez-Dunddel, D.; Ben-Zvi, I.; Stebbing, J.; McGuire, W.; Harris, W.; et al. Patient-derived xenografts effectively capture responses to oncology therapy in a heterogeneous cohort of patients with solid tumors. Ann. Oncol. 2017, 28, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.J.; Bhatia, S.; Van Court, B.; Darragh, L.; Serkova, N.; Karam, S.D. Intramucosal inoculation of squamous cell carcinoma cells in mice for tumor immune profiling and treatment response assessment. J. Vis. Exp. JoVE 2019. [Google Scholar] [CrossRef] [PubMed]
- A Pilot Clinical Study of Treatment Guided by Personalized Tumorgrafts in Patients with Advanced Cancer. Available online: https://www.ncbi.nlm.nih.gov/pubmed/?term=Mol+Cancer+Ther.+2011%3B10(8)%3A1311-6 (accessed on 21 November 2019).
- Dong, X.; Guan, J.; English, J.C.; Flint, J.; Yee, J.; Evans, K.; Murray, N.; Macaulay, C.; Ng, R.T.; Gout, P.W.; et al. Patient-derived first generation xenografts of non-small cell lung cancers: Promising tools for predicting drug responses for personalized chemotherapy. Clin. Cancer Res. 2010, 16, 1442–1451. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Wang, G.; Lin, Y.-C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.W.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.P.; Calvo, E.; Ordoñez, E.; Wick, M.J.; Viqueira, B.-R.; Lopez-Casas, P.P.; Bruckheimer, E.; Calles-Blanco, A.; Sidransky, D.; Hidalgo, M. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J. Clin. Oncol. 2012, 30, e45–e48. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Torras, S.; Vidal-Pla, A.; Miquel, R.; Almendro, V.; Fernández-Cruz, L.; Navarro, S.; Maurel, J.; Carbó, N.; Gascón, P.; Mazo, A. Characterization of human pancreatic orthotopic tumor xenografts suitable for drug screening. Cell Oncol. Dordr. 2011, 34, 511–521. [Google Scholar] [CrossRef]
- Facompre, N.D.; Sahu, V.; Montone, K.T.; Harmeyer, K.M.; Nakagawa, H.; Rustgi, A.K.; Weinstein, G.S.; Gimotty, P.A.; Basu, D. Barriers to generating PDX models of HPV-related head and neck cancer. Laryngoscope 2017, 127, 2777–2783. [Google Scholar] [CrossRef]
- Eirew, P.; Steif, A.; Khattra, J.; Ha, G.; Yap, D.; Farahani, H.; Gelmon, K.; Chia, S.; Mar, C.; Wan, A.; et al. Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution. Nature 2015, 518, 422–426. [Google Scholar] [CrossRef]
- Clappier, E.; Gerby, B.; Sigaux, F.; Delord, M.; Touzri, F.; Hernandez, L.; Ballerini, P.; Baruchel, A.; Pflumio, F.; Soulier, J. Clonal selection in xenografted human T cell acute lymphoblastic leukemia recapitulates gain of malignancy at relapse. J. Exp. Med. 2011, 208, 653–661. [Google Scholar] [CrossRef]
- John, T.; Kohler, D.; Pintilie, M.; Yanagawa, N.; Pham, N.-A.; Li, M.; Panchal, D.; Hui, F.; Meng, F.; Shepherd, F.A.; et al. The ability to form primary tumor xenografts is predictive of increased risk of disease recurrence in early-stage non-small cell lung cancer. Clin. Cancer Res. 2011, 17, 134–141. [Google Scholar] [CrossRef]
- Karamboulas, C.; Bruce, J.P.; Hope, A.J.; Meens, J.; Huang, S.H.; Erdmann, N.; Hyatt, E.; Pereira, K.; Goldstein, D.P.; Weinreb, I.; et al. Patient-derived xenografts for prognostication and personalized treatment for head and neck squamous cell carcinoma. Cell Rep. 2018, 25, 1318–1331. [Google Scholar] [CrossRef] [PubMed]
- Verhaegen, F.; Granton, P.; Tryggestad, E. Small animal radiotherapy research platforms. Phys. Med. Biol. 2011, 56, R55–R83. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.; Tofilon, P.J.; Camphausen, K. Preclinical models in radiation oncology. Radiat. Oncol. 2012, 7, 223. [Google Scholar] [CrossRef] [PubMed]
- Swick, A.D.; Prabakaran, P.J.; Miller, M.C.; Javaid, A.M.; Fisher, M.M.; Sampene, E.; Ong, I.M.; Hu, R.; Iida, M.; Nickel, K.P.; et al. Cotargeting mtorc and egfr signaling as a therapeutic strategy in hnscc. Mol. Cancer Ther. 2017, 16, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Kimple, R.J.; Smith, M.A.; Blitzer, G.C.; Torres, A.D.; Martin, J.A.; Yang, R.Z.; Peet, C.R.; Lorenz, L.D.; Nickel, K.P.; Klingelhutz, A.J.; et al. Enhanced radiation sensitivity in HPV-positive head and neck cancer. Cancer Res. 2013, 73, 4791–4800. [Google Scholar] [CrossRef] [PubMed]
- Brand, T.M.; Iida, M.; Corrigan, K.L.; Braverman, C.M.; Coan, J.P.; Flanigan, B.G.; Stein, A.P.; Salgia, R.; Rolff, J.; Kimple, R.J.; et al. The receptor tyrosine kinase AXL mediates nuclear translocation of the epidermal growth factor receptor. Sci. Signal 2017, 10, eaag1064. [Google Scholar] [CrossRef]
- Zhang, K.; Jones, L.; Lim, S.; Maher, C.A.; Adkins, D.; Lewis, J.; Kimple, R.J.; Fertig, E.J.; Chung, C.H.; Van Tine, B.A.; et al. Loss of Trop2 causes ErbB3 activation through a neuregulin-1-dependent mechanism in the mesenchymal subtype of HNSCC. Oncotarget 2014, 5, 9281–9294. [Google Scholar] [CrossRef]
- Spanos, W.C.; Nowicki, P.; Lee, D.W.; Hoover, A.; Hostager, B.; Gupta, A.; Anderson, M.E.; Lee, J.H. Immune response during therapy with cisplatin or radiation for human papillomavirus-related head and neck cancer. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 1137–1146. [Google Scholar] [CrossRef]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef]
- Demaria, S.; Golden, E.B.; Formenti, S.C. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015, 1, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Lhuillier, C.; Rudqvist, N.-P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [PubMed]
- Driehuis, E.; Spelier, S.; Beltrán Hernández, I.; de Bree, R.; M Willems, S.; Clevers, H.; Oliveira, S. Patient-derived head and neck cancer organoids recapitulate egfr expression levels of respective tissues and are responsive to egfr-targeted photodynamic therapy. J. Clin. Med. 2019, 8, 1880. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Osman, A.A.; Takahashi, Y.; Lindemann, A.; Patel, A.A.; Zhao, M.; Takahashi, H.; Myers, J.N. Head and neck cancer organoids established by modification of the CTOS method can be used to predict in vivo drug sensitivity. Oral Oncol. 2018, 87, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Ayuso, J.M.; Vitek, R.; Swick, A.D.; Skala, M.C.; Wisinski, K.B.; Kimple, R.J.; Lambert, P.F.; Beebe, D.J. Effects of culture method on response to EGFR therapy in head and neck squamous cell carcinoma cells. Sci. Rep. 2019, 9, 12480. [Google Scholar] [CrossRef]
- Pasch, C.A.; Favreau, P.F.; Yueh, A.E.; Babiarz, C.P.; Gillette, A.A.; Sharick, J.T.; Karim, M.R.; Nickel, K.P.; DeZeeuw, A.K.; Sprackling, C.M.; et al. Patient-derived cancer organoid cultures to predict sensitivity to chemotherapy and radiation. Clin. Cancer Res. J. Am. Assoc. Cancer Res. 2019, 25, 5376–5387. [Google Scholar] [CrossRef]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef]
- Yan, C.; Brunson, D.C.; Tang, Q.; Do, D.; Iftimia, N.A.; Moore, J.C.; Hayes, M.N.; Welker, A.M.; Garcia, E.G.; Dubash, T.D.; et al. Visualizing Engrafted Human Cancer and Therapy Responses in Immunodeficient Zebrafish. Cell 2019, 177, 1903–1914. [Google Scholar] [CrossRef]
- Hwang, M.; Yong, C.; Moretti, L.; Lu, B. Zebrafish as a model system to screen radiation modifiers. Curr. Genom. 2007, 8, 360–369. [Google Scholar]
- Abel, L.; Durmaz, A.; Hu, R.; Longhurst, C.; Scott, J.G.; Kimple, R.J. Impact of immediate cryopreservation on the establishment of patient derived xenografts from head and neck cancer patients. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Kuper, C.F.; Koornstra, P.J.; Hameleers, D.M.; Biewenga, J.; Spit, B.J.; Duijvestijn, A.M.; van Breda Vriesman, P.J.; Sminia, T. The role of nasopharyngeal lymphoid tissue. Immunol. Today 1992, 13, 219–224. [Google Scholar] [CrossRef]
- Pearson, T.; Greiner, D.L.; Shultz, L.D. Creation of “humanized” mice to study human immunity. Curr. Protoc. Immunol. 2008, 81, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Brehm, M.A.; Bridges, S.; Ferguson, S.; Kumar, P.; Mirochnitchenko, O.; Palucka, K.; Pelanda, R.; Sanders-Beer, B.; Shultz, L.D.; et al. Humanized immune system mouse models: Progress, challenges and opportunities. Nat. Immunol. 2019, 20, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, H.; Lindberg, M.F.; Donia, M.; Soderberg, E.M.V.; Andersen, R.; Keller, U.; Ny, L.; Svane, I.M.; Nilsson, L.M.; Nilsson, J.A. Clinical responses to adoptive T-cell transfer can be modeled in an autologous immune-humanized mouse model. Nat. Commun. 2017, 8, 707. [Google Scholar] [CrossRef]
- Rosato, R.R.; Davila-Gonzalez, D.; Choi, D.S.; Qian, W.; Chen, W.; Kozielski, A.J.; Wong, H.; Dave, B.; Chang, J.C. Evaluation of anti-PD-1-based therapy against triple-negative breast cancer patient-derived xenograft tumors engrafted in humanized mouse models. Breast Cancer Res. 2018, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yao, L.C.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.X.; Shi, W.; Ma, A.H.; De Vere White, R.W.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.J.; Bird, G.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.; Eagles, J.R.; Le, P.N.; et al. XactMice: Humanizing mouse bone marrow enables microenvironment reconstitution in a patient-derived xenograft model of head and neck cancer. Oncogene 2016, 35, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Lyons, B.L.; Burzenski, L.M.; Gott, B.; Chen, X.; Chaleff, S.; Kotb, M.; Gillies, S.D.; King, M.; Mangada, J.; et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J. Immunol. Baltim. Md 1950 2005, 174, 6477–6489. [Google Scholar]
- Brown, M.E.; Zhou, Y.; McIntosh, B.E.; Norman, I.G.; Lou, H.E.; Biermann, M.; Sullivan, J.A.; Kamp, T.J.; Thomson, J.A.; Anagnostopoulos, P.V.; et al. A humanized mouse model generated using surplus neonatal tissue. Stem Cell Rep. 2018, 10, 1175–1183. [Google Scholar] [CrossRef]
- Laing, S.T.; Griffey, S.M.; Moreno, M.E.; Stoddart, C.A. CD8-positive lymphocytes in graft-versus-host disease of humanized nod.cg-prkdc(scid)Il2rg(tm1wjl)/szj mice. J Comp. Pathol. 2015, 152, 238–242. [Google Scholar] [CrossRef]
- Ali, N.; Flutter, B.; Sanchez Rodriguez, R.; Sharif-Paghaleh, E.; Barber, L.D.; Lombardi, G.; Nestle, F.O. Xenogeneic graft-versus-host-disease in NOD-scid IL-2Rgammanull mice display a T-effector memory phenotype. PLoS ONE 2012, 7, e44219. [Google Scholar] [CrossRef]
- Wong, J.; Armour, E.; Kazanzides, P.; Iordachita, I.; Tryggestad, E.; Deng, H.; Matinfar, M.; Kennedy, C.; Liu, Z.; Chan, T.; et al. High-Resolution, small animal radiation research platform with X-ray tomographic guidance capabilities. Int. J. Radiat. Oncol. 2008, 71, 1591–1599. [Google Scholar] [CrossRef] [PubMed]
- Quantitative Concepts and Dosimetry in Radiobiology; ICRU: Bethesda, MD, USA, 1979.
- Ma, C.M.; Coffey, C.W.; DeWerd, L.A.; Liu, C.; Nath, R.; Seltzer, S.M.; Seuntjens, J.P. American Association of Physicists in Medicine AAPM protocol for 40–300 kV x-ray beam dosimetry in radiotherapy and radiobiology. Med. Phys. 2001, 28, 868–893. [Google Scholar] [CrossRef] [PubMed]
- Newton, J.; Oldham, M.; Thomas, A.; Li, Y.; Adamovics, J.; Kirsch, D.G.; Das, S. Commissioning a small-field biological irradiator using point, 2D, and 3D dosimetry techniques. Med. Phys. 2011, 38, 6754–6762. [Google Scholar] [CrossRef] [PubMed]
- Pidikiti, R.; Stojadinovic, S.; Speiser, M.; Song, K.H.; Hager, F.; Saha, D.; Solberg, T.D. Dosimetric characterization of an image-guided stereotactic small animal irradiator. Phys. Med. Biol. 2011, 56, 2585–2599. [Google Scholar] [CrossRef] [PubMed]
- Stone, H.B.; Bernhard, E.J.; Coleman, C.N.; Deye, J.; Capala, J.; Mitchell, J.B.; Brown, J.M. Preclinical data on efficacy of 10 drug-radiation combinations: Evaluations, concerns, and recommendations. Transl. Oncol. 2016, 9, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Chiu-Tsao, S.; Massillon-Jl, G.; Domingo-Muñoz, I.; Chan, M. SU-E-T-96: Energy dependence of the new gafchromic- Ebt3 film’s dose response-curve. Med. Phys. 2012, 39, 3724. [Google Scholar] [CrossRef]
- Gurtner, K.; Kryzmien, Z.; Koi, L.; Wang, M.; Benes, C.H.; Hering, S.; Willers, H.; Baumann, M.; Krause, M. Radioresistance of KRAS/TP53-mutated lung cancer can be overcome by radiation dose escalation or EGFR tyrosine kinase inhibition in vivo. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Helbig, L.; Koi, L.; Brüchner, K.; Gurtner, K.; Hess-Stumpp, H.; Unterschemmann, K.; Baumann, M.; Zips, D.; Yaromina, A. BAY 87-2243, a novel inhibitor of hypoxia-induced gene activation, improves local tumor control after fractionated irradiation in a schedule-dependent manner in head and neck human xenografts. Radiat. Oncol. Lond. Engl. 2014, 9, 207. [Google Scholar] [CrossRef]
- Morgan, M.A.; Parsels, L.A.; Maybaum, J.; Lawrence, T.S. Improving the efficacy of chemoradiation with targeted agents. Cancer Discov. 2014, 4, 280–291. [Google Scholar] [CrossRef]
- Lawrence, Y.R.; Vikram, B.; Dignam, J.J.; Chakravarti, A.; Machtay, M.; Freidlin, B.; Takebe, N.; Curran, W.J.; Bentzen, S.M.; Okunieff, P.; et al. NCI-RTOG translational program strategic guidelines for the early-stage development of radiosensitizers. J. Natl. Cancer Inst. 2013, 105, 11–24. [Google Scholar] [CrossRef]
- Blumenfeld, P.; Pfeffer, R.M.; Symon, Z.; Den, R.B.; Dicker, A.P.; Raben, D.; Lawrence, Y.R. The lag time in initiating clinical testing of new drugs in combination with radiation therapy, a significant barrier to progress? Br. J. Cancer 2014, 111, 1305–1309. [Google Scholar] [CrossRef] [PubMed]
- Higgins, G.S.; O’Cathail, S.M.; Muschel, R.J.; McKenna, W.G. Drug radiotherapy combinations: Review of previous failures and reasons for future optimism. Cancer Treat. Rev. 2015, 41, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Rosfjord, E.; Lucas, J.; Li, G.; Gerber, H.-P. Advances in patient-derived tumor xenografts: From target identification to predicting clinical response rates in oncology. Biochem. Pharmacol. 2014, 91, 135–143. [Google Scholar] [CrossRef]
- Zhou, S.; Kestell, P.; Paxton, J.W. Strain differences in the liver microsomal metabolism of the experimental anti-tumour agent 5,6-dimethylxanthenone-4-acetic acid in mice. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2002, 776, 231–236. [Google Scholar] [CrossRef]
- Terry, N.H.; Stratford, M.R.; Minchinton, A.I. Misonidazole toxicity and pharmacokinetics in mice: Dependence on strain and size. Eur. J. Cancer Clin. Oncol. 1985, 21, 845–851. [Google Scholar] [CrossRef]
- Visser, G.W.; Gorree, G.C.; Peters, G.J.; Herscheid, J.D. Tissue distribution of [18F]-5-fluorouracil in mice: Effects of route of administration, strain, tumour and dose. Cancer Chemother. Pharmacol. 1990, 26, 205–209. [Google Scholar] [CrossRef]
- Liang, H. Modeling antitumor activity in xenograft tumor treatment. Biom. J. Biom. Z 2005, 47, 358–368. [Google Scholar] [CrossRef]
- Zhang, Z. Semi-parametric regression model for survival data: Graphical visualization with R. Ann. Transl. Med. 2016, 4, 461. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Radiation Dose | Fractions | Schedule | Chemotherapy | Endpoint |
|---|---|---|---|---|
| 2–3 Gy/fraction | 5–10 fractions | daily × 1–2 weeks | +/− | Tumor growth delay, growth rate, time to tumor doubling |
| 2 Gy/fraction | 25–35 fractions | daily × 5–7 weeks | +/− | Cure rate, tumor control dose—50% |
| 5–10 Gy/fraction | 1–8 fractions | daily, 3 times/week | +/− | Tumor growth delay, growth rate, time to tumor doubling, cure rate, tumor control dose—50% |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosper, P.F.; Abel, L.; Lee, Y.-S.; Paz, C.; Kaushik, S.; Nickel, K.P.; Alexandridis, R.; Scott, J.G.; Bruce, J.Y.; Kimple, R.J. Patient Derived Models to Study Head and Neck Cancer Radiation Response. Cancers 2020, 12, 419. https://doi.org/10.3390/cancers12020419
Cosper PF, Abel L, Lee Y-S, Paz C, Kaushik S, Nickel KP, Alexandridis R, Scott JG, Bruce JY, Kimple RJ. Patient Derived Models to Study Head and Neck Cancer Radiation Response. Cancers. 2020; 12(2):419. https://doi.org/10.3390/cancers12020419
Chicago/Turabian StyleCosper, Pippa F., Lindsey Abel, Yong-Syu Lee, Cristina Paz, Saakshi Kaushik, Kwangok P. Nickel, Roxana Alexandridis, Jacob G. Scott, Justine Y. Bruce, and Randall J. Kimple. 2020. "Patient Derived Models to Study Head and Neck Cancer Radiation Response" Cancers 12, no. 2: 419. https://doi.org/10.3390/cancers12020419
APA StyleCosper, P. F., Abel, L., Lee, Y.-S., Paz, C., Kaushik, S., Nickel, K. P., Alexandridis, R., Scott, J. G., Bruce, J. Y., & Kimple, R. J. (2020). Patient Derived Models to Study Head and Neck Cancer Radiation Response. Cancers, 12(2), 419. https://doi.org/10.3390/cancers12020419