Photochemically-Induced Release of Lysosomal Sequestered Sunitinib: Obstacles for Therapeutic Efficacy





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Lysosomal Localization of Sunitinib and TPCS2a
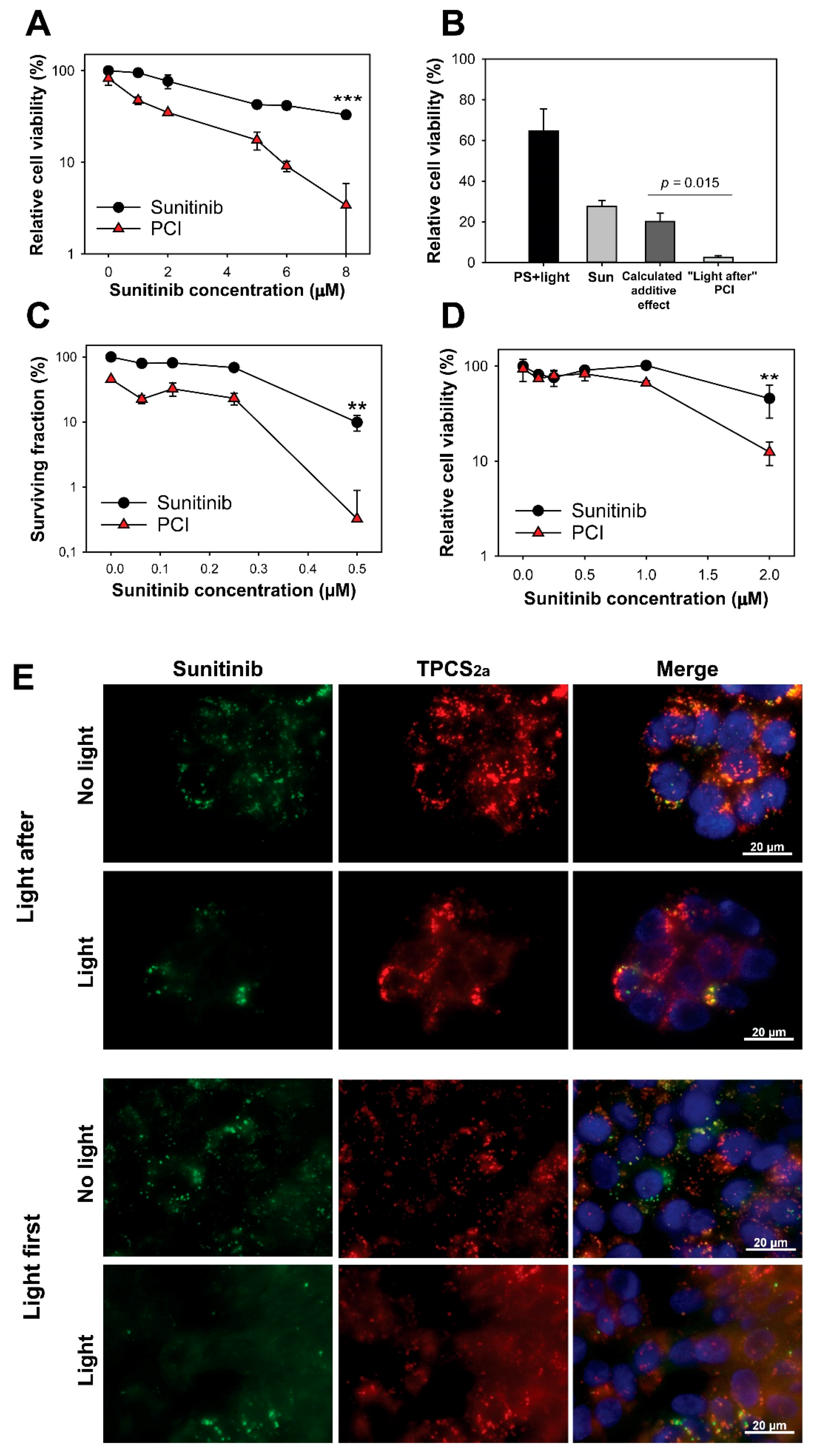
2.2. No Enhanced Toxicity by PCI “Light After” of Sunitinib
2.3. Sunitinib Is a Target for ROS-Mediated Photodamage
2.4. Synergistic Cytotoxicity by “Light First” Sunitinib-PCI
2.5. Acquired Resistance and Endo/Lysosomal Accumulation Following Prolonged Sunitinib Exposure
2.6. Photochemical Release of Sequestered Sunitinib Does Not Abolish Sunitinib Resistance in HT-29/SR
2.7. Modest HT-29 Tumor Growth Delay After Sunitinib-PCI in Athymic Mice
2.8. Poor CT26.WT Tumor Growth Delay After Sunitinib-PCI in Immunocompetent Mice
2.9. Tumor Tissue Response
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cultivation
4.2. Drugs and Chemicals
4.3. In Vitro Light Sources
4.4. In Vitro PCI Treatment
4.5. Treatment Efficacy In Vitro; Colony Formation Capacity, Viability and Proliferation
4.6. Intracellular Localization of TPCS2a and Sunitinib by Fluorescence Microscopy
4.7. Subcellular Localization of Sunitinib and TPCS2a
4.8. Cellular Accumulation of Sunitinib
4.9. Absorption and Fluorescence Spectroscopy of TPCS2a and Sunitinib
4.10. Animals
4.11. Tumor Grafts
4.12. In Vivo Experimental Design and Methods
4.13. Evaluation of Combination Therapy and Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- National Center for Biotechnology Information. PubChem Compound Database, Sunitinib, CID=5329102. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/5329102 (accessed on 11 October 2019).
- Le Tourneau, C.; Raymond, E.; Faivre, S. Sunitinib: A novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther. Clin. Risk Manag. 2007, 3, 341–348. [Google Scholar] [CrossRef]
- Finke, J.H.; Rini, B.; Ireland, J.; Rayman, P.; Richmond, A.; Golshayan, A.; Wood, L.; Elson, P.; Garcia, J.; Dreicer, R.; et al. Sunitinib reverses type-1 immune suppression and decreases T-regulatory cells in renal cell carcinoma patients. Clin. Cancer Res. 2008, 14, 6674–6682. [Google Scholar] [CrossRef]
- Ozao-Choy, J.; Ma, G.; Kao, J.; Wang, G.X.; Meseck, M.; Sung, M.; Schwartz, M.; Divino, C.M.; Pan, P.Y.; Chen, S.H. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and modulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009, 69, 2514–2522. [Google Scholar] [CrossRef]
- Gan, H.K.; Seruga, B.; Knox, J.J. Sunitinib in solid tumors. Expert. Opin. Investig. Drugs 2009, 18, 821–834. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomes as mediators of drug resistance in cancer. Drug Resist. Updat. 2016, 24, 23–33. [Google Scholar] [CrossRef]
- Gotink, K.J.; Broxterman, H.J.; Labots, M.; de Haas, R.R.; Dekker, H.; Honeywell, R.J.; Rudek, M.A.; Beerepoot, L.V.; Musters, R.J.; Jansen, G.; et al. Lysosomal sequestration of sunitinib: A novel mechanism of drug resistance. Clin. Cancer Res. 2011, 17, 7337–7346. [Google Scholar] [CrossRef]
- Kazmi, F.; Hensley, T.; Pope, C.; Funk, R.S.; Loewen, G.J.; Buckley, D.B.; Parkinson, A. Lysosomal sequestration (trapping) of lipophilic amine (cationic amphiphilic) drugs in immortalized human hepatocytes (Fa2N-4 cells). Drug Metab. Dispos. 2013, 41, 897–905. [Google Scholar] [CrossRef]
- Herlevsen, M.; Oxford, G.; Owens, C.R.; Conaway, M.; Theodorescu, D. Depletion of major vault protein increases doxorubicin sensitivity and nuclear accumulation and disrupts its sequestration in lysosomes. Mol. Cancer Ther. 2007, 6, 1804–1813. [Google Scholar] [CrossRef]
- Hurwitz, S.J.; Terashima, M.; Mizunuma, N.; Slapak, C.A. Vesicular anthracycline accumulation in doxorubicin-selected U-937 cells: Participation of lysosomes. Blood 1997, 89, 3745–3754. [Google Scholar] [CrossRef]
- Adar, Y.; Stark, M.; Bram, E.E.; Nowak-Sliwinska, P.; van den Bergh, H.; Szewczyk, G.; Sarna, T.; Skladanowski, A.; Griffioen, A.W.; Assaraf, Y.G. Imidazoacridinone-dependent lysosomal photodestruction: A pharmacological Trojan horse approach to eradicate multidrug-resistant cancers. Cell Death Dis. 2012, 3, e293. [Google Scholar] [CrossRef]
- Kaufmann, A.M.; Krise, J.P. Lysosomal sequestration of amine-containing drugs: Analysis and therapeutic implications. J. Pharm. Sci. 2007, 96, 729–746. [Google Scholar] [CrossRef]
- Berg, K.; Selbo, P.K.; Prasmickaite, L.; Tjelle, T.E.; Sandvig, K.; Moan, J.; Gaudernack, G.; Fodstad, O.; Kjolsrud, S.; Anholt, H.; et al. Photochemical internalization: A novel technology for delivery of macromolecules into cytosol. Cancer Res. 1999, 59, 1180–1183. [Google Scholar]
- Selbo, P.K.; Weyergang, A.; Hogset, A.; Norum, O.J.; Berstad, M.B.; Vikdal, M.; Berg, K. Photochemical internalization provides time- and space-controlled endolysosomal escape of therapeutic molecules. J. Control. Release 2010, 148, 2–12. [Google Scholar] [CrossRef]
- Sultan, A.A.; Jerjes, W.; Berg, K.; Hogset, A.; Mosse, C.A.; Hamoudi, R.; Hamdoon, Z.; Simeon, C.; Carnell, D.; Forster, M.; et al. Disulfonated tetraphenyl chlorin (TPCS2a)-induced photochemical internalisation of bleomycin in patients with solid malignancies: A phase 1, dose-escalation, first-in-man trial. Lancet Oncol. 2016, 17, 1217–1229. [Google Scholar] [CrossRef]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.; Angell-Petersen, E.; Hogset, A. Disulfonated tetraphenyl chlorin (TPCS2a), a novel photosensitizer developed for clinical utilization of photochemical internalization. Photochem. Photobiol. Sci. 2011, 10, 1637–1651. [Google Scholar] [CrossRef]
- Berg, K.; Høgset, A.; Prasmickaite, L.; Weyergang, A.; Bonsted, A.; Dietze, A.; Lou, P.-J.; Bown, S.; Norum, O.-J.; Møllergård, H.M.T.; et al. Photochemical internalization (PCI): A novel technology for activation of endocytosed therapeutic agents. Med. Laser Appl. 2006, 21, 239–250. [Google Scholar] [CrossRef]
- Prasmickaite, L.; Hogset, A.; Selbo, P.K.; Engesaeter, B.O.; Hellum, M.; Berg, K. Photochemical disruption of endocytic vesicles before delivery of drugs: A new strategy for cancer therapy. Brit. J. Cancer 2002, 86, 652–657. [Google Scholar] [CrossRef]
- Olsen, C.E.; Berg, K.; Selbo, P.K.; Weyergang, A. Circumvention of resistance to photodynamic therapy in doxorubicin-resistant sarcoma by photochemical internalization of gelonin. Free Radic. Biol. Med. 2013, 65, 1300–1309. [Google Scholar] [CrossRef]
- Lou, P.J.; Lai, P.S.; Shieh, M.J.; Macrobert, A.J.; Berg, K.; Bown, S.G. Reversal of doxorubicin resistance in breast cancer cells by photochemical internalization. Int. J. Cancer 2006, 119, 2692–2698. [Google Scholar] [CrossRef]
- Berg, K.; Dietze, A.; Kaalhus, O.; Hogset, A. Site-specific drug delivery by photochemical internalization enhances the antitumor effect of bleomycin. Clin. Cancer Res. 2005, 11, 8476–8485. [Google Scholar] [CrossRef]
- Nowak-Sliwinska, P.; Weiss, A.; van Beijnum, J.R.; Wong, T.J.; Kilarski, W.W.; Szewczyk, G.; Verheul, H.M.; Sarna, T.; van den Bergh, H.; Griffioen, A.W. Photoactivation of lysosomally sequestered sunitinib after angiostatic treatment causes vascular occlusion and enhances tumor growth inhibition. Cell Death Dis. 2015, 6, e1641. [Google Scholar] [CrossRef]
- Moan, J.; Sommer, S. Oxygen dependence of the photosensitizing effect of hematoporphyrin derivative in NHIK 3025 cells. Cancer Res. 1985, 45, 1608–1610. [Google Scholar]
- Weishaupt, K.R.; Gomer, C.J.; Dougherty, T.J. Identification of singlet oxygen as the cytotoxic agent in photoinactivation of a murine tumor. Cancer Res. 1976, 36, 2326–2329. [Google Scholar]
- Moan, J.; Berg, K. The photodegradation of porphyrins in cell can be used to estimate the lifetime of singlet oxygen. Photochem. Photobiol. 1991, 53, 549–553. [Google Scholar] [CrossRef]
- Shrestha, D.; Jenei, A.; Nagy, P.; Vereb, G.; Szollosi, J. Understanding FRET as a research tool for cellular studies. Int. J. Mol. Sci. 2015, 16, 6718–6756. [Google Scholar] [CrossRef]
- Ohtsuki, T.; Miki, S.; Kobayashi, S.; Haraguchi, T.; Nakata, E.; Hirakawa, K.; Sumita, K.; Watanabe, K.; Okazaki, S. The molecular mechanism of photochemical internalization of cell penetrating peptide-cargo-photosensitizer conjugates. Sci. Rep. 2015, 5, 18577. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.K.; Shouman, S.; El-Demerdash, E.; Elgendy, M.; Abdel-Naim, A.B. Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem. Biol. Interact. 2014, 217, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, S.; Cormerais, Y.; Dufies, M.; Grepin, R.; Colosetti, P.; Belaid, A.; Parola, J.; Martin, A.; Lacas-Gervais, S.; Mazure, N.M.; et al. Resistance to sunitinib in renal clear cell carcinoma results from sequestration in lysosomes and inhibition of the autophagic flux. Autophagy 2015, 11, 1891–1904. [Google Scholar] [CrossRef] [PubMed]
- Gotink, K.J.; Broxterman, H.J.; Honeywell, R.J.; Dekker, H.; de Haas, R.R.; Miles, K.M.; Adelaiye, R.; Griffioen, A.W.; Peters, G.J.; Pili, R.; et al. Acquired tumor cell resistance to sunitinib causes resistance in a HT-29 human colon cancer xenograft mouse model without affecting sunitinib biodistribution or the tumor microvasculature. Oncoscience 2014, 1, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Gotink, K.J.; Verheul, H.M. The Potential Role of Lysosomal Sequestration in Sunitinib Resistance of Renal Cell Cancer. J. Kidney Cancer VHL 2015, 2, 195–203. [Google Scholar] [CrossRef]
- Wiedmer, T.; Blank, A.; Pantasis, S.; Normand, L.; Bill, R.; Krebs, P.; Tschan, M.P.; Marinoni, I.; Perren, A. Autophagy Inhibition Improves Sunitinib Efficacy in Pancreatic Neuroendocrine Tumors via a Lysosome-dependent Mechanism. Mol. Cancer Ther. 2017, 16, 2502–2515. [Google Scholar] [CrossRef] [PubMed]
- Mendel, D.B.; Laird, A.D.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar] [PubMed]
- Norum, O.J.; Fremstedal, A.S.V.; Weyergang, A.; Golab, J.; Berg, K. Photochemical delivery of bleomycin induces T-cell activation of importance for curative effect and systemic anti-tumor immunity. J. Control. Release 2017, 268, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Fremstedal, A.S.; Skarpen, E.; Peng, Q.; Mohamedali, K.A.; Eng, M.S.; Cheung, L.H.; Rosenblum, M.G.; Waltenberger, J.; Berg, K. Light-enhanced VEGF121/rGel: A tumor targeted modality with vascular and immune-mediated efficacy. J. Control. Release 2018, 288, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef]
- Weyergang, A.; Cheung, L.H.; Rosenblum, M.G.; Mohamedali, K.A.; Peng, Q.; Waltenberger, J.; Berg, K. Photochemical internalization augments tumor vascular cytotoxicity and specificity of VEGF(121)/rGel fusion toxin. J. Control. Release 2014, 180, 1–9. [Google Scholar] [CrossRef]
- Vikdal, M.; Weyergang, A.; Selbo, P.K.; Berg, K. Vascular endothelial cells as targets for photochemical internalization (PCI). Photochem. Photobiol. 2013, 89, 1185–1192. [Google Scholar] [CrossRef]
- Lloyd, J.B. Lysosome membrane permeability: Implications for drug delivery. Adv. Drug Deliv. Rev. 2000, 41, 189–200. [Google Scholar] [CrossRef]
- Bareford, L.M.; Swaan, P.W. Endocytic mechanisms for targeted drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 748–758. [Google Scholar] [CrossRef]
- Appelqvist, H.; Waster, P.; Kagedal, K.; Ollinger, K. The lysosome: From waste bag to potential therapeutic target. J. Cell Biol. Mol. Sci. 2013, 5, 214–226. [Google Scholar] [CrossRef]
- Lilletvedt, M.; Tonnesen, H.H.; Hogset, A.; Sande, S.A.; Kristensen, S. Evaluation of physicochemical properties and aggregation of the photosensitizers TPCS2a and TPPS2a in aqueous media. Pharmazie 2011, 66, 325–333. [Google Scholar] [PubMed]
- Selbo, P.K.; Weyergang, A.; Bonsted, A.; Bown, S.G.; Berg, K. Photochemical internalization of therapeutic macromolecular agents: A novel strategy to kill multidrug-resistant cancer cells. J. Pharmacol. Exp. Ther. 2006, 319, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Weyergang, A.; Berstad, M.E.; Bull-Hansen, B.; Olsen, C.E.; Selbo, P.K.; Berg, K. Photochemical activation of drugs for the treatment of therapy-resistant cancers. Photochem. Photobiol. Sci. 2015, 14, 1465–1475. [Google Scholar] [CrossRef] [PubMed]
- Hammers, H.J.; Verheul, H.M.; Salumbides, B.; Sharma, R.; Rudek, M.; Jaspers, J.; Shah, P.; Ellis, L.; Shen, L.; Paesante, S.; et al. Reversible epithelial to mesenchymal transition and acquired resistance to sunitinib in patients with renal cell carcinoma: Evidence from a xenograft study. Mol. Cancer Ther. 2010, 9, 1525–1535. [Google Scholar] [CrossRef]
- Liang, W.; Ni, Y.; Chen, F. Tumor resistance to vascular disrupting agents: Mechanisms, imaging, and solutions. Oncotarget 2016, 7, 15444–15459. [Google Scholar] [CrossRef]
- Norum, O.J.; Gaustad, J.V.; Angell-Petersen, E.; Rofstad, E.K.; Peng, Q.; Giercksky, K.E.; Berg, K. Photochemical Internalization of Bleomycin is Superior to Photodynamic Therapy Due to the Therapeutic Effect in the Tumor Periphery. Photochem. Photobiol. 2009, 85, 740–749. [Google Scholar] [CrossRef]
- Van Hooren, L.; Georganaki, M.; Huang, H.; Mangsbo, S.M.; Dimberg, A. Sunitinib enhances the antitumor responses of agonistic CD40-antibody by reducing MDSCs and synergistically improving endothelial activation and T-cell recruitment. Oncotarget 2016, 7, 50277–50289. [Google Scholar] [CrossRef]
- Zhao, Q.; Guo, J.; Wang, G.; Chu, Y.; Hu, X. Suppression of immune regulatory cells with combined therapy of celecoxib and sunitinib in renal cell carcinoma. Oncotarget 2017, 8, 1668–1677. [Google Scholar] [CrossRef]
- Jaini, R.; Rayman, P.; Cohen, P.A.; Finke, J.H.; Tuohy, V.K. Combination of sunitinib with anti-tumor vaccination inhibits T cell priming and requires careful scheduling to achieve productive immunotherapy. Int. J. Cancer 2014, 134, 1695–1705. [Google Scholar] [CrossRef]
- Gu, Y.; Zhao, W.; Meng, F.; Qu, B.; Zhu, X.; Sun, Y.; Shu, Y.; Xu, Q. Sunitinib impairs the proliferation and function of human peripheral T cell and prevents T-cell-mediated immune response in mice. Clin. Immunol. 2010, 135, 55–62. [Google Scholar] [CrossRef]
- Franken, N.A.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nat. Protoc. 2006, 1, 2315–2319. [Google Scholar] [CrossRef] [PubMed]
- Merck KGaA. Biological Buffer Selection Guide: Buffer Reference Center. Available online: https://www.sigmaaldrich.com/life-science/core-bioreagents/biological-buffers/learning-center/buffer-reference-center.html (accessed on 28 January 2020).
- Ash, C.; Dubec, M.; Donne, K.; Bashford, T. Effect of wavelength and beam width on penetration in light-tissue interaction using computational methods. Lasers Med. Sci. 2017, 32, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Steel, G.G.; Peckham, M.J. Exploitable mechanisms in combined radiotherapy-chemotherapy: The concept of additivity. Int. J. Radiat. Oncol. Biol. Phys. 1979, 5, 85–91. [Google Scholar] [CrossRef]
- Selbo, P.K.; Kaalhus, O.; Sivam, G.; Berg, K. 5-Aminolevulinic acid-based photochemical internalization of the immunotoxin MOC31-gelonin generates synergistic cytotoxic effects in vitro. Photochem. Photobiol. 2001, 74, 303–310. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wong, J.J.W.; Berstad, M.B.; Fremstedal, A.S.V.; Berg, K.; Patzke, S.; Sørensen, V.; Peng, Q.; Selbo, P.K.; Weyergang, A. Photochemically-Induced Release of Lysosomal Sequestered Sunitinib: Obstacles for Therapeutic Efficacy. Cancers 2020, 12, 417. https://doi.org/10.3390/cancers12020417
Wong JJW, Berstad MB, Fremstedal ASV, Berg K, Patzke S, Sørensen V, Peng Q, Selbo PK, Weyergang A. Photochemically-Induced Release of Lysosomal Sequestered Sunitinib: Obstacles for Therapeutic Efficacy. Cancers. 2020; 12(2):417. https://doi.org/10.3390/cancers12020417
Chicago/Turabian StyleWong, Judith Jing Wen, Maria Brandal Berstad, Ane Sofie Viset Fremstedal, Kristian Berg, Sebastian Patzke, Vigdis Sørensen, Qian Peng, Pål Kristian Selbo, and Anette Weyergang. 2020. "Photochemically-Induced Release of Lysosomal Sequestered Sunitinib: Obstacles for Therapeutic Efficacy" Cancers 12, no. 2: 417. https://doi.org/10.3390/cancers12020417
APA StyleWong, J. J. W., Berstad, M. B., Fremstedal, A. S. V., Berg, K., Patzke, S., Sørensen, V., Peng, Q., Selbo, P. K., & Weyergang, A. (2020). Photochemically-Induced Release of Lysosomal Sequestered Sunitinib: Obstacles for Therapeutic Efficacy. Cancers, 12(2), 417. https://doi.org/10.3390/cancers12020417