PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation

Abstract
1. Introduction
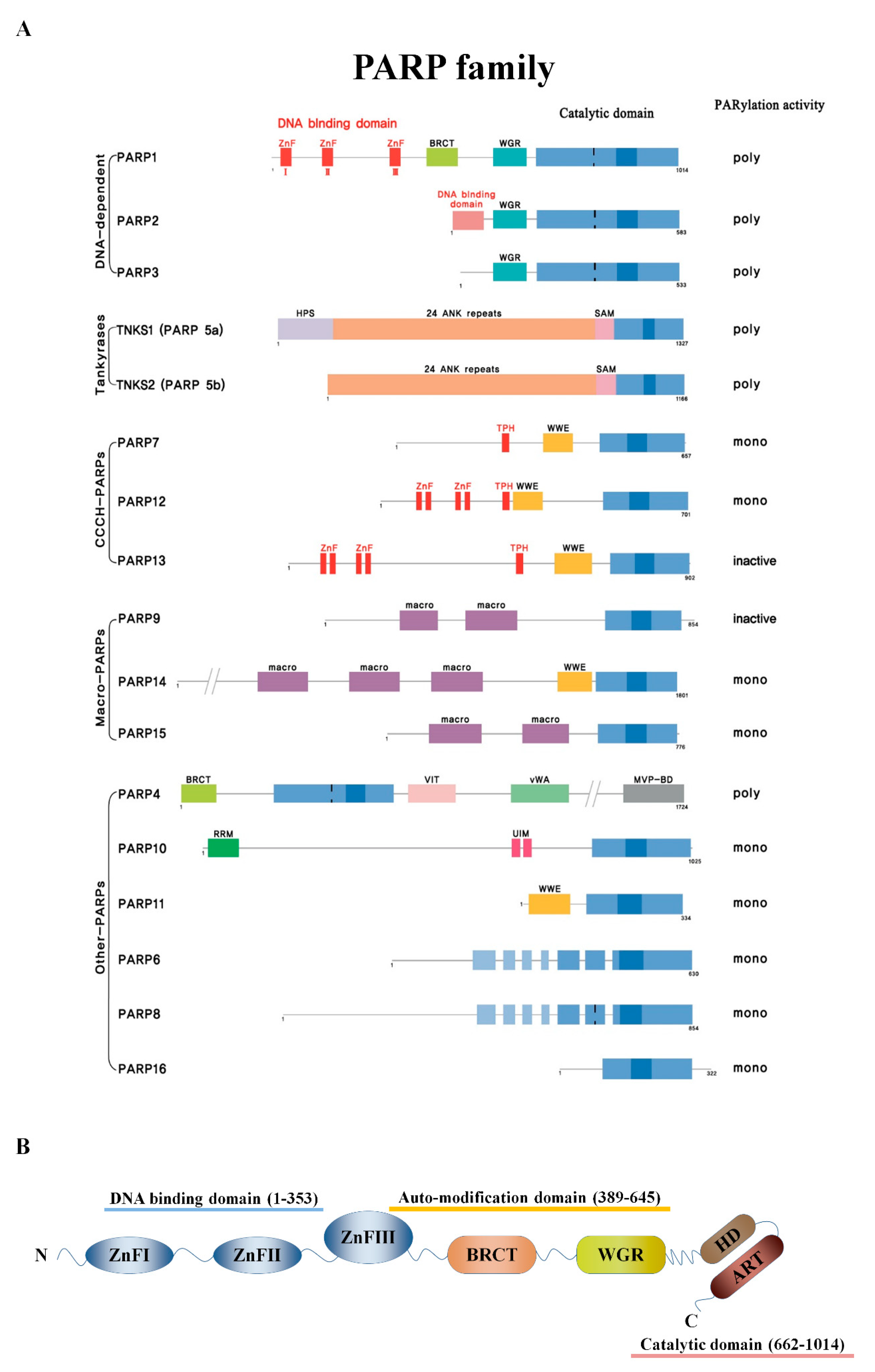
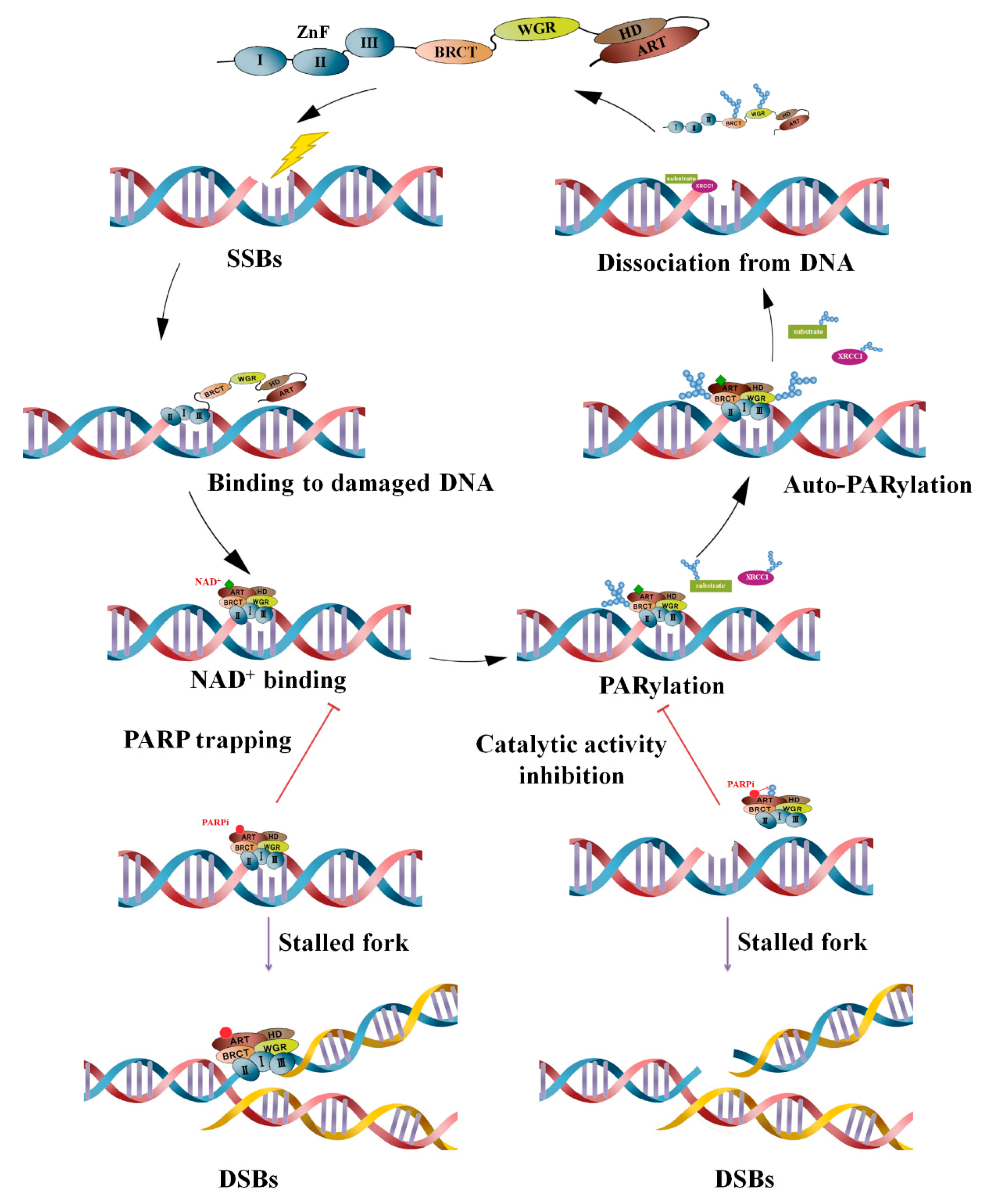
2. PARPs and PARylation
3. Clinical Development of PARP Inhibitors
4. Combination Effect of Conventional Chemotherapy according to the Mechanism of Action of PARP Inhibitors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Broustas, C.G.; Lieberman, H.B. DNA Damage Response Genes and the Development of Cancer Metastasis. Radiat. Res. 2014, 181, 111–130. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Perina, D.; Mikoc, A.; Ahel, J.; Cetkovic, H.; Žaja, R.; Ahel, I. Distribution of protein poly(ADP-ribosyl)ation systems across all domains of life. DNA Repair 2014, 23, 4–16. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Boil. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Chambon, P.; Weill, J.; Mandel, P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963, 11, 39–43. [Google Scholar] [CrossRef]
- O’Neil, N.J.; Bailey, M.L.; Hieter, P. Synthetic lethality and cancer. Nat. Rev. Genet. 2017, 18, 613–623. [Google Scholar] [CrossRef]
- Beijersbergen, R.L.; Wessels, L.F.; Bernards, R. Synthetic Lethality in Cancer Therapeutics. Annu. Rev. Cancer Boil. 2017, 1, 141–161. [Google Scholar] [CrossRef]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Tappenden, P.; Harnan, S.; Ren, S.; Thokala, P.; Wong, R.; Mukuria, C.; Green, C.; Pledge, S.; Tidy, J. Olaparib for Maintenance Treatment of BRCA 1 or 2 Mutated, Relapsed, Platinum-Sensitive Ovarian, Fallopian Tube and Peritoneal Cancer in People Whose Relapsed Disease has Responded to Platinum-Based Chemotherapy: An Evidence Review Group Perspective of a NICE Single Technology Appraisal. Pharmacoeconomics 2017, 35, 97–109. [Google Scholar]
- Pujade-Lauraine, É.; Ledermann, J.A.; Gebski, V.; Penson, R.T.; Oza, A.M.; Poveda, A.; Fujiwara, K.; Liu, J.; Lowe, E.S.; Bloomfield, R.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.; Das, B.B.; Renaud, A.; Zhang, Y.; Doroshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef]
- Murai, J.; Huang, S.Y.N.; Renaud, A.; Zhang, Y.; Ji, J.; Takeda, S.; Morris, J.; Teicher, B.; Doroshow, J.H.; Pommier, Y. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol. Cancer Ther. 2014, 13, 433–443. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; de Murcia, G. The PARP superfamily. Bioessays 2014, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Kraus, W.L. On PAR with PARP: Cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012, 26, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015, 58, 947–958. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Loeffler, P.A.; Cuneo, M.J.; Mueller, G.A.; Derose, E.F.; Gabel, S.A.; London, R.E. Structural studies of the PARP-1 BRCT domain. BMC Struct. Boil. 2011, 11, 37. [Google Scholar] [CrossRef]
- Eustermann, S.; Wu, W.-F.; Langelier, M.-F.; Yang, J.-C.; Easton, L.E.; Riccio, A.A.; Pascal, J.M.; Neuhaus, D. Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol. Cell 2015, 60, 742–754. [Google Scholar] [CrossRef]
- Langelier, M.F.; Servent, K.M.; Rogers, E.E.; Pascal, J.M. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J. Biol. Chem. 2008, 283, 4105–4114. [Google Scholar] [CrossRef]
- Langelier, M.-F.; Planck, J.L.; Roy, S.; Pascal, J.M. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K. Poly(ADP-ribose): An organizer of cellular architecture. J. Cell Boil. 2014, 205, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Schuhwerk, H.; Atteya, R.; Siniuk, K.; Wang, Z.-Q. PARPing for balance in the homeostasis of poly(ADP-ribosyl)ation. Semin. Cell Dev. Boil. 2017, 63, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Eisemann, T.; Pascal, J.M. Poly(ADP-ribose) polymerase enzymes and the maintenance of genome integrity. Cell. Mol. Life Sci. 2019, 77, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Kleine, H.; Poreba, E.; Lesniewicz, K.; Hassa, P.O.; Hottiger, M.O.; Litchfield, D.W.; Shilton, B.H.; Lüscher, B. Substrate-Assisted Catalysis by PARP10 Limits Its Activity to Mono-ADP-Ribosylation. Mol. Cell 2008, 32, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Kamaletdinova, T.; Fanaei-Kahrani, Z.; Wang, Z.-Q. The Enigmatic Function of PARP1: From PARylation Activity to PAR Readers. Cells 2019, 8, 1625. [Google Scholar] [CrossRef]
- Wei, H.; Yu, X. Functions of PARylation in DNA Damage Repair Pathways. Genom. Proteom. Bioinform. 2016, 14, 131–139. [Google Scholar] [CrossRef]
- Kim, I.K.; Stegeman, R.A.; Brosey, C.A.; Ellenberger, T. A quantitative assay reveals ligand specificity of the DNA scaffold repair protein XRCC1 and efficient disassembly of complexes of XRCC1 and the poly(ADP-ribose) polymerase 1 by poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 2015, 290, 3775–3783. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Lohani, A.; Evans, M.K.; Evans, M.K. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J. Biol. Chem. 2011, 286, 44679–44690. [Google Scholar] [CrossRef]
- Dantzer, F.; De La Rubia, G.; Murcia, J.M.-D.; Hostomsky, Z.; De Murcia, G.; Schreiber, V. Base excision repair is impaired in mammalian cells lacking Poly(ADP-ribose) polymerase-1. Biochemistry 2000, 39, 7559–7569. [Google Scholar] [CrossRef]
- Gyawali, B. The OlympiAD trial: Who won the gold? Ecancermedicalscience 2017, 11, ed75. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.-J.; Xu, R.-H.; Chin, K.; Lee, K.-W.; Park, S.H.; Rha, S.Y.; Shen, L.; Qin, S.; Xu, N.; Im, S.-A.; et al. Olaparib in combination with paclitaxel in patients with advanced gastric cancer who have progressed following first-line therapy (GOLD): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1637–1651. [Google Scholar] [CrossRef]
- Hopkins, T.A.; Shi, Y.; Rodriguez, L.E.; Solomon, L.R.; Donawho, C.K.; DiGiammarino, E.L.; Panchal, S.C.; Wilsbacher, J.L.; Gao, W.; Olson, A.M.; et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol. Cancer Res. 2015, 13, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Aoyagi-Scharber, M.; Wang, B. Trapping Poly(ADP-Ribose) Polymerase. J. Pharmacol. Exp. Ther. 2015, 353, 446–457. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; Lorusso, P.; Patel, M.R.; Domchek, S.M.; Balmaña, J.; Drew, Y.; Chen, L.-M.; et al. A Phase I–II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef]
- Coleman, R.L.; Oza, A.M.; Lorusso, M.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Hendrickson, A.E.W.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Matsumoto, K.; Tamura, K.; Yoshida, H.; Imai, Y.; Miyasaka, A.; Onoe, T.; Yamaguchi, S.; Shimizu, C.; Yonemori, K.; et al. Phase 1 dose-escalation study of single-agent veliparib in Japanese patients with advanced solid tumors. Cancer Sci. 2017, 108, 1834–1842. [Google Scholar] [CrossRef] [PubMed]
- Diéras, V.C.; Han, H.S.; Kaufman, B.; Wildiers, H.; Friedlander, M.; Ayoub, J.P.; Puhalla, S.L.; Bondarenko, I.; Campone, M.; Jakobsen, E.H.; et al. LBA9Phase III study of veliparib with carboplatin and paclitaxel in HER2-negative advanced/metastatic gBRCA-associated breast cancer. Ann. Oncol. 2019, 30, mdz394-008. [Google Scholar] [CrossRef]
- Oza, A.M.; Cibula, D.; Benzaquen, A.O.; Poole, C.; Mathijssen, R.H.J.; Sonke, G.S.; Colombo, N.; Spacek, J.; Vuylsteke, P.; Hirte, H.; et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: A randomised phase 2 trial. Lancet Oncol. 2015, 16, 87–97. [Google Scholar] [CrossRef]
- Ledermann, J.A.; Pujade-Lauraine, E. Olaparib as maintenance treatment for patients with platinum-sensitive relapsed ovarian cancer. Ther. Adv. Med Oncol. 2019, 11, 1758835919849753. [Google Scholar] [CrossRef]
- Matsumoto, K.; Nishimura, M.; Onoe, T.; Sakai, H.; Urakawa, Y.; Onda, T.; Yaegashi, N. PARP inhibitors for BRCA wild type ovarian cancer; gene alterations, homologous recombination deficiency and combination therapy. Jpn. J. Clin. Oncol. 2019, 49, 703–707. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 162, 454. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- McCann, K.E. Advances in the use of PARP inhibitors for BRCA1/2-associated breast cancer: Talazoparib. Futur. Oncol. 2019, 15, 1707–1715. [Google Scholar] [CrossRef]
- Litton, J.; Rugo, H.; Ettl, J.; Hurvitz, S.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.; Martin, M.; et al. Abstract GS6-07: EMBRACA: A phase 3 trial comparing talazoparib, an oral PARP inhibitor, to physician’s choice of therapy in patients with advanced breast cancer and a germline BRCA mutation. Gen. Sess. Abstr. 2018, 78, GS6-07. [Google Scholar]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Ben-Baruch, N.E.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Ramalingam, S.; Blais, N.; Mazieres, J.; Reck, M.; Jones, C.; Juhász, E.; Urban, L.; Orlov, S.; Barlesi, F.; Kio, E.; et al. A Randomized, Double-Blind, Phase 2 Trial of Veliparib (ABT-888) With Carboplatin and Paclitaxel in Previously Untreated Metastatic or Advanced Non-Small Cell Lung Cancer. Int. J. Radiat. Oncol. 2014, 90, S4–S5. [Google Scholar] [CrossRef]
- Strom, C.E.; Uhlen, M.; Szigyarto, C.A.-K.; Erixon, K.; Helleday, T.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Yi, M.; Dong, B.; Qin, S.; Chu, Q.; Wu, K.; Luo, S. Advances and perspectives of PARP inhibitors. Exp. Hematol. Oncol. 2019, 8, 1–12. [Google Scholar] [CrossRef]
- Rouleau, M.; Patel, A.; Hendzel, M.J.; Kaufmann, S.H.; Poirier, G.G. PARP inhibition: PARP1 and beyond. Nat. Rev. Cancer 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Murai, J.; Pommier, Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu. Rev. Cancer Boil. 2019, 3, 131–150. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, Y.; Pang, Y.; Pacak, K.; Yang, C. Double-barreled gun: Combination of PARP inhibitor with conventional chemotherapy. Pharmacol. Ther. 2018, 188, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Szabó, C. Therapeutic applications of PARP inhibitors: Anticancer therapy and beyond. Mol. Asp. Med. 2013, 34, 1217–1256. [Google Scholar] [CrossRef] [PubMed]
- Matsuno, Y.; Hyodo, M.; Fujimori, H.; Shimizu, A.; Yoshioka, K.-I. Sensitization of Cancer Cells to Radiation and Topoisomerase I Inhibitor Camptothecin Using Inhibitors of PARP and Other Signaling Molecules. Cancers 2018, 10, 364. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.A.; Lindeman, G.J.; Clemons, M.; Wildiers, H.; Chan, A.; McCarthy, N.J.; Singer, C.F.; Lowe, E.S.; Watkins, C.L.; Carmichael, J. Phase I trial of the oral PARP inhibitor olaparib in combination with paclitaxel for first- or second-line treatment of patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2013, 15, R88. [Google Scholar] [CrossRef]
- Ihnen, M.; Zu Eulenburg, C.; Kolarova, T.; Qi, J.W.; Manivong, K.; Chalukya, M.; Dering, J.; Anderson, L.; Ginther, C.; Meuter, A.; et al. Therapeutic potential of the poly(ADP-ribose) polymerase inhibitor rucaparib for the treatment of sporadic human ovarian cancer. Mol. Cancer Ther. 2013, 12, 1002–1015. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Monk, B.J. PARP inhibitor and chemotherapy combination trials for the treatment of advanced malignancies: Does a development pathway forward exist? Ann. Oncol. 2017, 28, 443–447. [Google Scholar] [CrossRef]
- Samol, J.; Ranson, M.; Scott, E.; Macpherson, E.; Carmichael, J.; Thomas, A.; Cassidy, J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Investig. New Drugs 2012, 30, 1493–1500. [Google Scholar] [CrossRef]
- Kunos, C.; Deng, W.; Dawson, D.; Lea, J.S.; Zanotti, K.M.; Gray, H.J.; Bender, D.P.; Guaglianone, P.P.; Carter, J.S.; Moore, K.N. A phase I-II evaluation of veliparib (NSC #737664), topotecan, and filgrastim or pegfilgrastim in the treatment of persistent or recurrent carcinoma of the uterine cervix: An NRG Oncology/Gynecologic Oncology Group study. Int. J. Gynecol. Cancer 2015, 25, 484–492. [Google Scholar]
- Lee, J.-M.; Peer, C.J.; Yu, M.; Amable, L.; Gordon, N.; Annunziata, C.M.; Houston, N.; Goey, A.K.L.; Sissung, T.M.; Parker, B.; et al. Sequence-Specific Pharmacokinetic and Pharmacodynamic Phase I/Ib Study of Olaparib Tablets and Carboplatin in Women’s Cancer. Clin. Cancer Res. 2017, 23, 1397. [Google Scholar] [CrossRef]
- Wesolowski, R.; Zhao, M.; Geyer, S.M.; Lustberg, M.B.; Mrozek, E.; Layman, R.M.; Macrae, E.M.; Zhang, J.; Hall, N.; Schregel, K.; et al. Phase I trial of the PARP inhibitor veliparib (V) in combination with carboplatin (C) in metastatic breast cancer (MBC). J. Clin. Oncol. 2014, 32, 1074. [Google Scholar] [CrossRef]
- Wilson, R.H.; Evans, T.J.; Middleton, M.R.; Molife, L.R.; Spicer, J.; Diéras, V.; Roxburgh, P.; Giordano, H.; Jaw-Tsai, S.; Goble, S.; et al. A phase I study of intravenous and oral rucaparib in combination with chemotherapy in patients with advanced solid tumours. Br. J. Cancer 2017, 116, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Halford, S.E.R.; Cruickshank, G.; Dunn, L.; Erridge, S.; Godfrey, L.; Herbert, C.; Jefferies, S.; Lopez, J.S.; McBain, C.; Pittman, M.; et al. Results of the OPARATIC trial: A phase I dose escalation study of olaparib in combination with temozolomide (TMZ) in patients with relapsed glioblastoma (GBM). J. Clin. Oncol. 2017, 35, 2022. [Google Scholar] [CrossRef]
- Isakoff, S.J.; Puhalla, S.; Domchek, S.M.; Friedlander, M.; Kaufman, B.; Robson, M.; Telli, M.L.; Diéras, V.; Han, H.S.; Garber, J.E.; et al. A randomized Phase II study of veliparib with temozolomide or carboplatin/paclitaxel versus placebo with carboplatin/paclitaxel in BRCA1/2 metastatic breast cancer: Design and rationale. Futur. Oncol. 2017, 13, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Villalona-Calero, M.A.; Duan, W.; Zhao, W.; Shilo, K.; Schaaf, L.J.; Thurmond, J.; Westman, J.A.; Marshall, J.; Xiaobai, L.; Ji, J.; et al. Veliparib Alone or in Combination with Mitomycin C in Patients with Solid Tumors With Functional Deficiency in Homologous Recombination Repair. J. Natl. Cancer Inst. 2016, 108, djv437. [Google Scholar] [CrossRef]
- Bendell, J.; O’Reilly, E.M.; Middleton, M.R.; Chau, I.; Hochster, H.; Fielding, A.; Burke, W.; Burris, H. Phase I study of olaparib plus gemcitabine in patients with advanced solid tumours and comparison with gemcitabine alone in patients with locally advanced/metastatic pancreatic cancer. Ann. Oncol. 2015, 26, 804–811. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Im, S.-A.; Lee, K.-W.; Cho, J.Y.; Song, E.-K.; Lee, K.H.; Kim, Y.H.; Park, J.O.; Chun, H.G.; Zang, D.Y.; et al. Randomized, Double-Blind Phase II Trial With Prospective Classification by ATM Protein Level to Evaluate the Efficacy and Tolerability of Olaparib Plus Paclitaxel in Patients With Recurrent or Metastatic Gastric Cancer. J. Clin. Oncol. 2015, 33, 3858–3865. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; De Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362. [Google Scholar] [CrossRef]
- Murai, J.; Zhang, Y.; Morris, J.; Ji, J.; Takeda, S.; Doroshow, J.H.; Pommier, Y. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 2014, 349, 408–416. [Google Scholar] [CrossRef]
- Patel, A.G.; Schneider, P.A.; Dai, N.T.; McDonald, J.S.; Poirier, G.G.; Kaufmann, S.H. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J. Biol. Chem. 2012, 287, 4198–4210. [Google Scholar] [CrossRef]
- Murai, J.; Pommier, Y. Classification of PARP Inhibitors Based on PARP Trapping and Catalytic Inhibition, and Rationale for Combinations with Topoisomerase I Inhibitors and Alkylating Agents. In PARP Inhibitors for Cancer Therapy; Curtin, N.J., Sharma, R.A., Eds.; Springer International Publishing: Cham, Switzerland, 2015; Volume 83, pp. 261–274. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
| Agents | Company | Target | Application in Clinics | Mean Half-Life (Hours) | Catalytic Inhibition (IC50 in Wild-Type DT40 Cells; nM) [13] | PARP Trapping Potency (Relative to Olaparib) [33] | Cytotoxicity (EC50 in BRCA2 Mutated Capan-1 Cells; nM) [34] | ||
|---|---|---|---|---|---|---|---|---|---|
| Indication | Clinical Trials Based on FDA Approval | Dosage | |||||||
| Olaparib | AstraZeneca | PARP1 PARP2 PARP3 | Maintenance treatment of germline BRCA-mutated advanced ovarian cancer. | Study 42 (NCT01078662) [35] | 300mg BID | 14.9 ± 8.2 | 6 | 1 | 259 |
| Maintenance treatment of recurrent serous ovarian cancer regardless of BRCA mutations | SOLO-2 (NCT01874353) [11] Study 19 (NCT00753545) [36] | ||||||||
| Treatment of germline BRCA-mutated HER2-negative locally advanced or metastatic breast cancers | OlympiAD (NCT02000622) [9] | ||||||||
| First-line maintenance treatment of germline BRCA-mutated metastatic pancreatic cancer | POLO trial (NCT02184195) [37] | ||||||||
| Rucaparib | Clovis Oncology | PARP1 PARP2 PARP3 | Treatment of germline and/or somatic BRCA-mutated advanced ovarian cancer | ARIEL2 (NCT01891344) [38] Study 10 (NCT01482715) [39] | 600mg BID | 18 ± 1 | 21 | 1 | 609 |
| Maintenance treatment in a platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer | ARIEL3 (NCT01968213) [40] | ||||||||
| Niraparib (MK4827) | Tesaro | PARP1 PARP2 | Maintenance treatment of platinum-sensitive, recurrent ovarian cancer | ENGOT-OV16/NOVA (NCT01847274) [41] | 300mg QD | 36 | 60 | 2 | 650 |
| Treatment of homologous recombination deficiency (HRD) positive advanced ovarian, fallopian tube, or primary peritoneal cancer | QUADRA (NCT02354586) [42] | ||||||||
| Talazoparib (BMN-673) | Pfizer | PARP1 PARP2 | Treatment of germline BRCA-mutated HER2-negative locally advanced or metastatic breast cancers | EMBRACA (NCT01945775) [43] | 1mg QD | 90 | 4 | 100 | 5 |
| Veliparib (ABT-888) | Abbott Laboratories | PARP1 PARP2 | Not yet approved for any indication, but in 2014. FDA awards orphan drug designation to Veliparib for advanced non-small cell lung cancer (NSCLC). In phase III trial, veliparib significantly improved progression-free survival in BRCA-mutated or HRD cohort compared to carboplatin plus paclitaxel (NCT02470585) [44]. In phase III trial of veliparib with carboplatin and paclitaxel in advanced HER2-negative breast cancer with germline BRCA mutation, median PFS in patients treated with veliparib plus carboplatin and paclitaxel was 14.5 months compared to 12.6 months in placebo plus carboplatin and paclitaxel (BROCADE3; NCT02163694) [45]. | 5.2 | 30 | <0.2 | >10,000 | ||
| Action Mechanism of PARP Inhibitors on the Combination Effects | Combined Chemotherapeutics | PARP Inhibitor | Tumor Type | Trial | Phase | Outcome | |
|---|---|---|---|---|---|---|---|
| Class of Agents | Chemotherapy Agents | ||||||
| Inhibition of PARP catalytic activity | Topoisomerase I inhibitors | Topotecan | Olaparib | Advanced solid tumors | NCT00516438 [67] | I | The Maximum Tolerated Dose (MTD) was determined as topotecan 1.0 mg/m2/day × 3 days plus olaparib 100 mg bid. |
| Veliparib | Recurrent cervix cancer | NCT01266447 [68] | II | Topotecan 0.6 mg/m2/day × 5 days plus veliparib 10 mg bid treatment resulted in 7% partial response (PR). | |||
| Platinum-based inhibitors | Carboplatin | Olaparib | Refractory or recurrent breast and ovarian cancer | NCT01237067 [69] | I | The MTD as olaparib 200 mg bid plus carboplatin AUC4; The responses including CRs and PRs was higher in BRCA mutation carriers compared with nonmutation carriers (68% vs 19%) | |
| Veliparib | HER2-negative metastatic breast cancer | NCT01251874 [70] | I | The MTD was established as veliparib 250 mg bid plus carboplatin AUC5. | |||
| Rucaparib | Advanced solid tumors | NCT01009190 [71] | I | The MTD for combination was established as 240 mg/day oral rucaparib and carboplatin AUC5. | |||
| PARP trapping | Alkylating agents | Temozolomide | Olaparib | Relapsed glioblastoma | NCT01390571 [72] | I | The temozolomide 75 mg/m2 daily plus olaparib 150 mg/day × 21 days treatment was well tolerated and encouraged 6 months progression-free survival rates. |
| Veliparib | Metastatic breast cancer and BRCA1/2 mutated breast cancer | NCT01506609 [73] | II | The responses in the BRCA mutation carriers showed a total response rate (RR) 25% (7/28) and clinical benefit rate (CBR) 50%. | |||
| Mitomycin C | Veliparib | Metastatic, unresectable or recurrent solid tumors | NCT01017640 [74] | I | Veliparib 200 mg bid after treated mitomycin C 10 mg/m2/day × 21 days was recommended for combination treatment. | ||
| Inhibition of PARP transcription cofactor function | Antimetabolite | Gemcitabine | Olaparib | Pancreatic cancer | NCT00515866 [75] | I | Olaparib 100 mg bid plus gemcitabine 600 mg/m2/week was tolerated and recommended for the phase II trial. |
| Unknown mechanism | taxanes | paclitaxel | Olaparib | Metastatic triple-negative breast cancer (TNBC) | NCT00707707 [64] | I | Olaparib 200 mg bid daily in combination with paclitaxel 90 mg/m2/week × 3 of 4 weeks was tolerated; The overall response rate (ORR) was 33.3%. |
| Advanced gastric cancer | NCT01063517 [76] | II | Olaparib 100 mg bid plus paclitaxel 80 mg/m2 treatment showed overall survival and progression-free survival benefit in ATM enriched phase II study. | ||||
| NCT01924533 [32] | III | In phase III study, median overall survival was 8.8 months in the weekly paclitaxel 80 mg/m2 + olaparib 100 mg bid vs 6.9 months in the paclitaxel + placebo group; HR 0.79, p = 0.026, without statistical significance. | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Min, A.; Im, S.-A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers 2020, 12, 394. https://doi.org/10.3390/cancers12020394
Min A, Im S-A. PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers. 2020; 12(2):394. https://doi.org/10.3390/cancers12020394
Chicago/Turabian StyleMin, Ahrum, and Seock-Ah Im. 2020. "PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation" Cancers 12, no. 2: 394. https://doi.org/10.3390/cancers12020394
APA StyleMin, A., & Im, S.-A. (2020). PARP Inhibitors as Therapeutics: Beyond Modulation of PARylation. Cancers, 12(2), 394. https://doi.org/10.3390/cancers12020394