Establishment of a Temperature-Sensitive Model of Oncogene-Induced Senescence in Angiosarcoma Cells

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods
2.1. Cells
2.2. Gene Microarray (GC)
2.3. Quantitative Reverse Transcription Polymerase Chain Reaction (RT-qPCR)
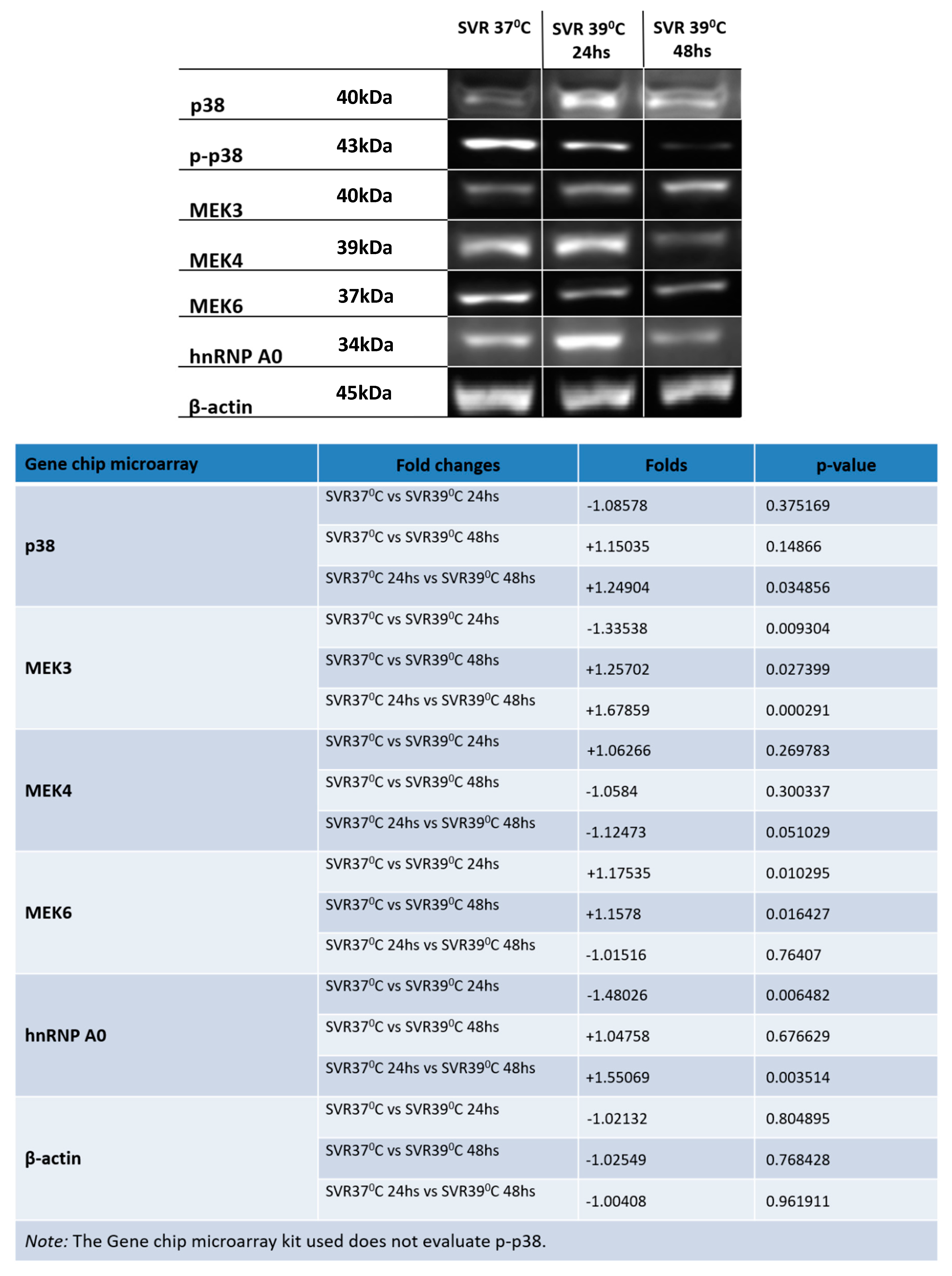
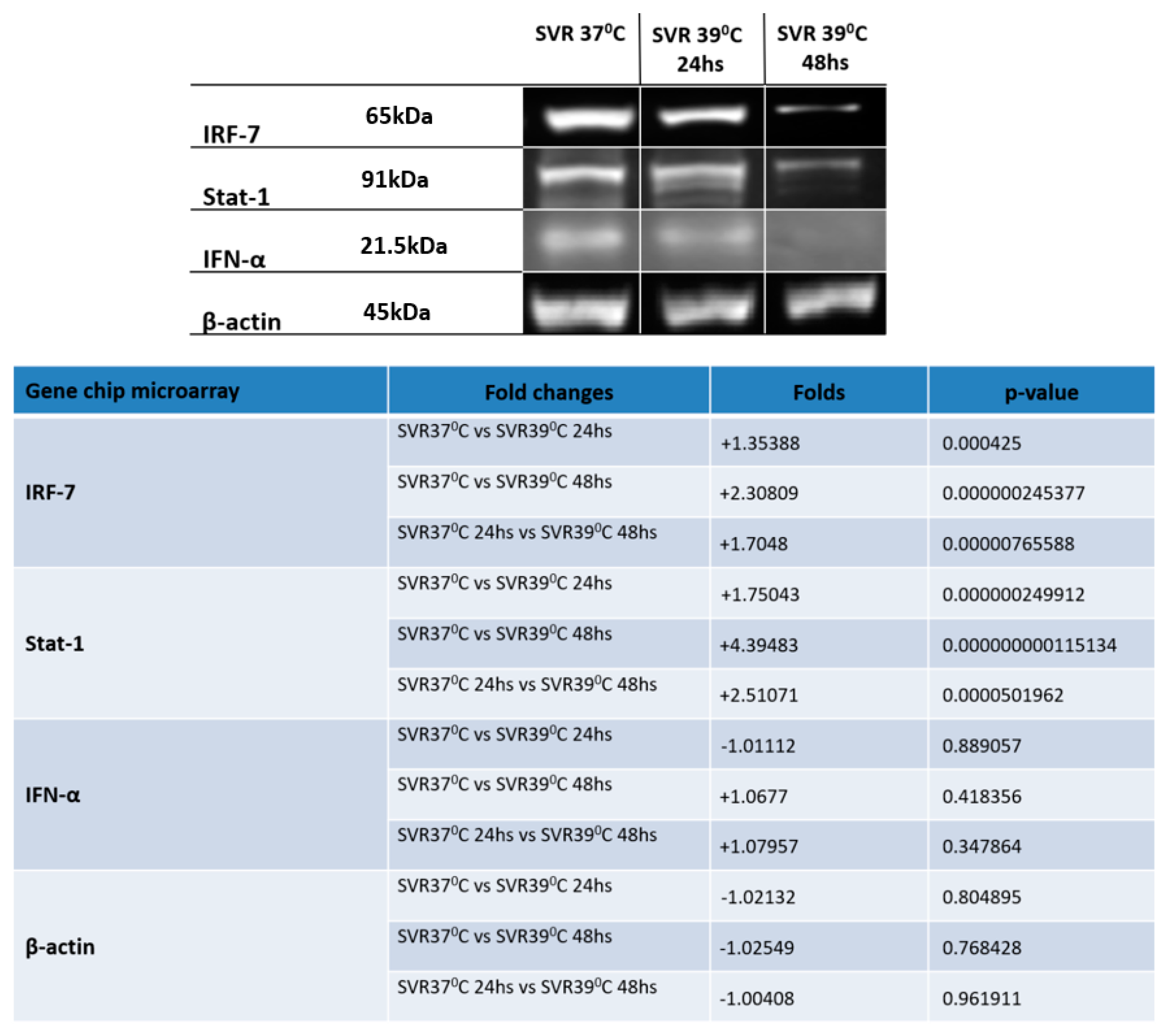
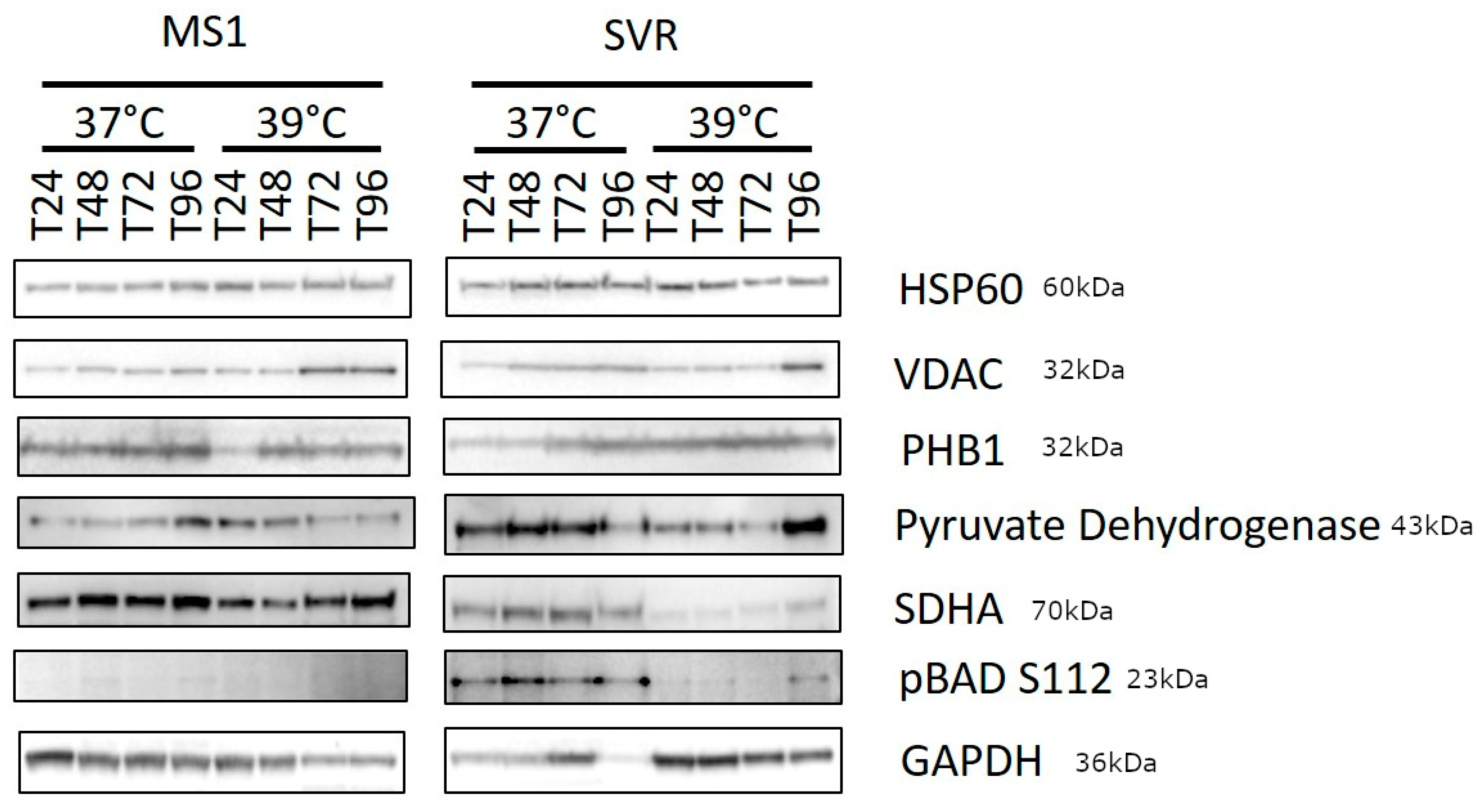
2.4. Western Blot (WB)
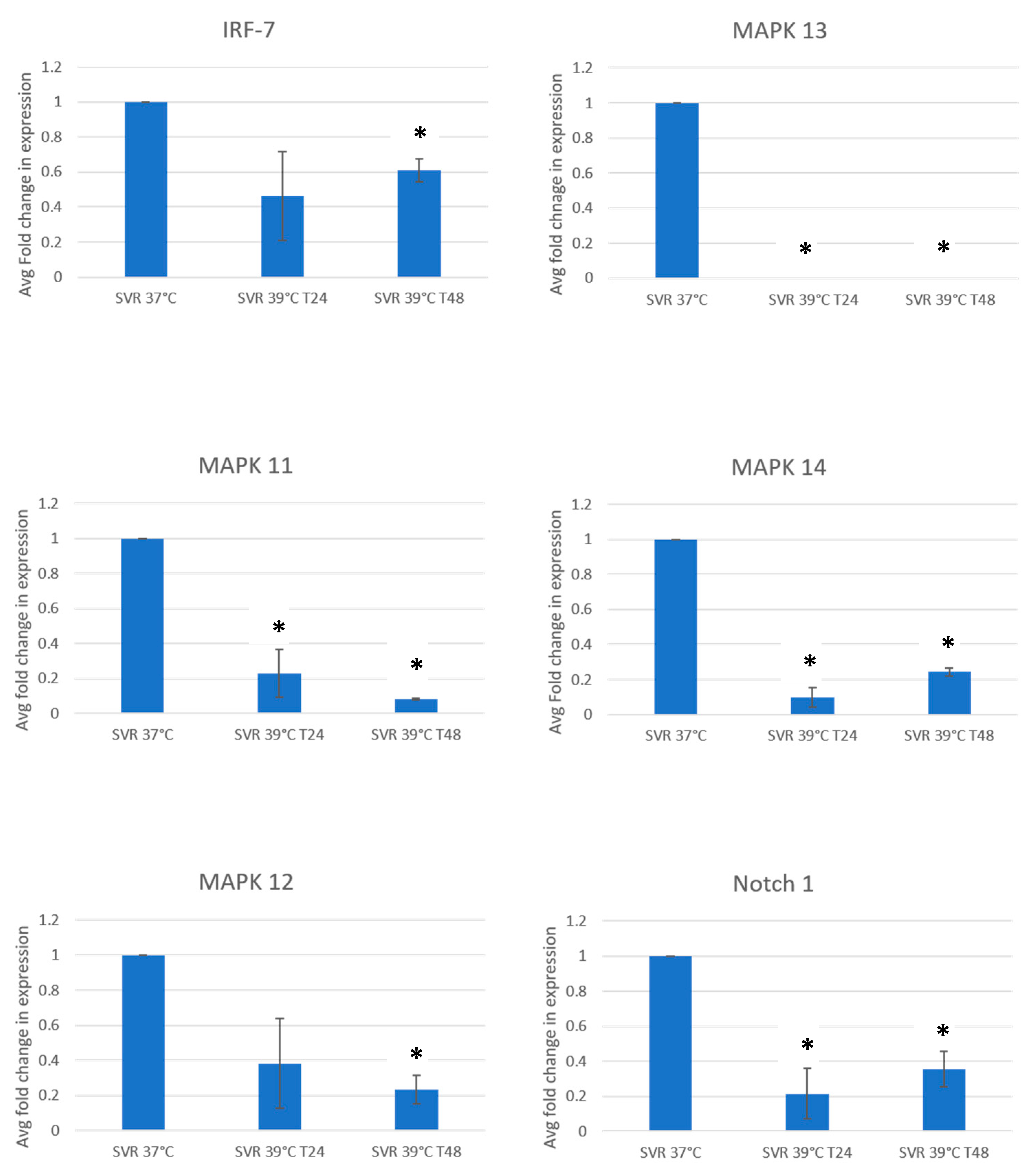
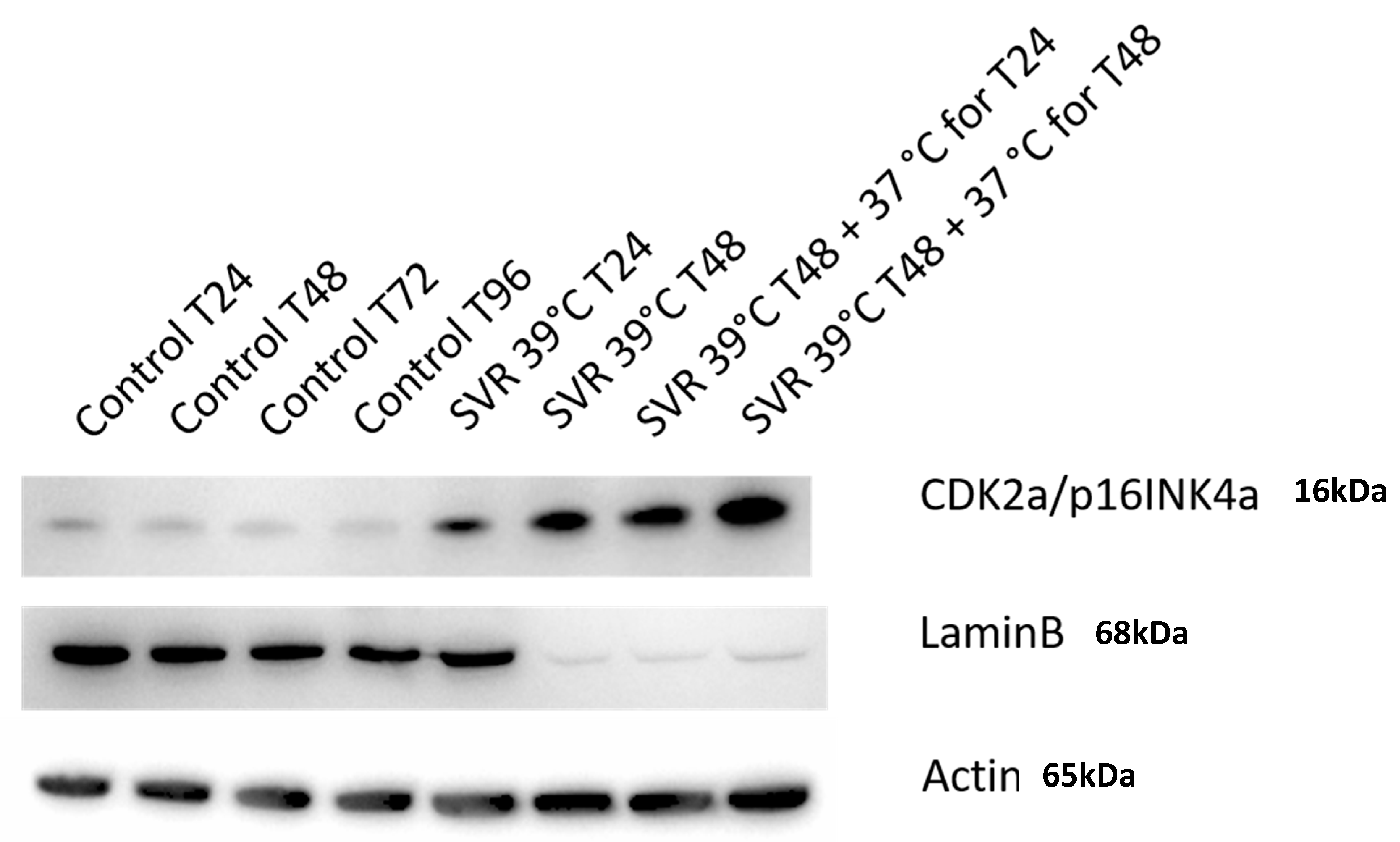
3. Results
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Barry, E.L.; Baron, J.A.; Grau, M.V.; Wallace, K.; Haile, R.W. K-ras mutations in incident sporadic colorectal adenomas. Cancer 2006, 106, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Spring, K.J. High prevalence of sessile serrated adenomas with BRAF mutations: A prospective study of patients undergoing colonoscopy. Gastroenterology 2006, 131, 1400–1407. [Google Scholar] [CrossRef]
- Thomas, N.E. BRAF somatic mutations in malignant melanoma and melanocytic naevi. Melanoma Res. 2006, 16, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Logie, A. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum. Mol. Genet. 2005, 14, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C. Multiple oncogenic mutations and clonal relationship in spatially distinct benign human epidermal tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 20780–20785. [Google Scholar] [CrossRef] [PubMed]
- Hafner, C. Oncogenic PIK3CA mutations occur in epidermal nevi and seborrheic keratoses with a characteristic mutation pattern. Proc. Natl. Acad. Sci. USA 2007, 104, 13450–13454. [Google Scholar] [CrossRef]
- Nakashima, M. The somatic GNAQ mutation c.548G > A (p.R183Q) is consistently found in Sturge-Weber syndrome. J. Hum. Genet. 2014, 59, 691–693. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Campisi, J. Senescent cells, tumor suppression, and organismal aging: Good citizens, bad neighbors. Cell 2005, 120, 513–522. [Google Scholar] [CrossRef]
- Campisi, J. Suppressing cancer: The importance of being senescent. Science 2005, 309, 886–887. [Google Scholar] [CrossRef]
- da Costa, A.; Bonner, M.; Arbiser, J.L. Comprehensive profiling of H-Ras signalling in angiosarcoma endothelium. Clin. Exp. Dermatol. 2017, 42, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Bayley, J.P.; Devilee, P.; Taschner, P.E. The SDH mutation database: An online resource for succinate dehydrogenase sequence variants involved in pheochromocytoma, paraganglioma and mitochondrial complex II deficiency. BMC Med. Genet. 2005, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef] [PubMed]
- Burnichon, N. SDHA is a tumor suppressor gene causing paraganglioma. Hum. Mol. Genet. 2010, 19, 3011–3020. [Google Scholar] [CrossRef]
- Tucker, M.A. A natural history of melanomas and dysplastic nevi: An atlas of lesions in melanoma-prone families. Cancer 2002, 94, 3192–3209. [Google Scholar] [CrossRef]
- Taube, J.M.; Begum, S.; Shi, C.; Eshleman, J.R.; Westra, W.H. Benign nodal nevi frequently harbor the activating V600E BRAF mutation. Am. J. Surg. Pathol. 2009, 33, 568–571. [Google Scholar] [CrossRef]
- Chesi, M. Frequent translocation t (4; 14) (p16.3; q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat. Genet. 1997, 16, 260–264. [Google Scholar] [CrossRef]
- Shayesteh, L. PIK3CA is implicated as an oncogene in ovarian cancer. Nat. Genet. 1999, 21, 99–102. [Google Scholar] [CrossRef]
- Meadows, K.N.; Bryant, P.; Vincent, P.A.; Pumiglia, K.M. Activated Ras induces a proangiogenic phenotype in primary endothelial cells. Oncogene 2004, 23, 192–200. [Google Scholar] [CrossRef]
- Stravopodis, D.J. Grade-dependent effects on cell cycle progression and apoptosis in response to doxorubicin in human bladder cancer cell lines. Int. J. Oncol. 2009, 34, 137–160. [Google Scholar]
- Tan, W. Sustained activation of c-Jun N-terminal and extracellular signal-regulated kinases in port-wine stain blood vessels. J. Am. Acad. Dermatol. 2014, 71, 964–968. [Google Scholar] [CrossRef] [PubMed]
- Sato, M. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 2000, 13, 539–548. [Google Scholar] [CrossRef]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q. ORF45 of Kaposi’s sarcoma-associated herpesvirus inhibits phosphorylation of interferon regulatory factor 7 by IKKepsilon and TBK1 as an alternative substrate. J. Virol. 2012, 86, 10162–10172. [Google Scholar] [CrossRef] [PubMed]
- Joo, C.H. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol. 2007, 81, 8282–8292. [Google Scholar] [CrossRef] [PubMed]
- Bidwell, B.N. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med. 2012, 18, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Li, Y. microRNA-762 promotes breast cancer cell proliferation and invasion by targeting IRF7 expression. Cell Prolif. 2015, 48, 643–649. [Google Scholar] [CrossRef]
- Li, Q.; Tainsky, M.A. Epigenetic silencing of IRF7 and/or IRF5 in lung cancer cells leads to increased sensitivity to oncolytic viruses. PLoS ONE 2011, 6, e28683. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kukkonen, S.; Martinez-Viedma Mdel, P.; Gupta, S.; Aldovini, A. Tat engagement of p38 MAP kinase and IRF7 pathways leads to activation of interferon-stimulated genes in antigen-presenting cells. Blood 2013, 121, 4090–4100. [Google Scholar] [CrossRef]
- Berezovska, O. Notch1 and amyloid precursor protein are competitive substrates for presenilin1-dependent gamma-secretase cleavage. J. Biol. Chem. 2001, 276, 30018–30023. [Google Scholar] [CrossRef]
- Berezovska, O. Rapid Notch1 nuclear translocation after ligand binding depends on presenilin-associated gamma-secretase activity. Ann. N. Y. Acad. Sci. 2000, 920, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Atorvastatin promotes presenilin-1 expression and Notch1 activity and increases neural progenitor cell proliferation after stroke. Stroke 2008, 39, 220–226. [Google Scholar] [CrossRef]
- Lleo, A. Notch1 competes with the amyloid precursor protein for gamma-secretase and down-regulates presenilin-1 gene expression. J. Biol. Chem. 2003, 278, 47370–47375. [Google Scholar] [CrossRef] [PubMed]
- Godin, C. Presenilin-1 is indirectly implicated in Notch1 cleavage. Neuroreport 2003, 14, 1613–1616. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y. Notch signaling regulates the lifespan of vascular endothelial cells via a p16-dependent pathway. PLoS ONE 2014, 9, e100359. [Google Scholar] [CrossRef] [PubMed]
- Howard, J.D. Notch signaling mediates melanoma-endothelial cell communication and melanoma cell migration. Pigment Cell Melanoma Res. 2013, 26, 697–707. [Google Scholar] [CrossRef]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef]
- Kim, W.Y.; Sharpless, N.E. The regulation of INK4/ARF in cancer and aging. Cell 2006, 127, 265–275. [Google Scholar] [CrossRef]
- Sharpless, N.E. INK4a/ARF: A multifunctional tumor suppressor locus. Mutat. Res. 2005, 576, 22–38. [Google Scholar] [CrossRef]
- Collado, M.; Blasco, M.A.; Serrano, M. Cellular senescence in cancer and aging. Cell 2007, 130, 223–233. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Velichko, A.K.; Petrova, N.V.; Razin, S.V.; Kantidze, O.L. Mechanism of heat stress-induced cellular senescence elucidates the exclusive vulnerability of early S-phase cells to mild genotoxic stress. Nucleic Acids Res. 2015, 43, 6309–6320. [Google Scholar] [CrossRef]
- Wang, A.S.; Ong, P.F.; Chojnowski, A.; Clavel, C.; Dreesen, O. Loss of lamin B1 is a biomarker to quantify cellular senescence in photoaged skin. Sci. Rep. 2017, 7, 15678. [Google Scholar] [CrossRef]
- Cotter, M.A.; Florell, S.R.; Leachman, S.A.; Grossman, D. Absence of Senescence-Associated β-Galactosidase Activity in Human Melanocytic Nevi In Vivo. J. Investig. Dermatol. 2007, 127, 2469–2471. [Google Scholar] [CrossRef]
- Ben-Hail, D. Novel Compounds Targeting the Mitochondrial Protein VDAC1 Inhibit Apoptosis and Protect against Mitochondrial Dysfunction. J. Biol. Chem. 2016, 291, 24986–25003. [Google Scholar] [CrossRef]
- Zaid, H.; Abu-Hamad, S.; Israelson, A.; Nathan, I.; Shoshan-Barmatz, V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005, 12, 751–760. [Google Scholar] [CrossRef]
- Stoklosa, T. BCR/ABL recruits p53 tumor suppressor protein to induce drug resistance. Cell Cycle 2004, 3, 1463–1472. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
da Costa, A.; Bonner, M.Y.; Rao, S.; Gilbert, L.; Sasaki, M.; Elsey, J.; MacKelfresh, J.; Arbiser, J.L. Establishment of a Temperature-Sensitive Model of Oncogene-Induced Senescence in Angiosarcoma Cells. Cancers 2020, 12, 395. https://doi.org/10.3390/cancers12020395
da Costa A, Bonner MY, Rao S, Gilbert L, Sasaki M, Elsey J, MacKelfresh J, Arbiser JL. Establishment of a Temperature-Sensitive Model of Oncogene-Induced Senescence in Angiosarcoma Cells. Cancers. 2020; 12(2):395. https://doi.org/10.3390/cancers12020395
Chicago/Turabian Styleda Costa, Adilson, Michael Y. Bonner, Shikha Rao, Linda Gilbert, Maiko Sasaki, Justin Elsey, Jamie MacKelfresh, and Jack L. Arbiser. 2020. "Establishment of a Temperature-Sensitive Model of Oncogene-Induced Senescence in Angiosarcoma Cells" Cancers 12, no. 2: 395. https://doi.org/10.3390/cancers12020395
APA Styleda Costa, A., Bonner, M. Y., Rao, S., Gilbert, L., Sasaki, M., Elsey, J., MacKelfresh, J., & Arbiser, J. L. (2020). Establishment of a Temperature-Sensitive Model of Oncogene-Induced Senescence in Angiosarcoma Cells. Cancers, 12(2), 395. https://doi.org/10.3390/cancers12020395