Plasma Next Generation Sequencing and Droplet Digital-qPCR-Based Quantification of Circulating Cell-Free RNA for Noninvasive Early Detection of Cancer

 , , ,
, , ,  ,
,  , ,
, ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Patients and Baseline Characteristics
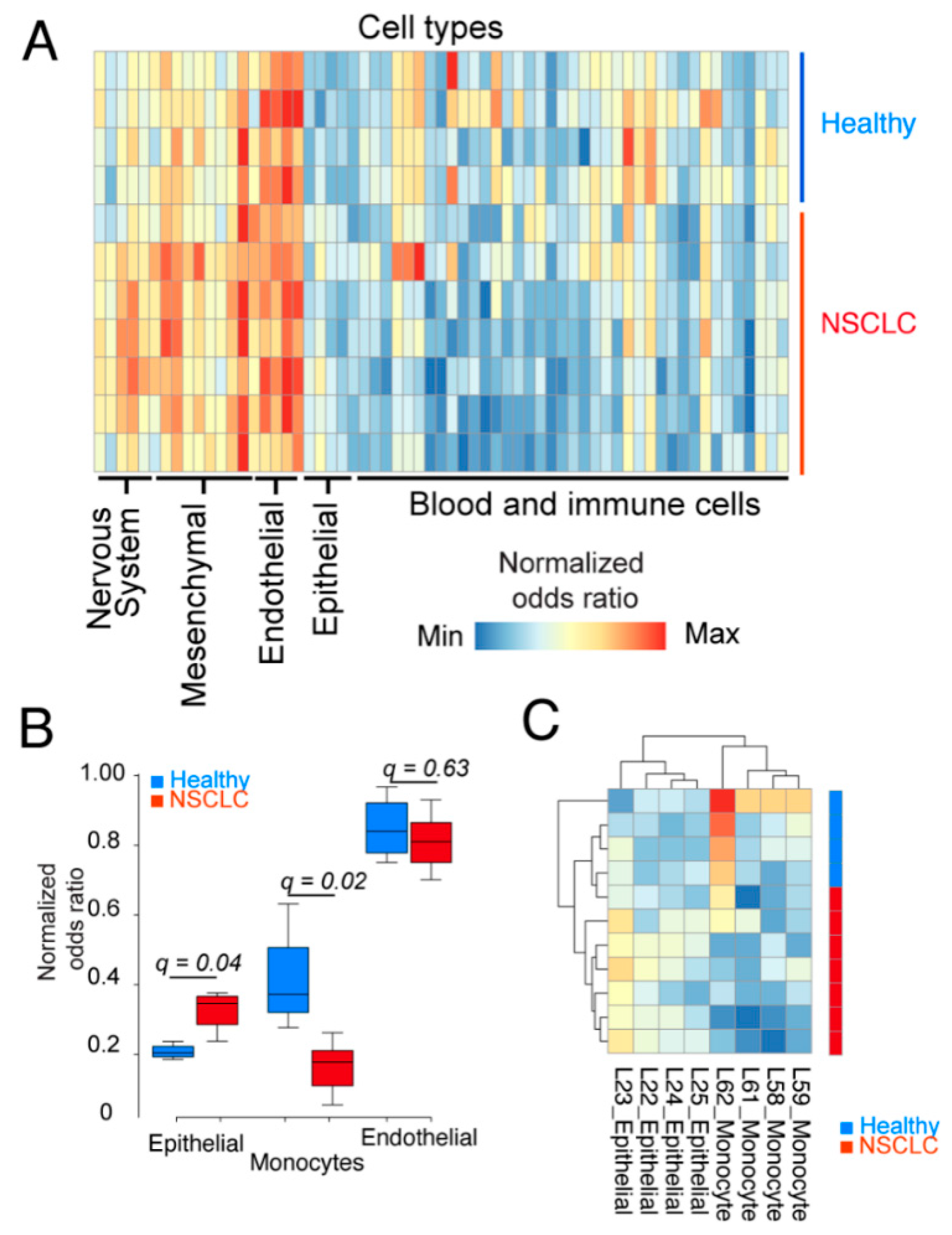
2.2. Epithelial Cells and Monocytes Differentiate Patient and Healthy cfRNA
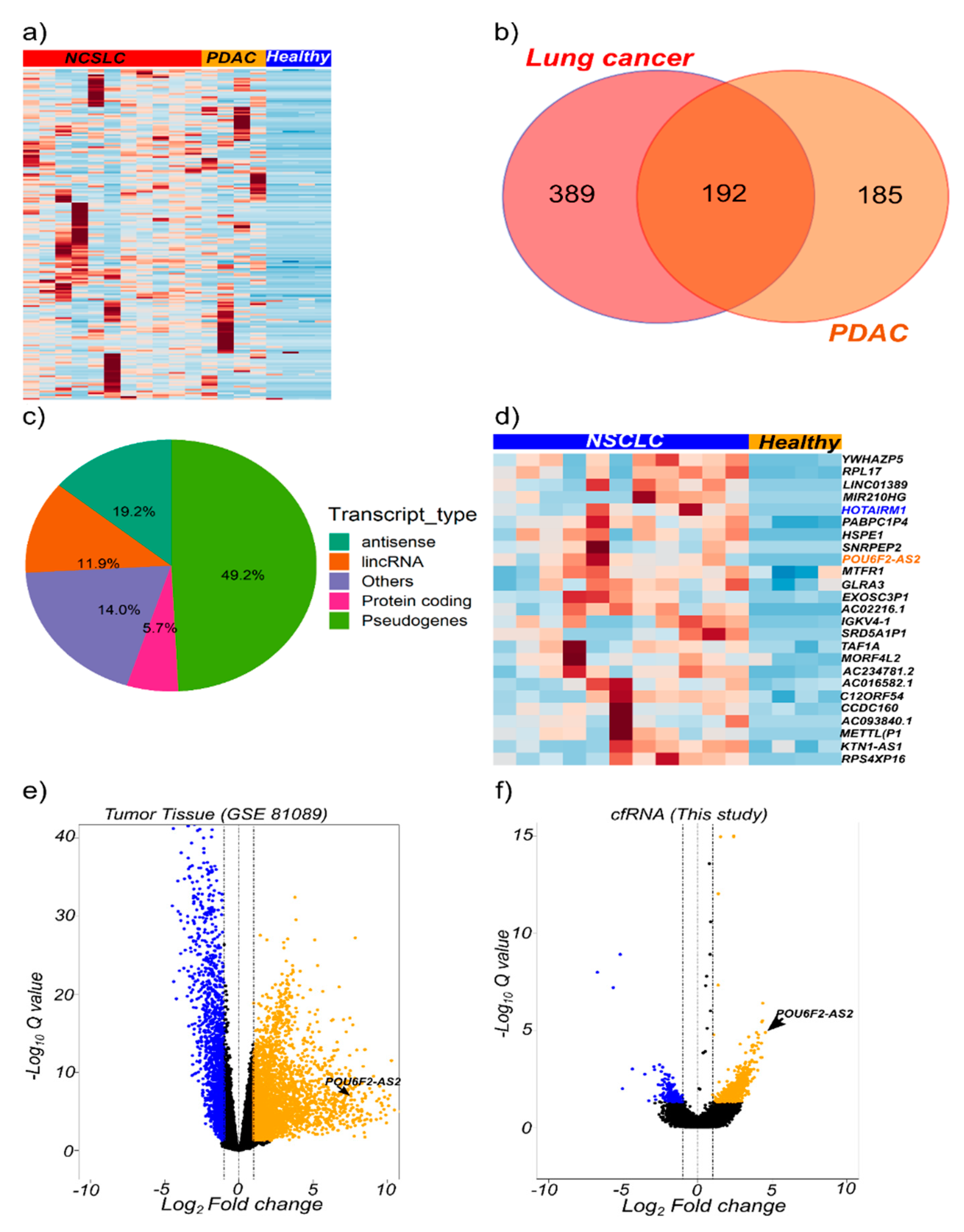
2.3. cfRNA Sequencing Identifies Common Transcripts Upregulated in Cancer Patients
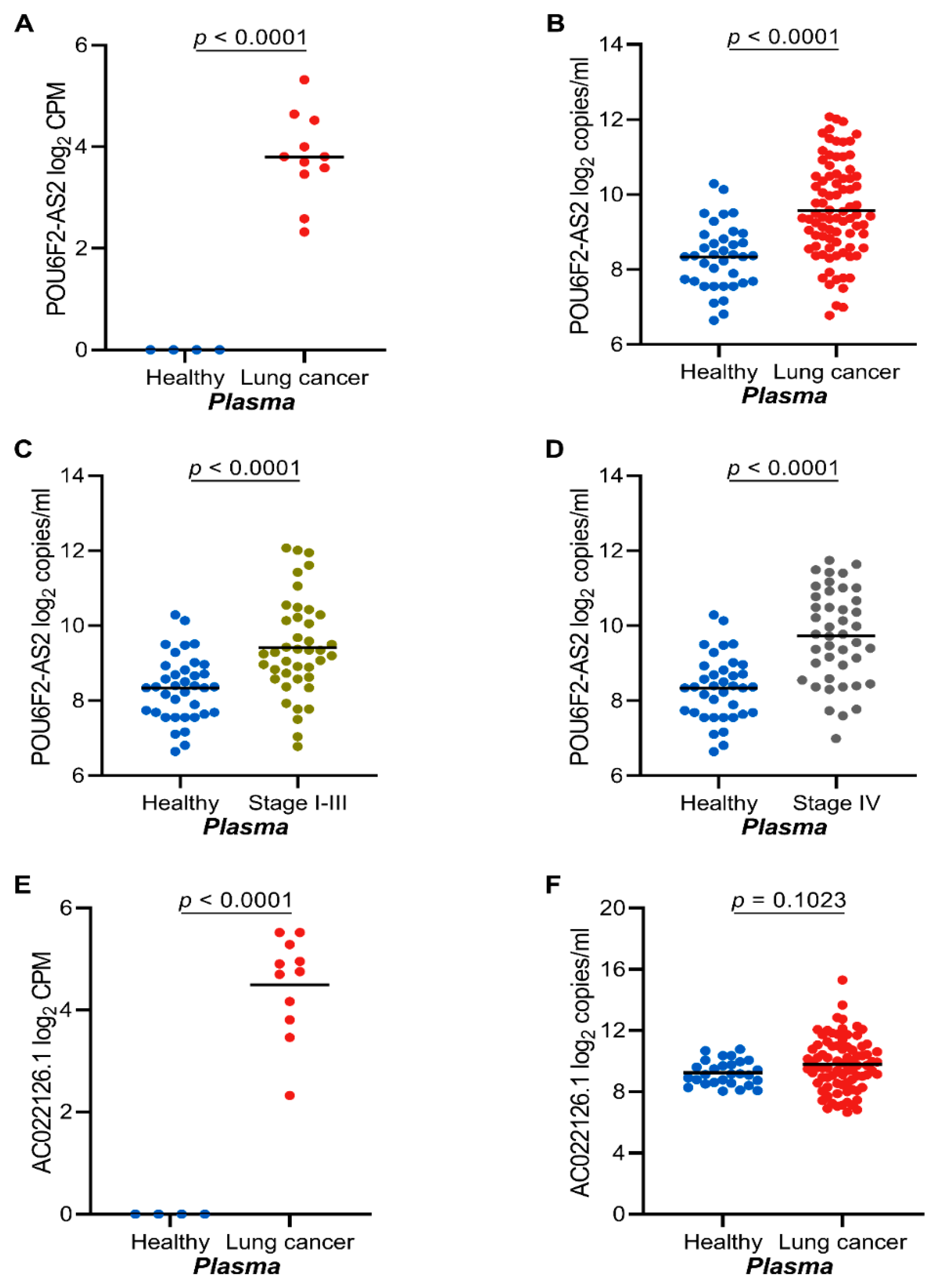
2.4. The lncRNAs POU6F2-AS2 and AC022126.1 Are Upregulated in Plasma from NSCLC Patients
2.5. High POU6F2-AS2 Levels Is Associated with Higher Cyfra 21.1 Levels
2.6. Serum cfRNA Concentration Correlates with POU6F2-AS2 Expression
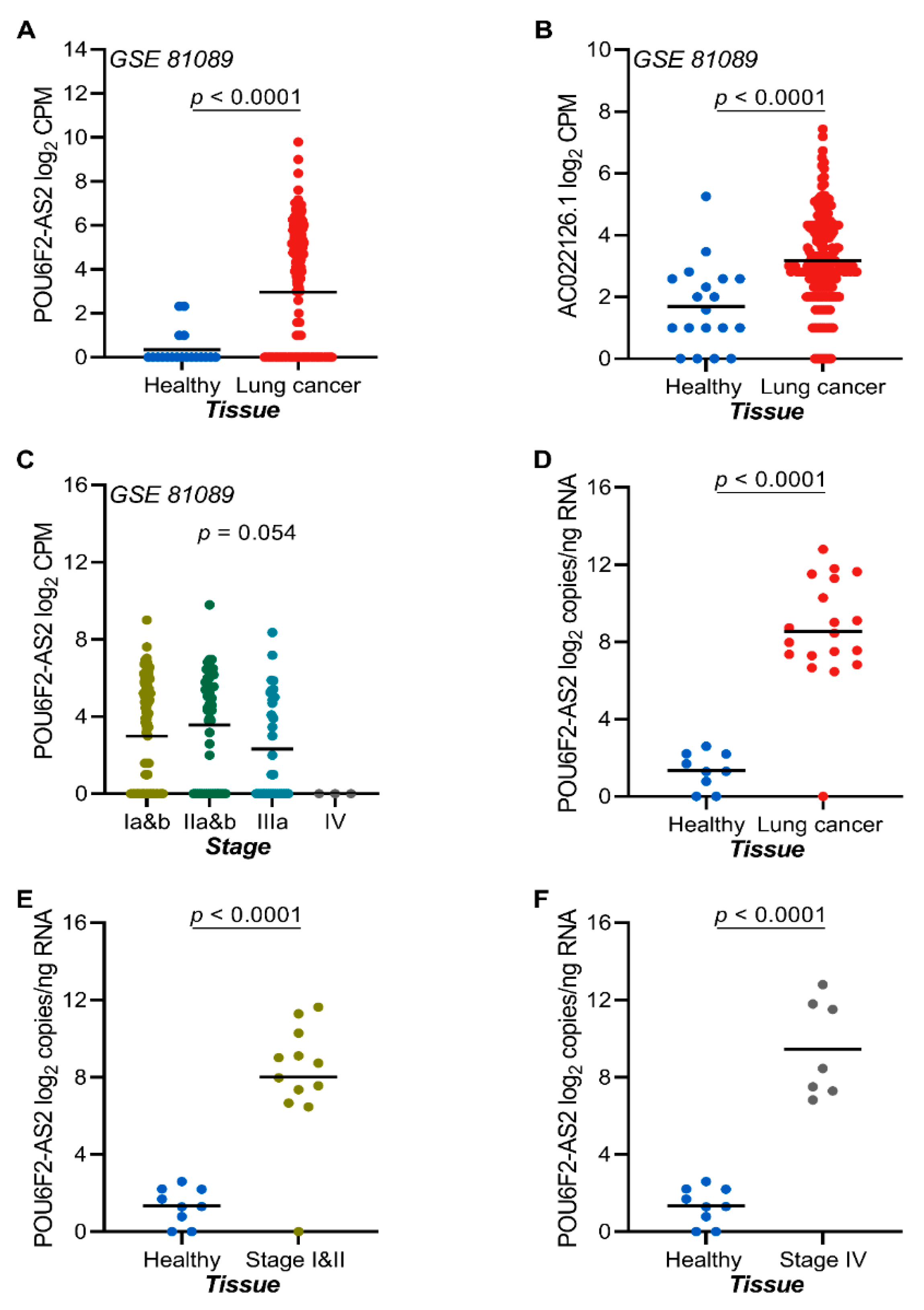
2.7. The lncRNAs POU6F2-AS2 and AC022126.1 Are Upregulated in NSCLC Tissue
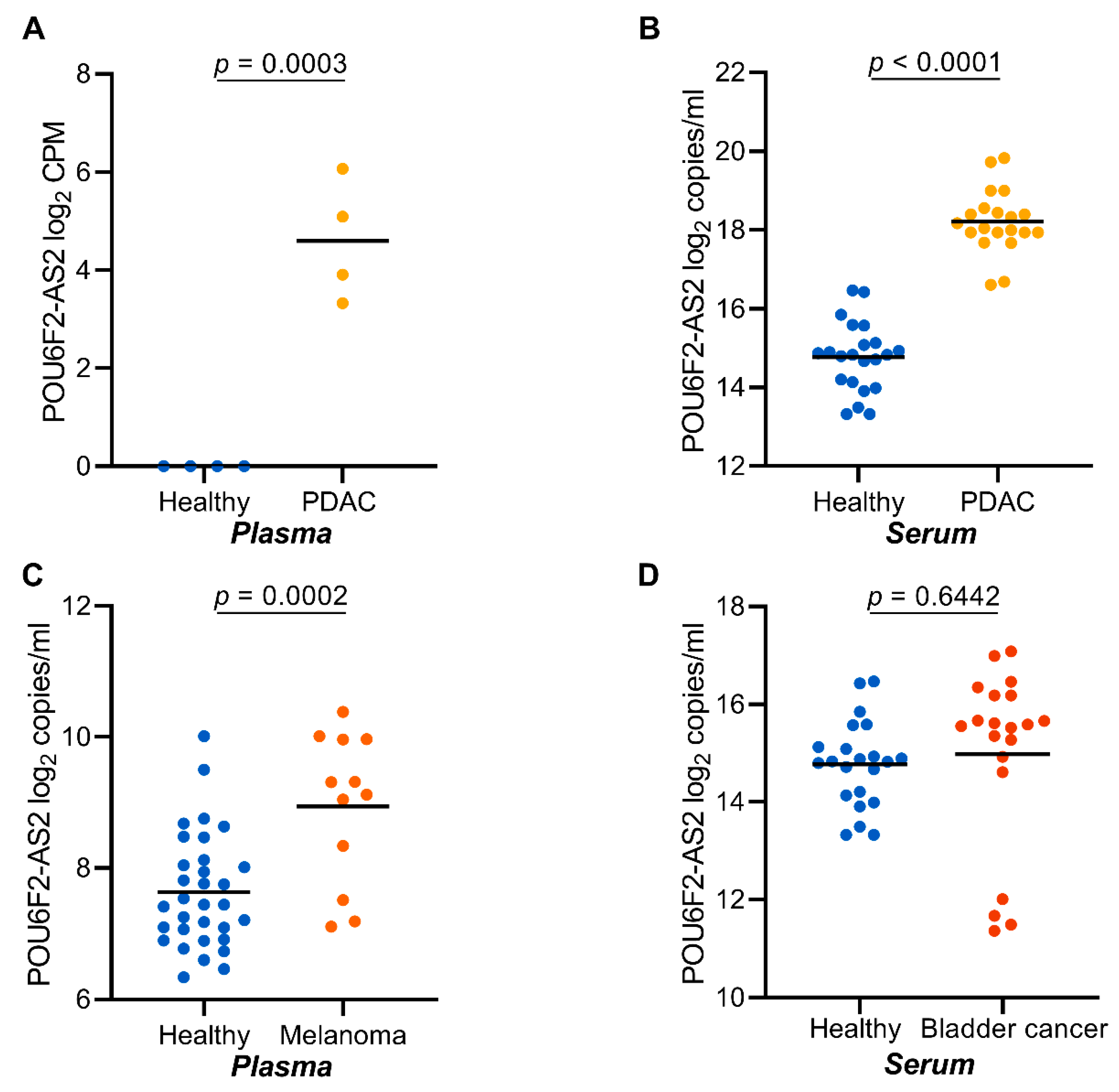
2.8. POU6F2-AS2 Is Upregulated in Additional Cancers
3. Discussion
4. Materials and Methods
4.1. Study Design and Patient Population
4.2. Blood Sampling and Plasma/Serum Preparation
4.3. cfRNA Isolation and Quantification
4.4. RNA Isolation from Tumor/Non-Tumor Tissue
4.5. RNA Isolation from FFPE Sections
4.6. In Silico Data Mining
4.7. Total cfRNA Sequencing
4.8. cfRNA Transcript Deconvolution
4.9. Transcript Quantification by RT-ddPCR
- C = copies per 20 µL reaction
- EV = elution volume
- ISV = cfRNA input volume
- TPV = total plasma/serum volume used of cfRNA isolation
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Herbst, R.S.; Morgensztern, D.; Boshoff, C. The biology and management of non-small cell lung cancer. Nature 2018, 553, 446–454. [Google Scholar] [CrossRef]
- Poroyko, V.; Mirzapoiazova, T.; Nam, A.; Mambetsariev, I.; Mambetsariev, B.; Wu, X.; Husain, A.; Vokes, E.E.; Wheeler, D.L.; Salgia, R. Exosomal mirnas species in the blood of small cell and non-small cell lung cancer patients. Oncotarget 2018, 9, 19793–19806. [Google Scholar] [CrossRef]
- Lu, S.; Kong, H.; Hou, Y.; Ge, D.; Huang, W.; Ou, J.; Yang, D.; Zhang, L.; Wu, G.; Song, Y.; et al. Two plasma microrna panels for diagnosis and subtype discrimination of lung cancer. Lung Cancer 2018, 123, 44–51. [Google Scholar] [CrossRef]
- Gridelli, C.; Rossi, A.; Carbone, D.P.; Guarize, J.; Karachaliou, N.; Mok, T.; Petrella, F.; Spaggiari, L.; Rosell, R. Non-small-cell lung cancer. Nat. Rev. Dis. Primers 2015, 1, 15009. [Google Scholar] [CrossRef]
- Lai, Y.; Wang, X.; Zeng, T.; Xing, S.; Dai, S.; Wang, J.; Chen, S.; Li, X.; Xie, Y.; Zhu, Y.; et al. Serum vegf levels in the early diagnosis and severity assessment of non-small cell lung cancer. J. Cancer 2018, 9, 1538–1547. [Google Scholar] [CrossRef]
- Scagliotti, G.; Hanna, N.; Fossella, F.; Sugarman, K.; Blatter, J.; Peterson, P.; Simms, L.; Shepherd, F.A. The differential efficacy of pemetrexed according to nsclc histology: A review of two phase iii studies. Oncology 2009, 14, 253–263. [Google Scholar] [CrossRef]
- Mok, T.S.; Wu, Y.L.; Thongprasert, S.; Yang, C.H.; Chu, D.T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (wjtog3405): An open label, randomised phase 3 trial. Lancet. Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Xing, S.; Zeng, T.; Xue, N.; He, Y.; Lai, Y.Z.; Li, H.L.; Huang, Q.; Chen, S.L.; Liu, W.L. Development and validation of tumor-educated blood platelets integrin alpha 2b (itga2b) rna for diagnosis and prognosis of non-small-cell lung cancer through rna-seq. Int. J. Biol. Sci. 2019, 15, 1977–1992. [Google Scholar] [CrossRef]
- Cassim, S.; Chepulis, L.; Keenan, R.; Kidd, J.; Firth, M.; Lawrenson, R. Patient and carer perceived barriers to early presentation and diagnosis of lung cancer: A systematic review. Bmc Cancer 2019, 19, 25. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.P. Screening with low-dose computed tomography reduced lung cancer mortality in high-risk patients. Ann. Intern. Med. 2011, 155, JC5-6. [Google Scholar] [CrossRef] [PubMed]
- Aberle, D.R.; Adams, A.M.; Berg, C.D.; Black, W.C.; Clapp, J.D.; Fagerstrom, R.M.; Gareen, I.F.; Gatsonis, C.; Marcus, P.M.; Sicks, J.D.; et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N. Engl. J. Med. 2011, 365, 395–409. [Google Scholar] [PubMed]
- Patz, E.F.; Pinsky, P.; Gatsonis, C.; Sicks, J.D.; Kramer, B.S.; Tammemagi, M.C.; Chiles, C.; Black, W.C.; Aberle, D.R.; Writ, N.O.M. Overdiagnosis in low-dose computed tomography screening for lung cancer. Jama Intern. Med. 2014, 174, 269–274. [Google Scholar] [CrossRef]
- Wiener, R.S.; Schwartz, L.M.; Woloshin, S.; Welch, H.G. Population-based risk for complications after transthoracic needle lung biopsy of a pulmonary nodule: An analysis of discharge records. Ann. Intern. Med. 2011, 155, 137–144. [Google Scholar] [CrossRef]
- Loke, S.Y.; Lee, A.S.G. The future of blood-based biomarkers for the early detection of breast cancer. Eur. J. Cancer 2018, 92, 54–68. [Google Scholar] [CrossRef]
- Ren, S.X.; Zhang, S.C.; Jiang, T.; He, Y.Y.; Ma, Z.Y.; Cai, H.R.; Xu, X.H.; Li, Y.; Cai, W.J.; Zhou, J.; et al. Early detection of lung cancer by using an autoantibody panel in chinese population. Oncoimmunology 2018, 7, e1384108. [Google Scholar] [CrossRef]
- Molina, R.; Marrades, R.M.; Auge, J.M.; Escudero, J.M.; Vinolas, N.; Reguart, N.; Ramirez, J.; Filella, X.; Molins, L.; Agusti, A. Assessment of a combined panel of six serum tumor markers for lung cancer. Am. J. Resp. Crit. Care 2016, 193, 427–437. [Google Scholar] [CrossRef]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in cancer staging, prognosis and treatment selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef]
- Maziak, W. Carcinoembryonic antigen (CEA) levels in hookah smokers, cigarette smokers and non-smokers--a comment. JPMA J. Pak. Med Assoc. 2008, 58, 155. [Google Scholar]
- Correa-Gallego, C.; Warshaw, A.L.; Fernandez-del Castillo, C. Fluid cea in ipmns: A useful test or the flip of a coin? Am. J. Gastroenterol. 2009, 104, 796–797. [Google Scholar] [CrossRef] [PubMed]
- Mensah, M.; Borzi, C.; Verri, C.; Suatoni, P.; Conte, D.; Pastorino, U.; Orazio, F.; Sozzi, G.; Boeri, M. Microrna based liquid biopsy: The experience of the plasma mirna signature classifier (msc) for lung cancer screening. J. Vis. Exp. JOVE 2017. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Hartmann, N.; Baeriswyl, L.; Andreasen, D.; Bernard, N.; Chen, C.; Cheo, D.; D’Andrade, P.; DeMayo, M.; Dennis, L.; et al. Evaluation of quantitative mirna expression platforms in the microrna quality control (mirqc) study. Nat. Methods 2014, 11, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Sozzi, G.; Boeri, M.; Rossi, M.; Verri, C.; Suatoni, P.; Bravi, F.; Roz, L.; Conte, D.; Grassi, M.; Sverzellati, N.; et al. Clinical utility of a plasma-based mirna signature classifier within computed tomography lung cancer screening: A correlative mild trial study. J. Clin. Oncol. 2014, 32, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Kopreski, M.S.; Benko, F.A.; Kwak, L.W.; Gocke, C.D. Detection of tumor messenger rna in the serum of patients with malignant melanoma. Clin. Cancer Res. 1999, 5, 1961–1965. [Google Scholar] [PubMed]
- Lo, K.W.; Lo, Y.M.D.; Leung, S.F.; Tsang, Y.S.; Chan, L.Y.S.; Johnson, P.J.; Hjelm, N.M.; Lee, J.C.K.; Huang, D.P. Analysis of cell-free epstein-barr virus-associated rna in the plasma of patients with nasopharyngeal carcinoma. Clin. Chem. 1999, 45, 1292–1294. [Google Scholar] [CrossRef]
- Chen, X.Q.; Bonnefoi, H.; Pelte, M.F.; Lyautey, J.; Lederrey, C.; Movarekhi, S.; Schaeffer, P.; Mulcahy, H.E.; Meyer, P.; Stroun, M.; et al. Telomerase rna as a detection marker in the serum of breast cancer patients. Clin. Cancer Res. 2000, 6, 3823–3826. [Google Scholar]
- Silva, J.M.; Dominguez, G.; Silva, J.; Garcia, J.M.; Sanchez, A.; Rodriguez, O.; Provencio, M.; Espana, P.; Bonilla, F. Detection of epithelial messenger rna in the plasma of breast cancer patients is associated with poor prognosis tumor characteristics. Clin. Cancer Res. 2001, 7, 2821–2825. [Google Scholar]
- Chuen, S.; Wong, C.; Fong, S. Quantification of plasma beta-catenin mrna in colorectal cancer and adenoma patients. Clin. Cancer Res. 2004, 10, 1613–1617, Erratum in 2006, 12, 996. [Google Scholar]
- Wong, S.C.C.; Lo, S.F.E.; Cheung, M.T.; Ng, K.O.E.; Tse, C.W.; Lai, B.S.P.; Lee, K.C.; Lo, Y.M.D. Quantification of plasma beta-catenin mrna in colorectal cancer and adenoma patients. Clin. Cancer Res. 2004, 10, 1613–1617. [Google Scholar] [CrossRef]
- Dasi, F.; Lledo, S.; Garcia-Granero, E.; Ripoll, R.; Marugan, M.; Tormo, M.; Garcia-Conde, J.; Alino, S.F. Real-time quantification in plasma of human telomerase reverse transcriptase (htert) mrna: A simple blood test to monitor disease in cancer patients. Lab Investig. 2001, 81, 767–769. [Google Scholar] [CrossRef] [PubMed]
- Miura, N.; Shiota, G.; Nakagawa, T.; Maeda, Y.; Sano, A.; Marumoto, A.; Kishimoto, Y.; Murawaki, Y.; Hasegawa, J. Sensitive detection of human telomerase reverse transcriptase mrna in the serum of patients with hepatocellular carcinoma. Oncology 2003, 64, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Sing, T.; Sander, O.; Beerenwinkel, N.; Lengauer, T. Rocr: Visualizing classifier performance in r. Bioinformatics 2005, 21, 3940–3941. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.D.; Kong, J.L.; Tang, J.L.; Wu, Y.H.; Xu, E.P.; Lai, M.D.; Zhang, H.H. Hotairm1 as a potential biomarker for diagnosis of colorectal cancer functions the role in the tumour suppressor. J. Cell Mol. Med. 2016, 20, 2036–2044. [Google Scholar] [CrossRef]
- Liu, J.; Sun, X.; Zhu, H.; Qin, Q.; Yang, X.; Sun, X. Long noncoding rna pou6f2-as2 is associated with oesophageal squamous cell carcinoma. J. Biochem. 2016, 160, 195–204. [Google Scholar] [CrossRef]
- Bazargan, M.; Lucas-Wright, A.; Jones, L.; Vargas, R.; Vadgama, J.V.; Evers-Manly, S.; Maxwell, A.E. Understanding perceived benefit of early cancer detection: Community-partnered research with african american women in south los angeles. J. Women’s Health 2015, 24, 755–761. [Google Scholar] [CrossRef]
- Zhu, W.Y.; Tan, L.L.; Wang, Z.Y.; Wang, S.J.; Xu, L.Y.; Yu, W.; Chen, Z.J.; Zhang, Y.K. Clinical characteristics and advantages of primary peripheral micro-sized lung adenocarcinoma over small-sized lung adenocarcinoma. Eur. J. Cardio-Thorac. Surg. 2016, 49, 1095–1102. [Google Scholar] [CrossRef][Green Version]
- Rubenstein, J.H.; Vakil, N.; Inadomi, J.M. The cost-effectiveness of biomarkers for predicting the development of oesophageal adenocarcinoma. Aliment. Pharmacol. Ther. 2005, 22, 135–146. [Google Scholar] [CrossRef]
- Midthun, D.E. Early detection of lung cancer. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Kwapisz, D. The first liquid biopsy test approved. Is it a new era of mutation testing for non-small cell lung cancer? Ann. Transl. Med. 2017, 5, 46. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. Egf receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef]
- Zeuschner, P.; Linxweiler, J.; Junker, K. Non-coding rnas as biomarkers in liquid biopsies with a special emphasis on extracellular vesicles in urological malignancies. Expert Rev. Mol. Diagn. 2019, 18, 1–17. [Google Scholar] [CrossRef]
- Jiang, N.; Pan, J.; Fang, S.; Zhou, C.; Han, Y.; Chen, J.; Meng, X.; Jin, X.; Gong, Z. Liquid biopsy: Circulating exosomal long noncoding rnas in cancer. Clinica chimica acta. Int. J. Clin. Chem. 2019, 495, 331–337. [Google Scholar]
- Collart, M.A. The ccr4-not complex is a key regulator of eukaryotic gene expression. Wiley interdisciplinary reviews. RNA 2016, 7, 438–454. [Google Scholar] [CrossRef]
- Li, Z.X.; Zhu, Q.N.; Zhang, H.B.; Hu, Y.; Wang, G.; Zhu, Y.S. Malat1: A potential biomarker in cancer. Cancer Manag. Res. 2018, 10, 6757–6768. [Google Scholar] [CrossRef]
- Ngo, T.T.M.; Moufarrej, M.N.; Rasmussen, M.L.H.; Camunas-Soler, J.; Pan, W.Y.; Okamoto, J.; Neff, N.F.; Liu, K.L.; Wong, R.J.; Downes, K.; et al. Noninvasive blood tests for fetal development predict gestational age and preterm delivery. Science 2018, 360, 1133–1136. [Google Scholar] [CrossRef]
- Mezheyeuski, A.; Bergsland, C.H.; Backman, M.; Djureinovic, D.; Sjoblom, T.; Bruun, J.; Micke, P. Multispectral imaging for quantitative and compartment-specific immune infiltrates reveals distinct immune profiles that classify lung cancer patients. J. Pathol. 2018, 244, 421–431. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for rna-seq data with deseq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. Limma powers differential expression analyses for rna-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Cima, I.; Kong, S.L.; Sengupta, D.; Tan, I.B.; Phyo, W.M.; Lee, D.; Hu, M.; Iliescu, C.; Alexander, I.; Goh, W.L.; et al. Tumor-derived circulating endothelial cell clusters in colorectal cancer. Sci. Transl. Med. 2016, 8, 345ra389. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.J.; Cima, I.; Choudhury, Y.; Wei, X.; Lim, J.C.; Thike, A.A.; Tan, M.H.; Tan, P.H. A five-gene reverse transcription-pcr assay for pre-operative classification of breast fibroepithelial lesions. Breast Cancer Res. BCR 2016, 18, 31. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | Discovery Cohort | Validation Cohort | ||||
|---|---|---|---|---|---|---|
| NSCLC | PDAC | NSCLC | PDAC | Melanoma | Bladder | |
| Total, n | 11 | 4 | 84 | 20 | 12 | 22 |
| Age | ||||||
| Median (SD) | 65 (8.0) | 66 (8.7) | 66 (9.2) | 70 (9.6) | 54 (13.5) | 70 (10.3) |
| Gender, n (%) | ||||||
| Male | 4 (36) | 2 (50) | 42 (50) | 11 (55) | 7 (60) | 18 (80) |
| Female | 7 (64) | 2 (50) | 42 (50) | 9 (45) | 5 (40) | 4 (20) |
| Smoking, n (%) | ||||||
| Never smoker | - | - | 5 (6) | - | - | - |
| Past smoker | - | - | 18 (21) | - | - | - |
| Current smoker | - | - | 14 (17) | - | - | - |
| Unknown | 11 (100) | 4 (100) | 47 (56) | 20 (100) | 12 (100) | 22 (100) |
| ECOG status | ||||||
| 0 | 8 (70) | - | 64 (76) | - | 10 (83) | - |
| 1 | 3 (30) | - | 16 (19) | - | 2 (17) | - |
| 2 | - | - | 1 (1) | - | - | - |
| Unknown | - | 4 (100) | 3 (4) | 20 (100) | - | 22 (100) |
| Stage | ||||||
| Early a | - | 1 (25) | 39 (46) | - | 6 (30) | |
| Late | 11 (100) | 3 (75) | 45 (54) | 20 (100) | 12 (100) | 16 (70) |
| CEA | ||||||
| <ULN | 1 (1) | - | 9 (11) | - | - | - |
| ≥ULN | 10 (99) | - | 32 (38) | - | - | - |
| Not done | - | 4 (100) | 43 (51) | 20 (100) | 12 (100) | 22 (100) |
| Cyfra21-1 | ||||||
| <ULN | 3 (30) | - | 5 (6) | - | - | - |
| ≥ULN | 8 (70) | - | 36 (43) | - | - | - |
| Not done | - | 4 (100) | 43 (51) | 20 (100) | 12 (100) | 22 (100) |
| CA 19.9 | ||||||
| <ULN | - | - | - | 5 (25) | - | - |
| ≥ULN | - | 2 (50) | - | 15 (75) | - | - |
| Not done | 11 (100) | 2 (50) | 84 (100) | - | 12 (100) | 22 (100) |
| LDH | ||||||
| <ULN | - | - | - | - | 10 (83) | - |
| ≥ULN | - | 3 (75) | - | - | 2 (17) | - |
| Not done | 11 (100) | 1 (25) | 84 (100) | 20 (100) | - | 22 (100) |
| Variable | N (%) | POU6F2-AS2 | AC022162.1 | ||
|---|---|---|---|---|---|
| n (%) | Mean | p | Mean | p | |
| Age | |||||
| <66 years | 38 (45.2) | 9.5 | 0.686 | 9.6 | 0.438 |
| ≥66 years | 46 (54.8) | 9.6 | 9.9 | ||
| Gender | |||||
| Male | 42 (50) | 9.8 | 0.058 | 9.9 | 0.430 |
| Female | 42 (50) | 9.3 | 9.6 | ||
| Smoking | |||||
| Never smoker | 5 (6) | 9.2 | 0.833 | 9.8 | 0.865 |
| Past smoker | 18 (21) | 9.5 | 10.1 | ||
| Current smoker | 14 (17) | 9.6 | 9.6 | ||
| Unknown | 47 (56) | 9.7 | 9.7 | ||
| ECOG status | |||||
| 0 | 64 (76) | 9.5 | 0.517 | 9.6 | 0.355 |
| 1 | 16 (19) | 9.9 | 10.3 | ||
| 2 | 1 (1) | 10.9 | 11.4 | ||
| Unknown | 3 (4) | 9.7 | 9.6 | ||
| Histology | |||||
| Adenocarcinoma | 23 (27) | 9.6 | 0.942 | 9.9 | 0.913 |
| Non-squamous NSCLC | 33 (39) | 9.5 | 9.6 | ||
| Squamous NSCLC | 21 (25) | 9.7 | 9.9 | ||
| Other | 7 (8) | 9.7 | 9.7 | ||
| Stage | |||||
| Early (I-II) | 33 (39) | 9.4 | 0.264 | 9.7 | 0.854 |
| Late (III-IV) | 51 (61) | 9.7 | 9.8 | ||
| CEA | |||||
| <ULN | 9 (11) | 9.2 | 0.371 | 9.1 | 0.467 |
| ≥ULN | 32 (38) | 9.8 | 9.8 | ||
| Not done | 43 (51) | 9.5 | 9.9 | ||
| Cyfra21-1 | |||||
| <ULN | 5 (6) | 8.2 | 0.017 | 8.3 | 0.151 |
| ≥ULN | 36 (43) | 9.9 | 9.9 | ||
| Not done | 43 (51) | 9.5 | 9.9 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metzenmacher, M.; Váraljai, R.; Hegedüs, B.; Cima, I.; Forster, J.; Schramm, A.; Scheffler, B.; Horn, P.A.; Klein, C.A.; Szarvas, T.; et al. Plasma Next Generation Sequencing and Droplet Digital-qPCR-Based Quantification of Circulating Cell-Free RNA for Noninvasive Early Detection of Cancer. Cancers 2020, 12, 353. https://doi.org/10.3390/cancers12020353
Metzenmacher M, Váraljai R, Hegedüs B, Cima I, Forster J, Schramm A, Scheffler B, Horn PA, Klein CA, Szarvas T, et al. Plasma Next Generation Sequencing and Droplet Digital-qPCR-Based Quantification of Circulating Cell-Free RNA for Noninvasive Early Detection of Cancer. Cancers. 2020; 12(2):353. https://doi.org/10.3390/cancers12020353
Chicago/Turabian StyleMetzenmacher, Martin, Renáta Váraljai, Balazs Hegedüs, Igor Cima, Jan Forster, Alexander Schramm, Björn Scheffler, Peter A. Horn, Christoph A. Klein, Tibor Szarvas, and et al. 2020. "Plasma Next Generation Sequencing and Droplet Digital-qPCR-Based Quantification of Circulating Cell-Free RNA for Noninvasive Early Detection of Cancer" Cancers 12, no. 2: 353. https://doi.org/10.3390/cancers12020353
APA StyleMetzenmacher, M., Váraljai, R., Hegedüs, B., Cima, I., Forster, J., Schramm, A., Scheffler, B., Horn, P. A., Klein, C. A., Szarvas, T., Reis, H., Bielefeld, N., Roesch, A., Aigner, C., Kunzmann, V., Wiesweg, M., Siveke, J. T., Schuler, M., & Lueong, S. S. (2020). Plasma Next Generation Sequencing and Droplet Digital-qPCR-Based Quantification of Circulating Cell-Free RNA for Noninvasive Early Detection of Cancer. Cancers, 12(2), 353. https://doi.org/10.3390/cancers12020353