GP130 Cytokines in Breast Cancer and Bone


{kind=link}
{kind=link}
Abstract
1. Introduction
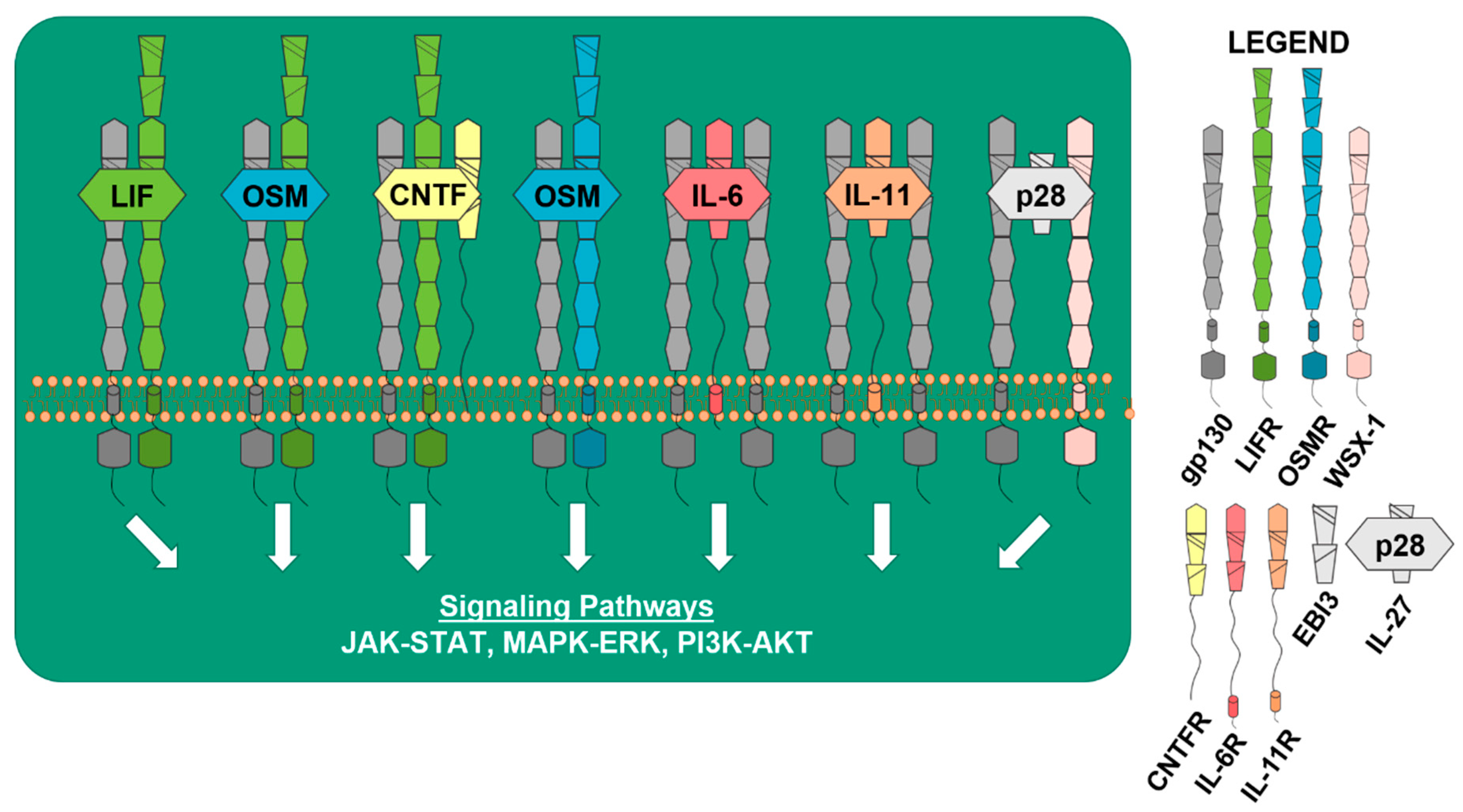
2. gp130 in Physiological Bone Remodeling
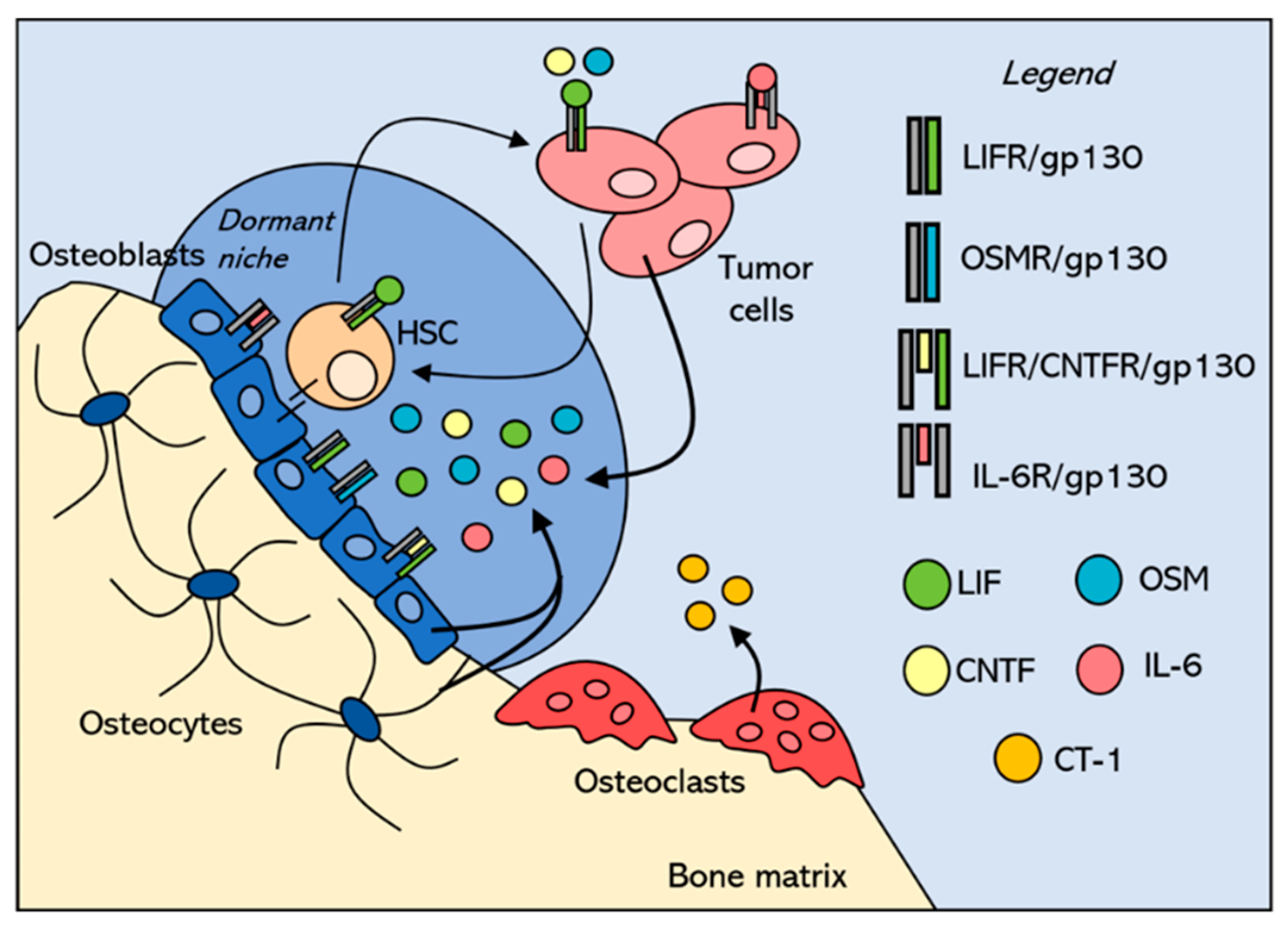
3. Tumor Niches within the Bone
4. gp130 Cytokines in Breast Cancer
5. IL-6 Cytokines and Cancer Stem Cells
6. Clinical Implications
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sterling, J.A.; Guelcher, S.A. Bone structural components regulating sites of tumor metastasis. Curr. Osteoporos. Rep. 2011, 9, 89–95. [Google Scholar] [CrossRef]
- Johnson, R.W.; Sowder, M.E.; Giaccia, A.J. Hypoxia and Bone Metastatic Disease. Curr. Osteoporos. Rep. 2017, 15, 231–238. [Google Scholar] [CrossRef]
- Shiozawa, Y.; Pedersen, E.A.; Havens, A.M.; Jung, Y.; Mishra, A.; Joseph, J.; Kim, J.K.; Patel, L.R.; Ying, C.; Ziegler, A.M.; et al. Human prostate cancer metastases target the hematopoietic stem cell niche to establish footholds in mouse bone marrow. J. Clin. Investig. 2011, 121, 1298–1312. [Google Scholar] [CrossRef]
- Mundy, G.R. Mechanisms of bone metastasis. Cancer 1997, 80, 1546–1556. [Google Scholar] [CrossRef]
- Johnson, R.W.; Sun, Y.; Ho, P.W.M.; Chan, A.S.M.; Johnson, J.A.; Pavlos, N.J.; Sims, N.A.; Martin, T.J. Parathyroid Hormone-Related Protein Negatively Regulates Tumor Cell Dormancy Genes in a PTHR1/Cyclic AMP-Independent Manner. Front. Endocrinol. 2018, 9, 241. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.D.; Nedjai, B.; Hurst, T.; Pennington, D.J. Cytokines and chemokines: At the crossroads of cell signalling and inflammatory disease. Biochimica Biophysica Acta (BBA) 2014, 1843, 2563–2582. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Basel, D.; Chow, S.-O.; Fong-Yee, C.; Kim, S.; Buttgereit, F.; Dunstan, C.R.; Zhou, H.; Seibel, M.J. Targeting IL-6 and RANKL signaling inhibits prostate cancer growth in bone. Clin. Exp. Metastasis 2014, 31, 921–933. [Google Scholar] [CrossRef]
- Sims, N.A. Cell-specific paracrine actions of IL-6 family cytokines from bone, marrow and muscle that control bone formation and resorption. Int. J. Biochem. Cell Biol. 2016, 79, 14–23. [Google Scholar] [CrossRef]
- Wang, X.; Lupardus, P.; LaPorte, S.L.; Garcia, K.C. Structural Biology of Shared Cytokine Receptors. Annu. Rev. Immunol. 2009, 27, 29–60. [Google Scholar] [CrossRef]
- Yoshida, K.; Taga, T.; Saito, M.; Suematsu, S.; Kumanogoh, A.; Tanaka, T.; Fujiwara, H.; Hirata, M.; Yamagami, T.; Nakahata, T.; et al. Targeted disruption of gp130, a common signal transducer for the interleukin 6 family of cytokines, leads to myocardial and hematological disorders. Proc. Nat. Acad. Sci. USA 1996, 93, 407–411. [Google Scholar] [CrossRef]
- Shin, H.-I.; Divieti, P.; Sims, N.A.; Kobayashi, T.; Miao, D.; Karaplis, A.C.; Baron, R.; Bringhurst, R.; Kronenberg, H.M. gp130-Mediated Signaling Is Necessary for Normal Osteoblastic Function in Vivo and in Vitro. Endocrinology 2004, 145, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, K.; Gao, Y.-H.; Yokose, S.; Kaji, Y.; Nakamura, T.; Suda, T.; Yoshida, K.; Taga, T.; Kishimoto, T.; Kataoka, H.; et al. Osteoclasts Are Present in gp130-Deficient Mice*. Endocrinology 1997, 138, 4959–4965. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Boulanger, M.J.; Chow, D.-c.; Brevnova, E.E.; Garcia, K.C. Hexameric Structure and Assembly of the Interleukin-6/IL-6 α-Receptor/gp130 Complex. Science 2003, 300, 2101–2104. [Google Scholar] [CrossRef] [PubMed]
- Taga, T.; Kishimoto, T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997, 15, 797–819. [Google Scholar] [CrossRef]
- Robledo, O.; Fourcin, M.; Chevalier, S.; Guillet, C.; Auguste, P.; Pouplard-Barthelaix, A.; Pennica, D.; Gascan, H. Signaling of the cardiotrophin-1 receptor. Evidence for a third receptor component. J. Biol. Chem. 1997, 272, 4855–4863. [Google Scholar] [CrossRef]
- Mosley, B.; De Imus, C.; Friend, D.; Boiani, N.; Thoma, B.; Park, L.S.; Cosman, D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 1996, 271, 32635–32643. [Google Scholar] [CrossRef]
- Pflanz, S.; Hibbert, L.; Mattson, J.; Rosales, R.; Vaisberg, E.; Bazan, J.F.; Phillips, J.H.; McClanahan, T.K.; de Waal Malefyt, R.; Kastelein, R.A. WSX-1 and glycoprotein 130 constitute a signal-transducing receptor for IL-27. J. Immunol. 2004, 172, 2225–2231. [Google Scholar] [CrossRef]
- Garbers, C.; Spudy, B.; Aparicio-Siegmund, S.; Waetzig, G.H.; Sommer, J.; Holscher, C.; Rose-John, S.; Grotzinger, J.; Lorenzen, I.; Scheller, J. An interleukin-6 receptor-dependent molecular switch mediates signal transduction of the IL-27 cytokine subunit p28 (IL-30) via a gp130 protein receptor homodimer. J. Biol. Chem. 2013, 288, 4346–4354. [Google Scholar] [CrossRef]
- Kourko, O.; Seaver, K.; Odoardi, N.; Basta, S.; Gee, K. IL-27, IL-30, and IL-35: A Cytokine Triumvirate in Cancer. Front. Oncol. 2019, 9, 969. [Google Scholar] [CrossRef]
- Stahl, N.; Boulton, T.G.; Farruggella, T.; Ip, N.Y.; Davis, S.; Witthuhn, B.A.; Quelle, F.W.; Silvennoinen, O.; Barbieri, G.; Pellegrini, S.; et al. Association and activation of Jak-Tyk kinases by CNTF-LIF-OSM-IL-6 beta receptor components. Science 1994, 263, 92–95. [Google Scholar] [CrossRef]
- Takahashi-Tezuka, M.; Yoshida, Y.; Fukada, T.; Ohtani, T.; Yamanaka, Y.; Nishida, K.; Nakajima, K.; Hibi, M.; Hirano, T. Gab1 acts as an adapter molecule linking the cytokine receptor gp130 to ERK mitogen-activated protein kinase. Mol. Cell. Biol. 1998, 18, 4109–4117. [Google Scholar] [CrossRef]
- Oh, H.; Fujio, Y.; Kunisada, K.; Hirota, H.; Matsui, H.; Kishimoto, T.; Yamauchi-Takihara, K. Activation of phosphatidylinositol 3-kinase through glycoprotein 130 induces protein kinase B and p70 S6 kinase phosphorylation in cardiac myocytes. J. Biol. Chem. 1998, 273, 9703–9710. [Google Scholar] [CrossRef]
- Fahmi, A.; Smart, N.; Punn, A.; Jabr, R.; Marber, M.; Heads, R. p42/p44-MAPK and PI3K are sufficient for IL-6 family cytokines/gp130 to signal to hypertrophy and survival in cardiomyocytes in the absence of JAK/STAT activation. Cell. Signal. 2013, 25, 898–909. [Google Scholar] [CrossRef]
- Chen, D.; Sun, Y.; Wei, Y.; Zhang, P.; Rezaeian, A.H.; Teruya-Feldstein, J.; Gupta, S.; Liang, H.; Lin, H.K.; Hung, M.C.; et al. LIFR is a breast cancer metastasis suppressor upstream of the Hippo-YAP pathway and a prognostic marker. Nat. Med. 2012, 18, 1511–1517. [Google Scholar] [CrossRef]
- Walker, E.C.; Johnson, R.W.; Hu, Y.; Brennan, H.J.; Poulton, I.J.; Zhang, J.G.; Jenkins, B.J.; Smyth, G.K.; Nicola, N.A.; Sims, N.A. Murine Oncostatin M Acts via Leukemia Inhibitory Factor Receptor to Phosphorylate Signal Transducer and Activator of Transcription 3 (STAT3) but Not STAT1, an Effect That Protects Bone Mass. J. Biol. Chem. 2016, 291, 21703–21716. [Google Scholar] [CrossRef]
- Rose, T.M.; Bruce, A.G. Oncostatin M is a member of a cytokine family that includes leukemia-inhibitory factor, granulocyte colony-stimulating factor, and interleukin 6. Proc. Natl. Acad. Sci. USA 1991, 88, 8641–8645. [Google Scholar] [CrossRef]
- Sims, N.A.; Gooi, J.H. Bone remodeling: Multiple cellular interactions required for coupling of bone formation and resorption. Semin. Cell Dev. Biol. 2008, 19, 444–451. [Google Scholar] [CrossRef]
- Bellido, T.; Borba, V.Z.C.; Roberson, P.; Manolagas, S.C. Activation of the Janus Kinase/STAT (Signal Transducer and Activator of Transcription) Signal Transduction Pathway by Interleukin-6-Type Cytokines Promotes Osteoblast Differentiation*. Endocrinology 1997, 138, 3666–3676. [Google Scholar] [CrossRef]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Solano, M.; Pompolo, S.; Fernandes, T.J.; Constable, M.J.; Nicholson, G.C.; Zhang, J.G.; Nicola, N.A.; et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J. Clin. Investig. 2010, 120, 582–592. [Google Scholar] [CrossRef]
- Walker, E.C.; McGregor, N.E.; Poulton, I.J.; Pompolo, S.; Allan, E.H.; Quinn, J.M.; Gillespie, M.T.; Martin, T.J.; Sims, N.A. Cardiotrophin-1 Is an Osteoclast-Derived Stimulus of Bone Formation Required for Normal Bone Remodeling. J. Bone Miner. Res. 2008, 23, 2025–2032. [Google Scholar] [CrossRef]
- Metcalf, D.; Gearing, D.P. Fatal syndrome in mice engrafted with cells producing high levels of the leukemia inhibitory factor. Proc. Natl. Acad. Sci. USA 1989, 86, 5948–5952. [Google Scholar] [CrossRef]
- McGregor, N.E.; Murat, M.; Elango, J.; Poulton, I.J.; Walker, E.C.; Crimeen-Irwin, B.; Ho, P.W.M.; Gooi, J.H.; Martin, T.J.; Sims, N.A. IL-6 exhibits both cis- and trans-signaling in osteocytes and osteoblasts, but only trans-signaling promotes bone formation and osteoclastogenesis. JBC 2019, 294, 7850–7863. [Google Scholar] [CrossRef]
- Ishimi, Y.; Miyaura, C.; Jin, C.H.; Akatsu, T.; Abe, E.; Nakamura, Y.; Yamaguchi, A.; Yoshiki, S.; Matsuda, T.; Hirano, T.; et al. IL-6 is produced by osteoblasts and induces bone resorption. J. Immunol. 1990, 145, 3297–3303. [Google Scholar]
- Liang, J.D.; Hock, J.M.; Sandusky, G.E.; Santerre, R.F.; Onyia, J.E. Immunohistochemical Localization of Selected Early Response Genes Expressed in Trabecular Bone of Young Rats Given hPTH 1-34. Calcified Tissue Int. 1999, 65, 369–373. [Google Scholar] [CrossRef]
- Romas, E.; Udagawa, N.; Zhou, H.; Tamura, T.; Saito, M.; Taga, T.; Hilton, D.J.; Suda, T.; Ng, K.W.; Martin, T.J. The role of gp130-mediated signals in osteoclast development: Regulation of interleukin 11 production by osteoblasts and distribution of its receptor in bone marrow cultures. J. Exp. Med. 1996, 183, 2581–2591. [Google Scholar] [CrossRef]
- Allan, E.H.; Hilton, D.J.; Brown, M.A.; Evely, R.S.; Yumita, S.; Metcalf, D.; Gough, N.M.; Ng, K.W.; Nicola, N.A.; Martin, T.J. Osteoblasts display receptors for and responses to leukemia-inhibitory factor. J. Cell. Physiol. 1990, 145, 110–119. [Google Scholar] [CrossRef]
- Ishimi, Y.; Abe, E.; Jin, C.H.; Miyaura, C.; Hong, M.H.; Oshida, M.; Kurosawa, H.; Yamaguchi, Y.; Tomida, M.; Hozumi, M.; et al. Leukemia inhibitory factor/differentiation-stimulating factor (LIF/D-factor): Regulation of its production and possible roles in bone metabolism. J. Cell. Physiol. 1992, 152, 71–78. [Google Scholar] [CrossRef]
- Liu, F.; Aubin, J.E.; Malaval, L. Expression of leukemia inhibitory factor (LIF)/interleukin-6 family cytokines and receptors during in vitro osteogenesis: Differential regulation by dexamethasone and LIF. Bone 2002, 31, 212–219. [Google Scholar] [CrossRef]
- McGregor, N.E.; Poulton, I.J.; Walker, E.C.; Pompolo, S.; Quinn, J.M.; Martin, T.J.; Sims, N.A. Ciliary neurotrophic factor inhibits bone formation and plays a sex-specific role in bone growth and remodeling. Calcified Tissue Int. 2010, 86, 261–270. [Google Scholar] [CrossRef]
- Hunt, L.C.; White, J. The Role of Leukemia Inhibitory Factor Receptor Signaling in Skeletal Muscle Growth, Injury and Disease. In Growth Factors and Cytokines in Skeletal Muscle Development, Growth, Regeneration and Disease; White, J., Smythe, G., Eds.; Springer International Publishing: Cham, Germany, 2016; pp. 45–59. [Google Scholar] [CrossRef]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an Endocrine Organ: Focus on Muscle-Derived Interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef]
- Johnson, R.W.; White, J.D.; Walker, E.C.; Martin, T.J.; Sims, N.A. Myokines (muscle-derived cytokines and chemokines) including ciliary neurotrophic factor (CNTF) inhibit osteoblast differentiation. Bone 2014, 64, 47–56. [Google Scholar] [CrossRef]
- Li, X.; Ominsky, M.S.; Niu, Q.T.; Sun, N.; Daugherty, B.; D’Agostin, D.; Kurahara, C.; Gao, Y.; Cao, J.; Gong, J.; et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008, 23, 860–869. [Google Scholar] [CrossRef]
- Ware, C.B.; Horowitz, M.C.; Renshaw, B.R.; Hunt, J.S.; Liggitt, D.; Koblar, S.A.; Gliniak, B.C.; McKenna, H.J.; Papayannopoulou, T.; Thoma, B.; et al. Targeted disruption of the low-affinity leukemia inhibitory factor receptor gene causes placental, skeletal, neural and metabolic defects and results in perinatal death. Development 1995, 121, 1283–1299. [Google Scholar]
- Poulton, I.J.; McGregor, N.E.; Pompolo, S.; Walker, E.C.; Sims, N.A. Contrasting roles of leukemia inhibitory factor in murine bone development and remodeling involve region-specific changes in vascularization. J. Bone Miner. Res. 2012, 27, 586–595. [Google Scholar] [CrossRef]
- Sims, N.A.; Jenkins, B.J.; Nakamura, A.; Quinn, J.M.; Li, R.; Gillespie, M.T.; Ernst, M.; Robb, L.; Martin, T.J. Interleukin-11 receptor signaling is required for normal bone remodeling. J. Bone Miner. Res. 2005, 20, 1093–1102. [Google Scholar] [CrossRef]
- Johnson, R.W.; Brennan, H.J.; Vrahnas, C.; Poulton, I.J.; McGregor, N.E.; Standal, T.; Walker, E.C.; Koh, T.T.; Nguyen, H.; Walsh, N.C.; et al. The Primary Function of gp130 Signaling in Osteoblasts Is To Maintain Bone Formation and Strength, Rather Than Promote Osteoclast Formation. J. Bone Miner. Res. 2014, 29, 1492–1505. [Google Scholar] [CrossRef]
- Tamura, T.; Udagawa, N.; Takahashi, N.; Miyaura, C.; Tanaka, S.; Yamada, Y.; Koishihara, Y.; Ohsugi, Y.; Kumaki, K.; Taga, T. Soluble interleukin-6 receptor triggers osteoclast formation by interleukin 6. Proc. Nat. Acad. Sci. USA 1993, 90, 11924–11928. [Google Scholar] [CrossRef]
- Richards, C.D.; Langdon, C.; Deschamps, P.; Pennica, D.; Shaughnessy, S.G. Stimulation of osteoclast differentiation in vitro by mouse oncostatin M, leukaemia inhibitory factor, cardiotrophin-1 and interleukin 6: Synergy with dexamethasone. Cytokine 2000, 12, 613–621. [Google Scholar] [CrossRef]
- Palmqvist, P.; Persson, E.; Conaway, H.H.; Lerner, U.H. IL-6, leukemia inhibitory factor, and oncostatin M stimulate bone resorption and regulate the expression of receptor activator of NF-kappa B ligand, osteoprotegerin, and receptor activator of NF-kappa B in mouse calvariae. J. Immunol. 2002, 169, 3353–3362. [Google Scholar] [CrossRef]
- Horwood, N.J.; Elliott, J.; Martin, T.J.; Gillespie, M.T. Osteotropic agents regulate the expression of osteoclast differentiation factor and osteoprotegerin in osteoblastic stromal cells. Endocrinology 1998, 139, 4743–4746. [Google Scholar] [CrossRef]
- Sims, N.A.; Walsh, N.C. GP130 cytokines and bone remodelling in health and disease. BMB Rep. 2010, 43, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; McGregor, N.E.; Brennan, H.J.; Crimeen-Irwin, B.; Poulton, I.J.; Martin, T.J.; Sims, N.A. Glycoprotein130 (Gp130)/interleukin-6 (IL-6) signalling in osteoclasts promotes bone formation in periosteal and trabecular bone. Bone 2015, 81, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.M.; Goss, G.A.; Sutherland, R.L.; Hilton, D.J.; Berndt, M.C.; Nicola, N.A.; Begley, C.G. Expression and function of members of the cytokine receptor superfamily on breast cancer cells. Oncogene 1997, 14, 661. [Google Scholar] [CrossRef] [PubMed]
- Nathan, R.W.; Leigh, C.M.; Peter, H.W. Oncostatin M suppresses oestrogen receptor-α expression and is associated with poor outcome in human breast cancer. Endocrine-Related Cancer 2012, 19, 181–195. [Google Scholar] [CrossRef]
- Selander, K.S.; Li, L.; Watson, L.; Merrell, M.; Dahmen, H.; Heinrich, P.C.; Muller-Newen, G.; Harris, K.W. Inhibition of gp130 signaling in breast cancer blocks constitutive activation of Stat3 and inhibits in vivo malignancy. Cancer Res. 2004, 64, 6924–6933. [Google Scholar] [CrossRef]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: The importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373. [Google Scholar] [CrossRef]
- Kiel, M.J.; Yilmaz, Ö.H.; Iwashita, T.; Yilmaz, O.H.; Terhorst, C.; Morrison, S.J. SLAM Family Receptors Distinguish Hematopoietic Stem and Progenitor Cells and Reveal Endothelial Niches for Stem Cells. Cell 2005, 121, 1109–1121. [Google Scholar] [CrossRef]
- Ding, L.; Saunders, T.L.; Enikolopov, G.; Morrison, S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature 2012, 481, 457–462. [Google Scholar] [CrossRef]
- Mendez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; Macarthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Li, W.; Johnson, S.A.; Shelley, W.C.; Yoder, M.C. Hematopoietic stem cell repopulating ability can be maintained in vitro by some primary endothelial cells. Exp. Hematol. 2004, 32, 1226–1237. [Google Scholar] [CrossRef]
- Kobayashi, H.; Butler, J.M.; O’Donnell, R.; Kobayashi, M.; Ding, B.S.; Bonner, B.; Chiu, V.K.; Nolan, D.J.; Shido, K.; Benjamin, L.; et al. Angiocrine factors from Akt-activated endothelial cells balance self-renewal and differentiation of haematopoietic stem cells. Nat. Cell Biol. 2010, 12, 1046–1056. [Google Scholar] [CrossRef]
- Omatsu, Y.; Sugiyama, T.; Kohara, H.; Kondoh, G.; Fujii, N.; Kohno, K.; Nagasawa, T. The essential functions of adipo-osteogenic progenitors as the hematopoietic stem and progenitor cell niche. Immunity 2010, 33, 387–399. [Google Scholar] [CrossRef]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Rattis, F.M.; DiMascio, L.N.; Congdon, K.L.; Pazianos, G.; Zhao, C.; Yoon, K.; Cook, J.M.; Willert, K.; Gaiano, N.; et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nat. Immunol. 2005, 6, 314. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Barbier, V.; Nowlan, B.; Jacobsen, R.N.; Forristal, C.E.; Patton, J.T.; Magnani, J.L.; Levesque, J.P. Vascular niche E-selectin regulates hematopoietic stem cell dormancy, self renewal and chemoresistance. Nat. Med. 2012, 18, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.; Yokota, T.; Xia, L.; Kincade, P.W.; McEver, R.P. Bone marrow dysfunction in mice lacking the cytokine receptor gp130 in endothelial cells. Blood 2005, 106, 4093–4101. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Calvi, L.M.; Adams, G.B.; Weibrecht, K.W.; Weber, J.M.; Olson, D.P.; Knight, M.C.; Martin, R.P.; Schipani, E.; Divieti, P.; Bringhurst, F.R.; et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature 2003, 425, 841. [Google Scholar] [CrossRef]
- Zhang, J.; Niu, C.; Ye, L.; Huang, H.; He, X.; Tong, W.-G.; Ross, J.; Haug, J.; Johnson, T.; Feng, J.Q.; et al. Identification of the haematopoietic stem cell niche and control of the niche size. Nature 2003, 425, 836. [Google Scholar] [CrossRef]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/Angiopoietin-1 Signaling Regulates Hematopoietic Stem Cell Quiescence in the Bone Marrow Niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Wang, H.; Yu, C.; Gao, X.; Welte, T.; Muscarella, A.M.; Tian, L.; Zhao, H.; Zhao, Z.; Du, S.; Tao, J.; et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer Cell 2015, 27, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078. [Google Scholar] [CrossRef] [PubMed]
- Iorns, E.; Ward, T.M.; Dean, S.; Jegg, A.; Thomas, D.; Murugaesu, N.; Sims, D.; Mitsopoulos, C.; Fenwick, K.; Kozarewa, I.; et al. Whole genome in vivo RNAi screening identifies the leukemia inhibitory factor receptor as a novel breast tumor suppressor. Breast Cancer Res. Treat. 2012, 135, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Boyle, P. Triple-negative breast cancer: Epidemiological considerations and recommendations. Ann. Oncol. 2012, 23 (Suppl. 6), vi7–12. [Google Scholar] [CrossRef]
- Allred, D.C.; Brown, P.; Medina, D. The origins of estrogen receptor alpha-positive and estrogen receptor alpha-negative human breast cancer. Breast Cancer Res. 2004, 6, 240–245. [Google Scholar] [CrossRef]
- Pauletti, G.; Godolphin, W.; Press, M.F.; Slamon, D.J. Detection and quantitation of HER-2/neu gene amplification in human breast cancer archival material using fluorescence in situ hybridization. Oncogene 1996, 13, 63–72. [Google Scholar]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.-y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef]
- Won, H.S.; Kim, Y.A.; Lee, J.S.; Jeon, E.K.; An, H.J.; Sun, D.S.; Ko, Y.H.; Kim, J.S. Soluble interleukin-6 receptor is a prognostic marker for relapse-free survival in estrogen receptor-positive breast cancer. Cancer Investig. 2013, 31, 516–521. [Google Scholar] [CrossRef]
- Li, H.; Xiao, H.; Lin, L.; Jou, D.; Kumari, V.; Lin, J.; Li, C. Drug design targeting protein-protein interactions (PPIs) using multiple ligand simultaneous docking (MLSD) and drug repositioning: Discovery of raloxifene and bazedoxifene as novel inhibitors of IL-6/GP130 interface. J. Med. Chem. 2014, 57, 632–641. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Yang, X.-Y.; Glass, O.; Lei, G.; Osada, T.; Dave, S.S.; Morse, M.A.; Clay, T.M.; Lyerly, H.K. HER2 overexpression elicits a proinflammatory IL-6 autocrine signaling loop that is critical for tumorigenesis. Cancer Res. 2011, 71, 4380–4391. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Kim, G.I.; Davis, A.; Malik, F.; Henry, N.L.; Ithimakin, S.; Quraishi, A.A.; Tawakkol, N.; D’Angelo, R.; Paulson, A.K.; et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol. Cell 2012, 47, 570–584. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, K.; Sahin, A.; Emami, K.; Hortobagyi, G.N.; Estrov, Z. Expression of leukemia inhibitory factor and its receptor in breast cancer: A potential autocrine and paracrine growth regulatory mechanism. Breast Cancer Res. Treat. 1998, 48, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, H.; Zhao, Y.; Wang, C.; Cheng, Z.; Tang, L.; Gao, Y.; Liu, F.; Li, J.; Li, Y.; et al. A mandatory role of nuclear PAK4-LIFR axis in breast-to-bone metastasis of ERα-positive breast cancer cells. Oncogene 2019, 38, 808–821. [Google Scholar] [CrossRef]
- Danforth, D.N., Jr.; Sgagias, M.K. Interleukin-1 alpha and interleukin-6 act additively to inhibit growth of MCF-7 breast cancer cells in vitro. Cancer Res. 1993, 53, 1538–1545. [Google Scholar]
- Morinaga, Y.; Suzuki, H.; Takatsuki, F.; Akiyama, Y.; Taniyama, T.; Matsushima, K.; Onozaki, K. Contribution of IL-6 to the antiproliferative effect of IL-1 and tumor necrosis factor on tumor cell lines. J. Immunol. 1989, 143, 3538–3542. [Google Scholar]
- Chiu, J.J.; Sgagias, M.K.; Cowan, K.H. Interleukin 6 acts as a paracrine growth factor in human mammary carcinoma cell lines. Clin. Cancer Res. 1996, 2, 215–221. [Google Scholar]
- Tamm, I.; Cardinale, I.; Krueger, J.; Murphy, J.S.; May, L.T.; Sehgal, P.B. Interleukin 6 decreases cell-cell association and increases motility of ductal breast carcinoma cells. J. Exp. Med. 1989, 170, 1649–1669. [Google Scholar] [CrossRef]
- Asgeirsson, K.S.; Olafsdottir, K.; Jonasson, J.G.; Ogmundsdottir, H.M. The effects of IL-6 on cell adhesion and e-cadherin expression in breast cancer. Cytokine 1998, 10, 720–728. [Google Scholar] [CrossRef]
- Badache, A.; Hynes, N.E. Interleukin 6 inhibits proliferation and, in cooperation with an epidermal growth factor receptor autocrine loop, increases migration of T47D breast cancer cells. Cancer Res. 2001, 61, 383–391. [Google Scholar] [PubMed]
- Johnston, P.G.; Rondinone, C.M.; Voeller, D.; Allegra, C.J. Identification of a Protein Factor Secreted by 3T3-L1 Preadipocytes Inhibitory for the Human MCF-7 Breast Cancer Cell Line. Cancer Res. 1992, 52, 6860–6865. [Google Scholar] [PubMed]
- Jiang, X.P.; Yang, D.C.; Elliott, R.L.; Head, J.F. Down-regulation of expression of interleukin-6 and its receptor results in growth inhibition of MCF-7 breast cancer cells. Anticancer Res. 2011, 31, 2899–2906. [Google Scholar] [PubMed]
- Nugoli, M.; Chuchana, P.; Vendrell, J.; Orsetti, B.; Ursule, L.; Nguyen, C.; Birnbaum, D.; Douzery, E.J.P.; Cohen, P.; Theillet, C. Genetic variability in MCF-7 sublines: Evidence of rapid genomic and RNA expression profile modifications. BMC Cancer 2003, 3, 13. [Google Scholar] [CrossRef]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.Y.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts Isolated from Common Sites of Breast Cancer Metastasis Enhance Cancer Cell Growth Rates and Invasiveness in an Interleukin-6–Dependent Manner. J. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef]
- Taguchi, Y.; Yamamoto, M.; Yamate, T.; Lin, S.C.; Mocharla, H.; DeTogni, P.; Nakayama, N.; Boyce, B.F.; Abe, E.; Manolagas, S.C. Interleukin-6-type cytokines stimulate mesenchymal progenitor differentiation toward the osteoblastic lineage. Proc. Assoc. Am. Phys. 1998, 110, 559–574. [Google Scholar]
- Wu, Q.; Zhou, X.; Huang, D.; Ji, Y.; Kang, F. IL-6 Enhances Osteocyte-Mediated Osteoclastogenesis by Promoting JAK2 and RANKL Activity In Vitro. Cell. Physiol. Biochem. 2017, 41, 1360–1369. [Google Scholar] [CrossRef]
- Bussard, K.M.; Venzon, D.J.; Mastro, A.M. Osteoblasts are a major source of inflammatory cytokines in the tumor microenvironment of bone metastatic breast cancer. J. Cell. Biochem. 2010, 111, 1138–1148. [Google Scholar] [CrossRef]
- Zheng, Y.; Chow, S.O.; Boernert, K.; Basel, D.; Mikuscheva, A.; Kim, S.; Fong-Yee, C.; Trivedi, T.; Buttgereit, F.; Sutherland, R.L.; et al. Direct Crosstalk Between Cancer and Osteoblast Lineage Cells Fuels Metastatic Growth in Bone via Auto-Amplification of IL-6 and RANKL Signaling Pathways. J. Bone Miner. Res. 2014, 29, 1938–1949. [Google Scholar] [CrossRef]
- Campbell, J.P.; Karolak, M.R.; Ma, Y.; Perrien, D.S.; Masood-Campbell, S.K.; Penner, N.L.; Munoz, S.A.; Zijlstra, A.; Yang, X.; Sterling, J.A.; et al. Stimulation of Host Bone Marrow Stromal Cells by Sympathetic Nerves Promotes Breast Cancer Bone Metastasis in Mice. PLoS Biol. 2012, 10, e1001363. [Google Scholar] [CrossRef]
- Luo, X.; Fu, Y.; Loza, A.J.; Murali, B.; Leahy, K.M.; Ruhland, M.K.; Gang, M.; Su, X.; Zamani, A.; Shi, Y.; et al. Stromal-Initiated Changes in the Bone Promote Metastatic Niche Development. Cell Rep. 2016, 14, 82–92. [Google Scholar] [CrossRef]
- Di Carlo, E. Interleukin-30: A novel microenvironmental hallmark of prostate cancer progression. Oncoimmunology 2014, 3, e27618. [Google Scholar] [CrossRef]
- Di Meo, S.; Airoldi, I.; Sorrentino, C.; Zorzoli, A.; Esposito, S.; Di Carlo, E. Interleukin-30 expression in prostate cancer and its draining lymph nodes correlates with advanced grade and stage. Clin. Cancer Res. 2014, 20, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Airoldi, I.; Cocco, C.; Sorrentino, C.; Angelucci, D.; Di Meo, S.; Manzoli, L.; Esposito, S.; Ribatti, D.; Bertolotto, M.; Iezzi, L.; et al. Interleukin-30 Promotes Breast Cancer Growth and Progression. Cancer Res. 2016, 76, 6218–6229. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Qu, J.; Jin, N.; Xu, J.; Lin, C.; Chen, Y.; Yang, X.; He, X.; Tang, S.; Lan, X.; et al. Feedback Activation of Leukemia Inhibitory Factor Receptor Limits Response to Histone Deacetylase Inhibitors in Breast Cancer. Cancer Cell 2016, 30, 459–473. [Google Scholar] [CrossRef]
- Kim, R.S.; Avivar-Valderas, A.; Estrada, Y.; Bragado, P.; Sosa, M.S.; Aguirre-Ghiso, J.A.; Segall, J.E. Dormancy signatures and metastasis in estrogen receptor positive and negative breast cancer. PLoS ONE 2012, 7, e35569. [Google Scholar] [CrossRef]
- Thomas, R.J.; Guise, T.A.; Yin, J.J.; Elliott, J.; Horwood, N.J.; Martin, T.J.; Gillespie, M.T. Breast Cancer Cells Interact with Osteoblasts to Support Osteoclast Formation1. Endocrinology 1999, 140, 4451–4458. [Google Scholar] [CrossRef]
- Woosley, A.N.; Dalton, A.C.; Hussey, G.S.; Howley, B.V.; Mohanty, B.K.; Grelet, S.; Dincman, T.; Bloos, S.; Olsen, S.K.; Howe, P.H. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene 2019. [Google Scholar] [CrossRef]
- Wang, X.J.; Qiao, Y.; Xiao, M.M.; Wang, L.; Chen, J.; Lv, W.; Xu, L.; Li, Y.; Wang, Y.; Tan, M.D.; et al. Opposing Roles of Acetylation and Phosphorylation in LIFR-Dependent Self-Renewal Growth Signaling in Mouse Embryonic Stem Cells. Cell Rep. 2017, 18, 933–946. [Google Scholar] [CrossRef]
- Schiemann, W.P.; Graves, L.M.; Baumann, H.; Morella, K.K.; Gearing, D.P.; Nielsen, M.D.; Krebs, E.G.; Nathanson, N.M. Phosphorylation of the human leukemia inhibitory factor (LIF) receptor by mitogen-activated protein kinase and the regulation of LIF receptor function by heterologous receptor activation. Proc. Nat. Acad. Sci. USA 1995, 92, 5361–5365. [Google Scholar] [CrossRef]
- Estrov, Z.; Samal, B.; Lapushin, R.; Kellokumpu-Lehtinen, P.; Sahin, A.A.; Kurzrock, R.; Talpaz, M.; Aggarwal, B.B. Leukemia Inhibitory Factor Binds to Human Breast Cancer Cells and Stimulates Their Proliferation. J. Interf. Cytok. Res. 1995, 15, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Kellokumpu-Lehtinen, P.; Talpaz, M.; Harris, D.; Van, Q.; Kurzrock, R.; Estrov, Z. Leukemia-inhibitory factor stimulates breast, kidney and prostate cancer cell proliferation by paracrine and autocrine pathways. Int. J. Cancer 1996, 66, 515–519. [Google Scholar] [CrossRef]
- Li, X.; Yang, Q.; Yu, H.; Wu, L.; Zhao, Y.; Zhang, C.; Yue, X.; Liu, Z.; Wu, H.; Haffty, B.G.; et al. LIF promotes tumorigenesis and metastasis of breast cancer through the AKT-mTOR pathway. Oncotarget 2014, 5, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.M.; Grant, S.L.; Gross, G.A.; Clouston, D.R.; Sutherland, R.L.; Begley, C.G. Oncostatin M induces the differentiation of breast cancer cells. Int. J. Cancer 1998, 75, 64–73. [Google Scholar] [CrossRef]
- Franken, N.A.P.; Rodermond, H.M.; Stap, J.; Haveman, J.; van Bree, C. Clonogenic assay of cells in vitro. Nature Protocols 2006, 1, 2315. [Google Scholar] [CrossRef]
- Liu, J.; Spence, M.; Wallace, P.; Forcier, K.; Hellstrom, I.; Vestal, R. Oncostatin M-specific receptor mediates inhibition of breast cancer cell growth and down-regulation of the c-myc proto-oncogene. Cell Growth Differ. 1997, 8, 667–676. [Google Scholar]
- Grant, S.L.; Douglas, A.M.; Goss, G.A.; Begley, C.G. Oncostatin M and leukemia inhibitory factor regulate the growth of normal human breast epithelial cells. Growth Factors 2001, 19, 153–162. [Google Scholar] [CrossRef]
- Li, C.; Ahlborn, T.E.; Kraemer, F.B.; Liu, J. Oncostatin M–induced growth inhibition and morphological changes of MDA-MB231 breast cancer cells are abolished by blocking the MEK/ERK signaling pathway. Breast Cancer Res. Treat. 2001, 66, 111–121. [Google Scholar] [CrossRef]
- Liu, J.; Hadjokas, N.; Mosley, B.; Estrov, Z.; Spence, M.J.; Vestal, R.E. Oncostatin M-specific receptor expression and function in regulating cell proliferation of normal and malignant mammary epithelial cells. Cytokine 1998, 10, 295–302. [Google Scholar] [CrossRef]
- West, N.R.; Murray, J.I.; Watson, P.H. Oncostatin-M promotes phenotypic changes associated with mesenchymal and stem cell-like differentiation in breast cancer. Oncogene 2013, 33, 1485. [Google Scholar] [CrossRef]
- Underhill-Day, N.; Heath, J.K. Oncostatin M (OSM) cytostasis of breast tumor cells: Characterization of an OSM receptor beta-specific kernel. Cancer Res. 2006, 66, 10891–10901. [Google Scholar] [CrossRef]
- Jorcyk, C.L.; Holzer, R.G.; Ryan, R.E. Oncostatin M induces cell detachment and enhances the metastatic capacity of T-47D human breast carcinoma cells. Cytokine 2006, 33, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Tawara, K.; Scott, H.; Emathinger, J.; Wolf, C.; LaJoie, D.; Hedeen, D.; Bond, L.; Montgomery, P.; Jorcyk, C. HIGH expression of OSM and IL-6 are associated with decreased breast cancer survival: Synergistic induction of IL-6 secretion by OSM and IL-1β. Oncotarget 2019, 10, 2068–2085. [Google Scholar] [CrossRef] [PubMed]
- Bolin, C.; Tawara, K.; Sutherland, C.; Redshaw, J.; Aranda, P.; Moselhy, J.; Anderson, R.; Jorcyk, C.L. Oncostatin M Promotes Mammary Tumor Metastasis to Bone and Osteolytic Bone Degradation. Genes Cancer 2012, 3, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Tawara, K.; Bolin, C.; Koncinsky, J.; Kadaba, S.; Covert, H.; Sutherland, C.; Bond, L.; Kronz, J.; Garbow, J.R.; Jorcyk, C.L. OSM potentiates preintravasation events, increases CTC counts, and promotes breast cancer metastasis to the lung. Breast Cancer Res. 2018, 20, 53. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Lapeire, L.; Hendrix, A.; Lambein, K.; Van Bockstal, M.; Braems, G.; Van Den Broecke, R.; Limame, R.; Mestdagh, P.; Vandesompele, J.; Vanhove, C.; et al. Cancer-associated adipose tissue promotes breast cancer progression by paracrine oncostatin M and Jak/STAT3 signaling. Cancer Res. 2014, 74, 6806–6819. [Google Scholar] [CrossRef]
- Tawara, K.; Scott, H.; Emathinger, J.; Ide, A.; Fox, R.; Greiner, D.; LaJoie, D.; Hedeen, D.; Nandakumar, M.; Oler, A.J.; et al. Co-Expression of VEGF and IL-6 Family Cytokines is Associated with Decreased Survival in HER2 Negative Breast Cancer Patients: Subtype-Specific IL-6 Family Cytokine-Mediated VEGF Secretion. Transl. Oncol. 2019, 12, 245–255. [Google Scholar] [CrossRef]
- Garbers, C.; Hermanns, H.M.; Schaper, F.; Müller-Newen, G.; Grötzinger, J.; Rose-John, S.; Scheller, J. Plasticity and cross-talk of Interleukin 6-type cytokines. Cytok. Growth Factor Rev. 2012, 23, 85–97. [Google Scholar] [CrossRef]
- Boulanger, M.J.; Garcia, K.C. Shared Cytokine Signaling Receptors: Structural Insights from the Gp130 System. In Advances in Protein Chemistry; Academic Press: New York, NY, USA, 2004; Vol. 68, pp. 107–146. [Google Scholar]
- Huynh, J.; Chand, A.; Gough, D.; Ernst, M. Therapeutically exploiting STAT3 activity in cancer—Using tissue repair as a road map. Nat. Rev. Cancer 2019, 19, 82–96. [Google Scholar] [CrossRef]
- Leslie, K.; Gao, S.P.; Berishaj, M.; Podsypanina, K.; Ho, H.; Ivashkiv, L.; Bromberg, J. Differential interleukin-6/Stat3 signaling as a function of cellular context mediates Ras-induced transformation. Breast Cancer Res. 2010, 12, R80. [Google Scholar] [CrossRef] [PubMed]
- Liang, F.; Ren, C.; Wang, J.; Wang, S.; Yang, L.; Han, X.; Chen, Y.; Tong, G.; Yang, G. The crosstalk between STAT3 and p53/RAS signaling controls cancer cell metastasis and cisplatin resistance via the Slug/MAPK/PI3K/AKT-mediated regulation of EMT and autophagy. Oncogenesis 2019, 8, 59. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Tawara, K.; Oxford, J.T.; Jorcyk, C.L. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: Potential of anti-IL-6 therapies. Cancer Manag. Res. 2011, 3, 177–189. [Google Scholar] [CrossRef]
- Helbig, G.; Christopherson, K.W., 2nd; Bhat-Nakshatri, P.; Kumar, S.; Kishimoto, H.; Miller, K.D.; Broxmeyer, H.E.; Nakshatri, H. NF-κB promotes breast cancer cell migration and metastasis by inducing the expression of the chemokine receptor CXCR4. J. Biol. Chem. 2003, 278, 21631–21638. [Google Scholar] [CrossRef]
- Sansone, P.; Storci, G.; Tavolari, S.; Guarnieri, T.; Giovannini, C.; Taffurelli, M.; Ceccarelli, C.; Santini, D.; Paterini, P.; Marcu, K.B.; et al. IL-6 triggers malignant features in mammospheres from human ductal breast carcinoma and normal mammary gland. J. Clin. Investig. 2007, 117, 3988–4002. [Google Scholar] [CrossRef]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Junk, D.J.; Bryson, B.L.; Smigiel, J.M.; Parameswaran, N.; Bartel, C.A.; Jackson, M.W. Oncostatin M promotes cancer cell plasticity through cooperative STAT3-SMAD3 signaling. Oncogene 2017, 36, 4001–4013. [Google Scholar] [CrossRef]
- Viswanadhapalli, S.; Luo, Y.; Sareddy, G.R.; Santhamma, B.; Zhou, M.; Li, M.; Ma, S.; Sonavane, R.; Pratap, U.P.; Altwegg, K.A.; et al. EC359: A First-in-Class Small-Molecule Inhibitor for Targeting Oncogenic LIFR Signaling in Triple-Negative Breast Cancer. Mol. Cancer Ther. 2019, 18, 1341–1354. [Google Scholar] [CrossRef]
- Kleffel, S.; Schatton, T. Tumor Dormancy and Cancer Stem Cells: Two Sides of the Same Coin? In Systems Biology of Tumor Dormancy; Enderling, H., Almog, N., Hlatky, L., Eds.; Springer New York: New York, NY, USA, 2013; pp. 145–179. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R.; Badve, S.; Nakshatri, H. CD44+/CD24−breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006, 8, R59. [Google Scholar] [CrossRef] [PubMed]
- Patel, L.R.; Camacho, D.F.; Shiozawa, Y.; Pienta, K.J.; Taichman, R.S. Mechanisms of cancer cell metastasis to the bone: A multistep process. Future Oncol. 2011, 7, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Ayob, A.Z.; Ramasamy, T.S. Cancer stem cells as key drivers of tumour progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.M.; McBride, W.H.; Pajonk, F. The Response of CD24 −/low /CD44 + Breast Cancer–Initiating Cells to Radiation. JNCI 2006, 98, 1777–1785. [Google Scholar] [CrossRef]
- Li, X.; Lewis, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.-F.; Hilsenbeck, S.G.; Pavlick, A.; Zhang, X.; Chamness, G.C.; et al. Intrinsic Resistance of Tumorigenic Breast Cancer Cells to Chemotherapy. JNC 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Moitra, K.; Lou, H.; Dean, M. Multidrug Efflux Pumps and Cancer Stem Cells: Insights Into Multidrug Resistance and Therapeutic Development. Clin. Pharmacol. Ther. 2011, 89, 491–502. [Google Scholar] [CrossRef]
- Scheel, C.; Eaton, E.N.; Li, S.H.; Chaffer, C.L.; Reinhardt, F.; Kah, K.J.; Bell, G.; Guo, W.; Rubin, J.; Richardson, A.L.; et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 2011, 145, 926–940. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–615. [Google Scholar] [CrossRef]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef]
- Zardawi, S.J.; O’Toole, S.A.; Sutherland, R.L.; Musgrove, E.A. Dysregulation of Hedgehog, Wnt and Notch signalling pathways in breast cancer. Histol. Histopathol. 2009, 24, 385–398. [Google Scholar] [CrossRef]
- Boulanger, C.A.; Wagner, K.-U.; Smith, G.H. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGF-β1 expression. Oncogene 2004, 24, 552. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.B.; Spence, K.; Anderson, E.; Howell, A.; Okano, H.; Potten, C.S. A putative human breast stem cell population is enriched for steroid receptor-positive cells. Dev. Biol. 2005, 277, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Dontu, G.; Abdallah, W.M.; Foley, J.M.; Jackson, K.W.; Clarke, M.F.; Kawamura, M.J.; Wicha, M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 2003, 17, 1253–1270. [Google Scholar] [CrossRef] [PubMed]
- Kritikou, E.A.; Sharkey, A.; Abell, K.; Came, P.J.; Anderson, E.; Clarkson, R.W.E.; Watson, C.J. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development 2003, 130, 3459–3468. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Ernst, M.; Novak, U.; Nicholson, S.E.; Layton, J.E.; Dunn, A.R. The carboxyl-terminal domains of gp130-related cytokine receptors are necessary for suppressing embryonic stem cell differentiation. Involvement of STAT3. J. Biol. Chem. 1999, 274, 9729–9737. [Google Scholar] [CrossRef]
- Hirai, H.; Karian, P.; Kikyo, N. Regulation of embryonic stem cell self-renewal and pluripotency by leukaemia inhibitory factor. Biochem. J. 2011, 438, 11–23. [Google Scholar] [CrossRef]
- Bourillot, P.Y.; Aksoy, I.; Schreiber, V.; Wianny, F.; Schulz, H.; Hummel, O.; Hubner, N.; Savatier, P. Novel STAT3 target genes exert distinct roles in the inhibition of mesoderm and endoderm differentiation in cooperation with Nanog. Stem Cells 2009, 27, 1760–1771. [Google Scholar] [CrossRef]
- Yoshida, K.; Chambers, I.; Nichols, J.; Smith, A.; Saito, M.; Yasukawa, K.; Shoyab, M.; Taga, T.; Kishimoto, T. Maintenance of the pluripotential phenotype of embryonic stem cells through direct activation of gp130 signalling pathways. Mechanisms Dev. 1994, 45, 163–171. [Google Scholar] [CrossRef]
- Matsuda, T.; Nakamura, T.; Nakao, K.; Arai, T.; Katsuki, M.; Heike, T.; Yokota, T. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 1999, 18, 4261–4269. [Google Scholar] [CrossRef]
- Niwa, H.; Burdon, T.; Chambers, I.; Smith, A. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 1998, 12, 2048–2060. [Google Scholar] [CrossRef]
- Conover, J.C.; Ip, N.Y.; Poueymirou, W.T.; Bates, B.; Goldfarb, M.P.; DeChiara, T.M.; Yancopoulos, G.D. Ciliary neurotrophic factor maintains the pluripotentiality of embryonic stem cells. Development 1993, 119, 559–565. [Google Scholar] [PubMed]
- Rose, T.M.; Weiford, D.M.; Gunderson, N.L.; Bruce, A.G. Oncostatin M (OSM) inhibits the differentiation of pluripotent embryonic stem cells in vitro. Cytokine 1994, 6, 48–54. [Google Scholar] [CrossRef]
- Pennica, D.; Shaw, K.J.; Swanson, T.A.; Moore, M.W.; Shelton, D.L.; Zioncheck, K.A.; Rosenthal, A.; Taga, T.; Paoni, N.F.; Wood, W.I. Cardiotrophin-1. Biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J. Biol. Chem. 1995, 270, 10915–10922. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gao, W.; Lytle, N.K.; Huang, P.; Yuan, X.; Dann, A.M.; Ridinger-Saison, M.; DelGiorno, K.E.; Antal, C.E.; Liang, G.; et al. Targeting LIF-mediated paracrine interaction for pancreatic cancer therapy and monitoring. Nature 2019, 569, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.R.; Parvani, J.G.; Tamagno, I.; Junk, D.J.; Bryson, B.L.; Cheon, H.J.; Stark, G.R.; Jackson, M.W. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019, 21, 54. [Google Scholar] [CrossRef] [PubMed]
- Dethlefsen, C.; Hojfeldt, G.; Hojman, P. The role of intratumoral and systemic IL-6 in breast cancer. Breast Cancer Res. Treat. 2013, 138, 657–664. [Google Scholar] [CrossRef]
- Kotowicz, B.; Fuksiewicz, M.; Jonska-Gmyrek, J.; Bidzinski, M.; Kowalska, M. The assessment of the prognostic value of tumor markers and cytokines as SCCAg, CYFRA 21.1, IL-6, VEGF and sTNF receptors in patients with squamous cell cervical cancer, particularly with early stage of the disease. Tumour Biol. 2016, 37, 1271–1278. [Google Scholar] [CrossRef]
- Chung, Y.C.; Chang, Y.F. Serum interleukin-6 levels reflect the disease status of colorectal cancer. J. Surg. Oncol. 2003, 83, 222–226. [Google Scholar] [CrossRef]
- Chen, M.F.; Chen, P.T.; Lu, M.S.; Lin, P.Y.; Chen, W.C.; Lee, K.D. IL-6 expression predicts treatment response and outcome in squamous cell carcinoma of the esophagus. Mol. Cancer 2013, 12, 26. [Google Scholar] [CrossRef]
- Jinno, T.; Kawano, S.; Maruse, Y.; Matsubara, R.; Goto, Y.; Sakamoto, T.; Hashiguchi, Y.; Kaneko, N.; Tanaka, H.; Kitamura, R.; et al. Increased expression of interleukin-6 predicts poor response to chemoradiotherapy and unfavorable prognosis in oral squamous cell carcinoma. Oncol. Rep. 2015, 33, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Riedel, F.; Zaiss, I.; Herzog, D.; Gotte, K.; Naim, R.; Hormann, K. Serum levels of interleukin-6 in patients with primary head and neck squamous cell carcinoma. Anticancer Res. 2005, 25, 2761–2765. [Google Scholar] [PubMed]
- Maccio, A.; Madeddu, C. The role of interleukin-6 in the evolution of ovarian cancer: Clinical and prognostic implications--a review. J. Mol. Med. 2013, 91, 1355–1368. [Google Scholar] [CrossRef] [PubMed]
- Sanguinete, M.M.M.; Oliveira, P.H.; Martins-Filho, A.; Micheli, D.C.; Tavares-Murta, B.M.; Murta, E.F.C.; Nomelini, R.S. Serum IL-6 and IL-8 Correlate with Prognostic Factors in Ovarian Cancer. Immunol. Investig. 2017, 46, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Mitsunaga, S.; Ikeda, M.; Shimizu, S.; Ohno, I.; Takahashi, H.; Furuse, J.; Inagaki, M.; Higashi, S.; Kato, H.; et al. Characterization of patients with advanced pancreatic cancer and high serum interleukin-6 levels. Pancreas 2015, 44, 756–763. [Google Scholar] [CrossRef]
- Culig, Z.; Puhr, M. Interleukin-6: A multifunctional targetable cytokine in human prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 52–58. [Google Scholar] [CrossRef]
- Altundag, O.; Altundag, K.; Gunduz, E. Interleukin-6 and C-reactive protein in metastatic renal cell carcinoma. J. Clin. Oncol. 2005, 23, 1044. [Google Scholar] [CrossRef]
- Chang, Q.; Bournazou, E.; Sansone, P.; Berishaj, M.; Gao, S.P.; Daly, L.; Wels, J.; Theilen, T.; Granitto, S.; Zhang, X.; et al. The IL-6/JAK/Stat3 feed-forward loop drives tumorigenesis and metastasis. Neoplasia 2013, 15, 848–862. [Google Scholar] [CrossRef]
- Deisseroth, A.; Ko, C.W.; Nie, L.; Zirkelbach, J.F.; Zhao, L.; Bullock, J.; Mehrotra, N.; Del Valle, P.; Saber, H.; Sheth, C.; et al. FDA approval: Siltuximab for the treatment of patients with multicentric Castleman disease. Clin. Cancer Res. 2015, 21, 950–954. [Google Scholar] [CrossRef]
- Shimabukuro-Vornhagen, A.; Godel, P.; Subklewe, M.; Stemmler, H.J.; Schlosser, H.A.; Schlaak, M.; Kochanek, M.; Boll, B.; von Bergwelt-Baildon, M.S. Cytokine release syndrome. J. Immunother. Cancer 2018, 6, 56. [Google Scholar] [CrossRef]
- Abreu, T.R.; Fonseca, N.A.; Goncalves, N.; Moreira, J.N. Current challenges and emerging opportunities of CAR-T cell therapies. J. Contr. Release 2019, 319, 246–261. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.B.; Salama, A.K.S. A review of cancer immunotherapy toxicity. CA Cancer J. Clin. 2020. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nature Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra225. [Google Scholar] [CrossRef]
- Wang, Z.; Han, W. Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy. Biomarker Res. 2018, 6, 4. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.-A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Lee, Y.G.; Chu, H.; Lu, Y.; Leamon, C.P.; Srinivasarao, M.; Putt, K.S.; Low, P.S. Regulation of CAR T cell-mediated cytokine release syndrome-like toxicity using low molecular weight adapters. Nat. Commun. 2019, 10, 2681. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- Honjo, O.; Kubo, T.; Sugaya, F.; Nishizaka, T.; Kato, K.; Hirohashi, Y.; Takahashi, H.; Torigoe, T. Severe cytokine release syndrome resulting in purpura fulminans despite successful response to nivolumab therapy in a patient with pleomorphic carcinoma of the lung: A case report. J. Immunother. Cancer 2019, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, C.E.; Iriyama, C.; Rose-Zerilli, M.J.; Nagelkerke, S.Q.; Hussain, K.; Ganderton, R.; Lee, C.; Machado, L.R.; Hollox, E.J.; Parker, H.; et al. Evaluation of High-Throughput Genomic Assays for the Fc Gamma Receptor Locus. PLoS ONE 2015, 10, e0142379. [Google Scholar] [CrossRef] [PubMed]
- Dahal, L.N.; Roghanian, A.; Beers, S.A.; Cragg, M.S. FcgammaR requirements leading to successful immunotherapy. Immunol. Rev. 2015, 268, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Alakhras, N.S.; Qiu, J.; Rocha, G.V.; Witcher, D.R.; Koester, A.; You, J.; Schaer, D.A.; Holmgaard, R.B.; Driscoll, K.; Willy, J.A.; et al. FcgammaRIIIa-dependent IFN-gamma release in whole blood assay is predictive of therapeutic IgG1 antibodies safety. mAbs 2018, 10, 913–921. [Google Scholar] [CrossRef]
- Hussain, K.; Hargreaves, C.E.; Rowley, T.F.; Sopp, J.M.; Latham, K.V.; Bhatta, P.; Sherington, J.; Cutler, R.M.; Humphreys, D.P.; Glennie, M.J.; et al. Impact of Human FcγR Gene Polymorphisms on IgG-Triggered Cytokine Release: Critical Importance of Cell Assay Format. Front. Immunol. 2019, 10, 390. [Google Scholar] [CrossRef]
- Lee, C.S.; Ashton-Key, M.; Cogliatti, S.; Rondeau, S.; Schmitz, S.F.; Ghielmini, M.; Cragg, M.S.; Johnson, P. Expression of the inhibitory Fc gamma receptor IIB (FCGR2B, CD32B) on follicular lymphoma cells lowers the response rate to rituximab monotherapy (SAKK 35/98). Br. J. Haematol. 2015, 168, 145–148. [Google Scholar] [CrossRef]
- Yu, X.; Marshall, M.J.E.; Cragg, M.S.; Crispin, M. Improving Antibody-Based Cancer Therapeutics Through Glycan Engineering. BioDrugs 2017, 31, 151–166. [Google Scholar] [CrossRef]
- Musolino, A.; Naldi, N.; Bortesi, B.; Pezzuolo, D.; Capelletti, M.; Missale, G.; Laccabue, D.; Zerbini, A.; Camisa, R.; Bisagni, G.; et al. Immunoglobulin G fragment C receptor polymorphisms and clinical efficacy of trastuzumab-based therapy in patients with HER-2/neu-positive metastatic breast cancer. J. Clin. Oncol. 2008, 26, 1789–1796. [Google Scholar] [CrossRef]
- Junttila, T.T.; Parsons, K.; Olsson, C.; Lu, Y.; Xin, Y.; Theriault, J.; Crocker, L.; Pabonan, O.; Baginski, T.; Meng, G.; et al. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010, 70, 4481–4489. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omokehinde, T.; Johnson, R.W. GP130 Cytokines in Breast Cancer and Bone. Cancers 2020, 12, 326. https://doi.org/10.3390/cancers12020326
Omokehinde T, Johnson RW. GP130 Cytokines in Breast Cancer and Bone. Cancers. 2020; 12(2):326. https://doi.org/10.3390/cancers12020326
Chicago/Turabian StyleOmokehinde, Tolu, and Rachelle W. Johnson. 2020. "GP130 Cytokines in Breast Cancer and Bone" Cancers 12, no. 2: 326. https://doi.org/10.3390/cancers12020326
APA StyleOmokehinde, T., & Johnson, R. W. (2020). GP130 Cytokines in Breast Cancer and Bone. Cancers, 12(2), 326. https://doi.org/10.3390/cancers12020326