Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment


Simple Summary
Abstract
1. Introduction
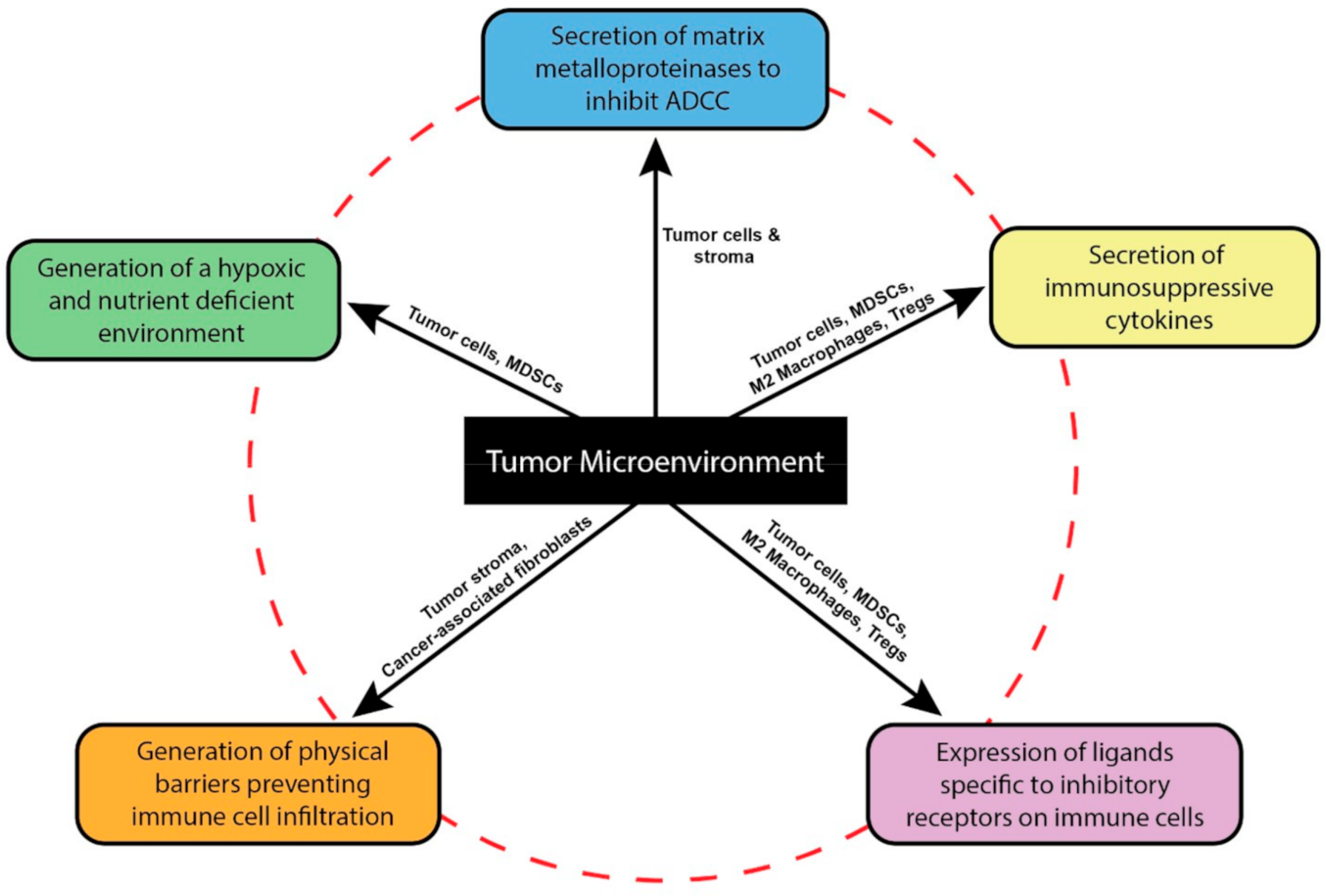
2. The Tumor Microenvironment Dysregulates NK Cell Functions
3. Strategies to Overcome the Tumor Microenvironment
3.1. CAR-NK Cell Therapy
3.2. Dominant Negative and Switch Receptors
3.3. Enhanced NK Receptors and Genetic Deletions
3.4. NK Checkpoint Inhibitors
3.4.1. NKG2A
3.4.2. KIRs
3.4.3. TIM-3
3.4.4. TIGIT
3.5. Engagers
4. Discussion
5. Conclusions and Future Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Kerbauy, L.N.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Brudno, J.N.; Kochenderfer, J.N. Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 2019, 34, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Zakrzewski, J.; James, S.; Sadelain, M. Posttransplant chimeric antigen receptor therapy. Blood 2018, 131, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Yu, W.H.; Poulaki, V.; Tsokos, M.; Stamenkovic, I. Matrix metalloproteinase-7-mediated cleavage of Fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001, 61, 577–581. [Google Scholar]
- Rossi, G.R.; Trindade, E.S.; Souza-Fonseca-Guimaraes, F. Tumor Microenvironment-Associated Extracellular Matrix Components Regulate NK Cell Function. Front. Immunol. 2020, 11, 73. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Clark, R.A.F.; McCoy, G.A.; Folkvord, J.M.; McPherson, J.M. TGF-β1 stimulates cultured human fibroblasts to proliferate and produce tissue-like fibroplasia: A fibronectin matrix-dependent event. J. Cell. Physiol. 1997, 170, 69–80. [Google Scholar] [CrossRef]
- Veglia, F.; Perego, M.; Gabrilovich, D.I. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Marvel, D.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the tumor microenvironment: Expect the unexpected. J. Clin. Investig. 2015, 125, 3356–3364. [Google Scholar] [CrossRef]
- Bruno, A.; Mortara, L.; Baci, D.; Noonan, D.M.; Albini, A. Myeloid Derived Suppressor Cells Interactions With Natural Killer Cells and Pro-angiogenic Activities: Roles in Tumor Progression. Front. Immunol. 2019, 10, 771. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-β1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Burga, R.A.; Thorn, M.; Point, G.R.; Guha, P.; Nguyen, C.T.; Licata, L.A.; DeMatteo, R.P.; Ayala, A.; Espat, N.J.; Junghans, R.P.; et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol. Immunother. 2015, 64, 817–829. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Kuang, W.; Zhou, Q.; Zhang, Y. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int. J. Mol. Med. 2018, 42, 3395–3403. [Google Scholar] [CrossRef] [PubMed]
- Nuñez, S.Y.; Ziblat, A.; Secchiari, F.; Torres, N.I.; Sierra, J.M.; Iraolagoitia, X.L.R.; Araya, R.E.; Domaica, C.I.; Fuertes, M.B.; Zwirner, N.W. Human M2 Macrophages Limit NK Cell Effector Functions through Secretion of TGF-β and Engagement of CD85j. J. Immunol. 2017, 200, 1008–1015. [Google Scholar] [CrossRef]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A Distinct Macrophage Population Mediates Metastatic Breast Cancer Cell Extravasation, Establishment and Growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef]
- Smyth, M.J.; Teng, M.W.L.; Swann, J.; Kyparissoudis, K.; Godfrey, D.I.; Hayakawa, Y. CD4+CD25+ T Regulatory Cells Suppress NK Cell-Mediated Immunotherapy of Cancer. J. Immunol. 2006, 176, 1582–1587. [Google Scholar] [CrossRef]
- Chang, W.-C.; Ling-Hui, C.; Wen-Chun, C.; Huang, P.-S.; Sheu, B.-C.; Huang, S.-C. Regulatory T Cells Suppress Natural Killer Cell Immunity in Patients With Human Cervical Carcinoma. Int. J. Gynecol. Cancer 2016, 26, 156–162. [Google Scholar] [CrossRef]
- Janco, J.M.T.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef]
- Harimoto, H.; Shimizu, M.; Nakagawa, Y.; Nakatsuka, K.; Wakabayashi, A.; Sakamoto, C.; Takahashi, H. Inactivation of tumor-specific CD8+ CTLs by tumor-infiltrating tolerogenic dendritic cells. Immunol. Cell Biol. 2013, 91, 545–555. [Google Scholar] [CrossRef] [PubMed]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Zhang, Y.; Rosenblatt, J.D. B cell regulation of the anti-tumor response and role in carcinogenesis. J. Immunother. Cancer 2016, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Melaiu, O.; Lucarini, V.; Cifaldi, L.; Fruci, D. Influence of the Tumor Microenvironment on NK Cell Function in Solid Tumors. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef]
- Nayyar, G.; Chu, Y.; Cairo, M.S. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 2019, 9, 51. [Google Scholar] [CrossRef]
- Hu, J.; Bernatchez, C.; Zhang, L.; Xia, X.; Kleinerman, E.S.; Hung, M.-C.; Hwu, P.; Li, S. Induction of NKG2D Ligands on Solid Tumors Requires Tumor-Specific CD8+ T Cells and Histone Acetyltransferases. Cancer Immunol. Res. 2017, 5, 300–311. [Google Scholar] [CrossRef]
- Schönfeld, K.; Sahm, C.; Zhang, C.; Naundorf, S.; Brendel, C.; Odendahl, M.; Nowakowska, P.; Bönig, H.; Köhl, U.; Kloess, S.; et al. Selective Inhibition of Tumor Growth by Clonal NK Cells Expressing an ErbB2/HER2-Specific Chimeric Antigen Receptor. Mol. Ther. 2015, 23, 330–338. [Google Scholar] [CrossRef]
- Zhang, C.; Oberoi, P.; Oelsner, S.; Waldmann, A.; Lindner, A.; Tonn, T.; Wels, W.S. Chimeric Antigen Receptor-Engineered NK-92 Cells: An Off-the-Shelf Cellular Therapeutic for Targeted Elimination of Cancer Cells and Induction of Protective Antitumor Immunity. Front. Immunol. 2017, 8, 533. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef]
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.-S.; et al. Novel Human NK Cell Line Carrying CAR Targeting EGFRvIII Induces Antitumor Effects in Glioblastoma Cells. Anticancer. Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.-H.; Grandi, P.; et al. CAR-Engineered NK Cells Targeting Wild-Type EGFR and EGFRvIII Enhance Killing of Glioblastoma and Patient-Derived Glioblastoma Stem Cells. Sci. Rep. 2015, 5, 11483. [Google Scholar] [CrossRef] [PubMed]
- Sahm, C.; Schönfeld, K.; Wels, W. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.M.; Sotillo, E.; Majzner, R.G. CAR T Cell Therapy for Neuroblastoma. Front. Immunol. 2018, 9, 2380. [Google Scholar] [CrossRef]
- Esser, R.; Müller, T.; Stefes, D.; Kloess, S.; Seidel, D.; Gillies, S.D.; Aperlo-Iffland, C.; Huston, J.S.; Uherek, C.; Schönfeld, K.; et al. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell. Mol. Med. 2012, 16, 569–581. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W.; et al. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. OncoImmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [PubMed]
- Specht, J.M.; Lee, S.; Turtle, C.; Berger, C.; Veatch, J.; Gooley, T.; Mullane, E.; Chaney, C.; Riddell, S.; Maloney, D.G. Phase I study of immunotherapy for advanced ROR1+ malignancies with autologous ROR1-specific chimeric antigen receptor-modified (CAR)-T cells. J. Clin. Oncol. 2018, 36, TPS79. [Google Scholar] [CrossRef]
- Park, H.; Awasthi, A.; Ayello, J.; Chu, Y.; Riddell, S.; Rosenblum, J.; Lee, D.S.; Cairo, M.S. ROR1-Specific Chimeric Antigen Receptor (CAR) NK Cell Immunotherapy for High Risk Neuroblastomas and Sarcomas. Biol. Blood Marrow Transpl. 2017, 23, S136–S137. [Google Scholar] [CrossRef][Green Version]
- Xie, G.; Dong, H.; Liang, Y.; Ham, J.D.; Romee, R.; Chen, J. CAR-NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020, 59, 102975. [Google Scholar] [CrossRef]
- Wang, W.; Jiang, J.; Jiang, J. CAR-NK for tumor immunotherapy: Clinical transformation and future prospects. Cancer Lett. 2020, 472, 175–180. [Google Scholar] [CrossRef]
- Oh, S.; Lee, J.H.; Kwack, K.; Choi, S.-W. Natural Killer Cell Therapy: A New Treatment Paradigm for Solid Tumors. Cancers 2019, 11, 1534. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, A.; Huntington, N.D. You Have Got a Fast CAR: Chimeric Antigen Receptor NK Cells in Cancer Therapy. Cancers 2020, 12, 706. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-Based Activating Chimeric Antigen Receptor for NK Cell Tumor Immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Meazza, R.; Azzarone, B.; Orengo, A.M.; Ferrini, S. Role of Common-Gamma Chain Cytokines in NK Cell Development and Function: Perspectives for Immunotherapy. J. Biomed. Biotechnol. 2011, 2011, 1–16. [Google Scholar] [CrossRef]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef]
- Juillerat, A.; Marechal, A.; Filhol, J.M.; Valogne, Y.; Valton, J.; Duclert, A.; Duchateau, P.; Poirot, L. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 2017, 7, srep39833. [Google Scholar] [CrossRef]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-β and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef]
- Friedman, E.; Gold, L.I.; Klimstra, D.; Zeng, Z.S.; Winawer, S.; Cohen, A. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol. Biomark. Prev. 1995, 4, 549–554. [Google Scholar]
- Park, H.; Bang, J.; Nam, A.; Park, J.E.; Jin, M.H.; Bang, Y.; Oh, D.-Y. The prognostic role of soluble TGF-beta and its dynamics in unresectable pancreatic cancer treated with chemotherapy. Cancer Med. 2019, 9, 43–51. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Gao, S.; Zhao, W.-C.; Qiu, B.-A.; Xia, N.-X.; Zhang, P.-J.; Fan, Z. Transforming growth factor-β and peripheral regulatory cells are negatively correlated with the overall survival of hepatocellular carcinoma. World J. Gastroenterol. 2018, 24, 2733–2740. [Google Scholar] [CrossRef] [PubMed]
- Lazarova, M.; Steinle, A. Impairment of NKG2D-Mediated Tumor Immunity by TGF-β. Front. Immunol. 2019, 10, 2689. [Google Scholar] [CrossRef] [PubMed]
- Dahmani, A.; Delisle, J.-S. TGF-β in T Cell Biology: Implications for Cancer Immunotherapy. Cancers 2018, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Trotta, R.; Col, J.D.; Yu, J.; Ciarlariello, D.; Thomas, B.; Zhang, X.; Allard, J.; Wei, M.; Mao, H.; Byrd, J.C.; et al. TGF-β Utilizes SMAD3 to Inhibit CD16-Mediated IFN-γ Production and Antibody-Dependent Cellular Cytotoxicity in Human NK Cells1. J. Immunol. 2008, 181, 3784–3792. [Google Scholar] [CrossRef]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-Derived Transforming Growth Factor- Down-Regulates NKG2D Thereby Inhibiting Natural Killer Cell Antitumor Reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef]
- Castriconi, R.; Cantoni, C.; Della Chiesa, M.; Vitale, M.; Marcenaro, E.; Conte, R.; Biassoni, R.; Bottino, C.; Moretta, L.; Moretta, A. Transforming growth factor 1 inhibits expression of NKp30 and NKG2D receptors: Consequences for the NK-mediated killing of dendritic cells. Proc. Natl. Acad. Sci. USA 2003, 100, 4120–4125. [Google Scholar] [CrossRef]
- Lee, J.-C.; Lee, K.-M.; Kim, D.-W.; Heo, D.S. Elevated TGF-β1 Secretion and Down-Modulation of NKG2D Underlies Impaired NK Cytotoxicity in Cancer Patients. J. Immunol. 2004, 172, 7335–7340. [Google Scholar] [CrossRef]
- Friese, M.A.; Wischhusen, J.; Wick, W.; Weiler, M.; Eisele, G.; Steinle, A.; Weller, M. RNA Interference Targeting Transforming Growth Factor-β Enhances NKG2D-Mediated Antiglioma Immune Response, Inhibits Glioma Cell Migration and Invasiveness, and Abrogates TumorigenicityIn vivo. Cancer Res. 2004, 64, 7596–7603. [Google Scholar] [CrossRef]
- Yvon, E.S.; Burga, R.; Powell, A.; Cruz, C.R.; Fernandes, R.; Barese, C.; Nguyen, T.; Abdel-Baki, M.S.; Bollard, C.M. Cord blood natural killer cells expressing a dominant negative TGF-β receptor: Implications for adoptive immunotherapy for glioblastoma. Cytotherapy 2017, 19, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Liu, H.; Shi, W.; Wang, Z.; Sun, S.; Zhang, G.; Hu, Y.; Liu, T.; Jiao, S. Blocking transforming growth factor-β signaling pathway augments antitumor effect of adoptive NK-92 cell therapy. Int. Immunopharmacol. 2013, 17, 198–204. [Google Scholar] [CrossRef]
- Burga, R.A.; Yvon, E.; Chorvinsky, E.; Fernandes, R.; Cruz, C.R.Y.; Bollard, C.M. Engineering the TGFβ Receptor to Enhance the Therapeutic Potential of Natural Killer Cells as an Immunotherapy for Neuroblastoma. Clin. Cancer Res. 2019, 25, 4400–4412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guo, L.; Song, Y.; Zhang, Y.; Lin, D.; Hu, B.; Mei, Y.; Sandikin, D.; Liu, H. Augmented anti-tumor activity of NK-92 cells expressing chimeric receptors of TGF-βR II and NKG2D. Cancer Immunol. Immunother. 2017, 66, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Raffaghello, L.; Prigione, I.; Airoldi, I.; Camoriano, M.; Levreri, I.; Gambini, C.; Pende, D.; Steinle, A.; Ferrone, S.; Pistoia, V. Downregulation and/or Release of NKG2D Ligands as Immune Evasion Strategy of Human Neuroblastoma. Neoplasia 2004, 6, 558–568. [Google Scholar] [CrossRef]
- Bi, J.; Tian, Z. NK Cell Dysfunction and Checkpoint Immunotherapy. Front. Immunol. 2019, 10, 1999. [Google Scholar] [CrossRef]
- Dasgupta, S.; Bhattacharya-Chatterjee, M.; O’Malley, B.W.; Chatterjee, S.K. Inhibition of NK Cell Activity through TGF-β1 by Down-Regulation of NKG2D in a Murine Model of Head and Neck Cancer. J. Immunol. 2005, 175, 5541–5550. [Google Scholar] [CrossRef]
- Duan, S.; Guo, W.; Xu, Z.; He, Y.; Liang, C.; Mo, Y.; Wang, Y.; Xiong, F.; Guo, C.; Li, Y.; et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Mol. Cancer 2019, 18, 1–14. [Google Scholar] [CrossRef]
- Mistry, A.R.; O’Callaghan, C.A. Regulation of ligands for the activating receptor NKG2D. Immunology 2007, 121, 439–447. [Google Scholar] [CrossRef]
- Harrison, D.; Phillips, J.H.; Lanier, L.L. Involvement of a metalloprotease in spontaneous and phorbol ester-induced release of natural killer cell-associated Fc gamma RIII (CD16-II). J. Immunol. 1991, 147, 3459–3465. [Google Scholar]
- Liu, Y.; Cheng, Y.; Xu, Y.; Wang, Z.; Du, X.; Li, C.; Peng, J.; Gao, L.; Liang, X.; Ma, C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene 2017, 36, 6143–6153. [Google Scholar] [CrossRef]
- Stojanovic, A.; Cerwenka, A. Natural Killer Cells and Solid Tumors. J. Innate Immun. 2011, 3, 355–364. [Google Scholar] [CrossRef]
- Parihar, R.; Rivas, C.H.; Huynh, M.; Omer, B.; Lapteva, N.; Metelitsa, L.S.; Gottschalk, S.M.; Rooney, C.M. NK Cells Expressing a Chimeric Activating Receptor Eliminate MDSCs and Rescue Impaired CAR-T Cell Activity against Solid Tumors. Cancer Immunol. Res. 2019, 7, 363–375. [Google Scholar] [CrossRef]
- Chang, Y.-H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A Chimeric Receptor with NKG2D Specificity Enhances Natural Killer Cell Activation and Killing of Tumor Cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Santana, V.M.; Barfield, R.C. Anti-GD2 Antibody Therapy for GD2-Expressing Tumors. Curr. Cancer Drug Targets 2010, 10, 200–209. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P.; Keating, G.M.; Scott, L.J. Trastuzumab. Drugs 2010, 70, 215–239. [Google Scholar] [CrossRef]
- García-Foncillas, J.; Díaz-Rubio, E. Progress in metastatic colorectal cancer: Growing role of cetuximab to optimize clinical outcome. Clin. Transl. Oncol. 2010, 12, 533–542. [Google Scholar] [CrossRef]
- Winter, M.; Hancock, B.W. Ten years of rituximab in NHL. Expert Opin. Drug Saf. 2009, 8, 223–235. [Google Scholar] [CrossRef]
- Albertini, M.R.; Hank, J.A.; Sondel, P.M. Native and genetically engineered anti-disialoganglioside monoclonal antibody treatment of melanoma. Cancer Chemother. Biol. Response Modif. Annu. 2005, 22, 789–797. [Google Scholar] [CrossRef]
- Yang, R.K.; Sondel, P.M. Anti-GD2 Strategy in the Treatment of Neuroblastoma. Drugs Futur. 2010, 35, 665. [Google Scholar] [CrossRef] [PubMed]
- Parihar, R.; Nadella, P.; Lewis, A.; Jensen, R.; De Hoff, C.; Dierksheide, J.E.; VanBuskirk, A.M.; Magro, C.M.; Young, N.C.; Shapiro, C.L.; et al. A Phase I Study of Interleukin 12 with Trastuzumab in Patients with Human Epidermal Growth Factor Receptor-2-Overexpressing Malignancies: Analysis of Sustained Interferon Production in a Subset of Patients. Clin. Cancer Res. 2004, 10, 5027–5037. [Google Scholar] [CrossRef] [PubMed]
- Fleming, G.F.; Meropol, N.J.; Rosner, G.L.; Hollis, D.R.; Carson, W.E.; Caligiuri, M.; Mortimer, J.; Tkaczuk, K.; Parihar, R.; Schilsky, R.L.; et al. A phase I trial of escalating doses of trastuzumab combined with daily subcutaneous interleukin 2: Report of cancer and leukemia group B 9661. Clin. Cancer Res. 2002, 8, 3718–3727. [Google Scholar] [PubMed]
- Snyder, K.M.; Hullsiek, R.; Mishra, H.K.; Mendez, D.C.; Li, Y.; Rogich, A.; Kaufman, D.S.; Wu, J.; Walcheck, B. Expression of a Recombinant High Affinity IgG Fc Receptor by Engineered NK Cells as a Docking Platform for Therapeutic mAbs to Target Cancer Cells. Front. Immunol. 2018, 9, 2873. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.-F.; et al. Pluripotent stem cell–derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef]
- Pomeroy, E.J.; Hunzeker, J.T.; Kluesner, M.G.; Lahr, W.S.; Smeester, B.A.; Crosby, M.R.; Lonetree, C.-L.; Yamamoto, K.; Bendzick, L.; Miller, J.S.; et al. A Genetically Engineered Primary Human Natural Killer Cell Platform for Cancer Immunotherapy. Mol. Ther. 2020, 28, 52–63. [Google Scholar] [CrossRef]
- Savan, R.; Chan, T.; Young, H.A. Lentiviral gene transduction in human and mouse NK cell lines. Adv. Struct. Saf. Stud. 2010, 612, 209–221. [Google Scholar] [CrossRef]
- Matosevic, S. Viral and Nonviral Engineering of Natural Killer Cells as Emerging Adoptive Cancer Immunotherapies. J. Immunol. Res. 2018, 2018, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Afolabi, L.O.; Adeshakin, A.O.; Sani, M.M.; Bi, J.; Wan, X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology 2019, 158, 63–69. [Google Scholar] [CrossRef]
- Carlsten, M.; Childs, R.W. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front. Immunol. 2015, 6, 266. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immunol. 2020, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e13. [Google Scholar] [CrossRef]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA Class-I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Pende, D.; Tripodi, G.; Tambussi, G.; Viale, O.; Orengo, A.; Barbaresi, M.; Merli, A.; Ciccone, E. Identification of four subsets of human CD3-CD16+ natural killer (NK) cells by the expression of clonally distributed functional surface molecules: Correlation between subset assignment of NK clones and ability to mediate specific alloantigen recognition. J. Exp. Med. 1990, 172, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Sola, C.; Chanuc, F.; Thielens, A.; Fuseri, N.; Morel, Y.; Bléry, M.; André, P.; Vivier, E.; Graziano, R.; Romagne, F.; et al. Anti-tumoral efficacy of therapeutic human anti-KIR antibody (Lirilumab/BMS-986015/IPH2102) in a preclinical xenograft tumor model. J. Immunother. Cancer 2013, 1, P40. [Google Scholar] [CrossRef]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef]
- Zhang, C.-X.; Huang, D.-J.; Baloche, V.; Zhang, L.; Xu, J.-X.; Li, B.-W.; Zhao, X.-R.; He, J.; Mai, H.-Q.; Chen, Q.-Y.; et al. Galectin-9 promotes a suppressive microenvironment in human cancer by enhancing STING degradation. Oncogenesis 2020, 9, 1–14. [Google Scholar] [CrossRef]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef]
- Farkas, A.M.; Audenet, F.; Anastos, H.; Galsky, M.; Sfakianos, J.; Bhardwaj, N. Tim-3 and TIGIT mark Natural Killer cells susceptible to effector dysfunction in human bladder cancer. J. Immunol. 2018, 200, 124.14. [Google Scholar]
- Da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-Cell Exhaustion in Advanced Melanoma by Tim-3 Blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef]
- Xu, L.; Huang, Y.; Tan, L.; Yu, W.; Chen, D.; Lu, C.; He, J.; Wu, G.; Liu, X.; Zhang, Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int. Immunopharmacol. 2015, 29, 635–641. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xu, C. Immune checkpoint signaling and cancer immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Liu, T.; Wang, Z.; Ma, M.; Lei, J.; Zhang, Z.; Fu, S.; Fu, Y.; Hu, Q.; Ding, H.; et al. Expression of the Inhibitory Receptor TIGIT Is Up-Regulated Specifically on NK Cells With CD226 Activating Receptor From HIV-Infected Individuals. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.J.; Evert, J.S.H.-V.; Van Der Meer, J.M.; Mekers, V.; Rezaeifard, S.; Korman, A.J.; De Jonge, P.K.; Cany, J.; Woestenenk, R.; Schaap, N.P.; et al. TIGIT blockade enhances functionality of peritoneal NK cells with altered expression of DNAM-1/TIGIT/CD96 checkpoint molecules in ovarian cancer. OncoImmunology 2020, 9, 1843247. [Google Scholar] [CrossRef] [PubMed]
- Judge, S.J.; Darrow, M.A.; Thorpe, S.W.; Gingrich, A.A.; O’Donnell, E.F.; Bellini, A.R.; Sturgill, I.R.; Vick, L.V.; Dunai, C.; Stoffel, K.M.; et al. Analysis of tumor-infiltrating NK and T cells highlights IL-15 stimulation and TIGIT blockade as a combination immunotherapy strategy for soft tissue sarcomas. J. Immunother. Cancer 2020, 8, e001355. [Google Scholar] [CrossRef]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16xCD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef]
- Sarhan, D.; Brandt, L.; Felices, M.; Guldevall, K.; Lenvik, T.; Hinderlie, P.; Curtsinger, J.; Warlick, E.; Spellman, S.R.; Blazar, B.R.; et al. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018, 2, 1459–1469. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.L.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef]
- Vallera, D.A.; Ferrone, S.; Kodal, B.; Hinderlie, P.; Bendzick, L.; Ettestad, B.; Hallstrom, C.; Zorko, N.A.; Rao, A.; Fujioka, N.; et al. NK-Cell-Mediated Targeting of Various Solid Tumors Using a B7-H3 Tri-Specific Killer Engager In Vitro and In Vivo. Cancers 2020, 12, 2659. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Taras, E.; Miller, J.S.; Vallera, D.A. Enhanced ADCC and NK Cell Activation of an Anticarcinoma Bispecific Antibody by Genetic Insertion of a Modified IL-15 Cross-linker. Mol. Ther. 2016, 24, 1312–1322. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Wang, G.; Huang, D.-S.; Sui, M.; Xu, Y. Cancer Immunotherapy Based on Natural Killer Cells: Current Progress and New Opportunities. Front. Immunol. 2019, 10, 1205. [Google Scholar] [CrossRef] [PubMed]
- Minetto, P.; Guolo, F.; Pesce, S.; Greppi, M.; Obino, V.; Ferretti, E.; Sivori, S.; Genova, C.; Lemoli, R.M.; Marcenaro, E. Harnessing NK Cells for Cancer Treatment. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Lapteva, N.; Szmania, S.M.; Van Rhee, F.; Rooney, C.M. Clinical Grade Purification and Expansion of Natural Killer Cells. Crit. Rev. Oncog. 2013, 19, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kaufman, D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. Methods Mol. Biol. 2019, 2048, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.; Berg, M.; Davidsonmoncada, J.K.; Tian, X.; Cullis, H.; Childs, R.W. A novel method to expand large numbers of CD56+ natural killer cells from a minute fraction of selectively accessed cryopreserved cord blood for immunotherapy after transplantation. Cytotherapy 2015, 17, 1582–1593. [Google Scholar] [CrossRef]
- Ojo, E.O.; Sharma, A.A.; Liu, R.; Moreton, S.; Checkley-Luttge, M.-A.; Gupta, K.; Lee, G.; Lee, D.A.; Otegbeye, F.; Sekaly, R.-P.; et al. Membrane bound IL-21 based NK cell feeder cells drive robust expansion and metabolic activation of NK cells. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; Denhollander, N.; Gupta, V.; Wang, X.-H.; Chaboureau, A.; Viswanathan, S.; Keating, A. A phase I trial of NK-92 cells for refractory hematological malignancies relapsing after autologous hematopoietic cell transplantation shows safety and evidence of efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef]
- Matsuo, Y.; Drexler, H.G. Immunoprofiling of cell lines derived from natural killer-cell and natural killer-like T-cell leukemia–lymphoma. Leuk. Res. 2003, 27, 935–945. [Google Scholar] [CrossRef]
- Jochems, C.; Hodge, J.W.; Fantini, M.; Fujii, R.; Ii, Y.M.M.; Greiner, J.W.; Padget, M.R.; Tritsch, S.R.; Tsang, K.Y.; Campbell, K.S.; et al. An NK cell line (haNK) expressing high levels of granzyme and engineered to express the high affinity CD16 allele. Oncotarget 2016, 7, 86359–86373. [Google Scholar] [CrossRef]
- Zhang, J.; Zheng, H.; Diao, Y. Natural Killer Cells and Current Applications of Chimeric Antigen Receptor-Modified NK-92 Cells in Tumor Immunotherapy. Int. J. Mol. Sci. 2019, 20, 317. [Google Scholar] [CrossRef] [PubMed]
- Borst, L.; Van Der Burg, S.H.; Van Hall, T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer Res. 2020, 26, 5549–5556. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; DeSantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tian, Z.-G.; Zhang, C. Chimeric antigen receptor (CAR)-transduced natural killer cells in tumor immunotherapy. Acta Pharm. Ther. Sin. 2017, 39, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Janelle, V. The Bumpy Road to CAR Activation. Mol. Ther. 2019, 27, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Poli, A.; Michel, T.; Thérésine, M.; Andrès, E.; Hentges, F.; Zimmer, J. CD56brightnatural killer (NK) cells: An important NK cell subset. Immunology 2008, 126, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.A.; Rosario, M.; Romee, R.; Berrien-Elliott, M.M.; Schneider, S.E.; Leong, J.W.; Sullivan, R.P.; Jewell, B.A.; Becker-Hapak, M.; Schappe, T.; et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J. Clin. Investig. 2017, 127, 4042–4058. [Google Scholar] [CrossRef]
- Gunesch, J.T.; Dixon, A.L.; Ebrahim, T.A.; Berrien-Elliott, M.M.; Tatineni, S.; Kumar, T.; Hegewisch-Solloa, E.; Fehniger, T.A.; Mace, E.M. CD56 regulates human NK cell cytotoxicity through Pyk2. eLife 2020, 9. [Google Scholar] [CrossRef]
- De Jonge, K.; Ebering, A.; Nassiri, S.; Hajjami, H.M.-E.; Ouertatani-Sakouhi, H.; Baumgaertner, P.; Speiser, D.E. Circulating CD56bright NK cells inversely correlate with survival of melanoma patients. Sci. Rep. 2019, 9, 4487. [Google Scholar] [CrossRef]
- Vahedi, F.; Nham, T.; Poznanski, S.M.; Chew, M.V.; Shenouda, M.M.; Lee, D.; Ashkar, A. Ex Vivo Expanded Human NK Cells Survive and Proliferate in Humanized Mice with Autologous Human Immune Cells. Sci. Rep. 2017, 7, 12083. [Google Scholar] [CrossRef]
- Hoogi, S.; Eisenberg, V.; Mayer, S.; Shamul, A.; Barliya, T.; Cohen, C.J. A TIGIT-based chimeric co-stimulatory switch receptor improves T-cell anti-tumor function. J. Immunother. Cancer 2019, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ranganathan, R.; Jiang, S.; Fang, C.; Sun, J.; Kim, S.; Newick, K.; Lo, A.; June, C.H.; Zhao, Y.; et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016, 76, 1578–1590. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019, 9, 2064–2078. [Google Scholar] [PubMed]
- Sambi, M.; Bagheri, L.; Szewczuk, M.R. Current Challenges in Cancer Immunotherapy: Multimodal Approaches to Improve Efficacy and Patient Response Rates. J. Oncol. 2019, 2019, 1–12. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived Suppressor Cells Hinder the Anti-Cancer Activity of Immune Checkpoint Inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Trombetta, A.C.; Soldano, S.; Contini, P.; Tomatis, V.; Ruaro, B.; Paolino, S.; Brizzolara, R.; Montagna, P.; Sulli, A.; Pizzorni, C.; et al. A circulating cell population showing both M1 and M2 monocyte/macrophage surface markers characterizes systemic sclerosis patients with lung involvement. Respir. Res. 2018, 19, 186. [Google Scholar] [CrossRef]
- Kararoudi, M.N.; Dolatshad, H.; Trikha, P.; Hussain, S.-R.A.; Elmas, E.; Foltz, J.A.; Moseman, J.E.; Thakkar, A.; Nakkula, R.J.; Lamb, M.; et al. Generation of Knock-out Primary and Expanded Human NK Cells Using Cas9 Ribonucleoproteins. J. Vis. Exp. 2018, e58237. [Google Scholar] [CrossRef]
- Kitada, T.; DiAndreth, B.; Teague, B.; Weisst, R. Programming gene and engineered-cell therapies with synthetic biology. Science 2018, 359, eaad1067. [Google Scholar] [CrossRef]
- Kakarla, S.; Chow, K.K.H.; Mata, M.; Shaffer, D.R.; Song, X.-T.; Wu, M.-F.; Liu, H.; Wang, L.L.; Rowley, D.R.; Pfizenmaier, K.; et al. Antitumor Effects of Chimeric Receptor Engineered Human T Cells Directed to Tumor Stroma. Mol. Ther. 2013, 21, 1611–1620. [Google Scholar] [CrossRef]
- Tran, E.; Chinnasamy, D.; Yu, Z.; Morgan, R.A.; Lee, C.-C.R.; Restifo, N.P.; Rosenberg, S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013, 210, 1125–1135. [Google Scholar] [CrossRef]
- Schuberth, P.C.; Hagedorn, C.; Jensen, S.M.; Gulati, P.; Broek, M.V.D.; Mischo, A.; Soltermann, A.; Jüngel, A.; Belaunzaran, O.M.; Stahel, R.A.; et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013, 11, 187. [Google Scholar] [CrossRef] [PubMed]
- Brodin, P.; Lakshmikanth, T.; Johansson, S.; Kärre, K.; Höglund, P. The strength of inhibitory input during education quantitatively tunes the functional responsiveness of individual natural killer cells. Blood 2009, 113, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Carisey, A.F.; Mace, E.M.; Saeed, M.B.; Davis, D.M.; Orange, J.S. Nanoscale Dynamism of Actin Enables Secretory Function in Cytolytic Cells. Curr. Biol. 2018, 28, 489–502.e9. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, M.R.; Tsun, A.; Stinchcombe, J.C.; Griffiths, G.M. The Strength of T Cell Receptor Signal Controls the Polarization of Cytotoxic Machinery to the Immunological Synapse. Immunology 2009, 31, 621–631. [Google Scholar] [CrossRef]
- Beal, A.M.; Anikeeva, N.; Varma, R.; Cameron, T.O.; Vasiliver-Shamis, G.; Norris, P.J.; Dustin, M.L.; Sykulev, Y. Kinetics of Early T Cell Receptor Signaling Regulate the Pathway of Lytic Granule Delivery to the Secretory Domain. Immunology 2009, 31, 632–642. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; Van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Suarez, E.R.; Chang, D.-K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef]
- Ng, Y.Y.; Tay, J.C.; Wang, S. CXCR1 Expression to Improve Anti-Cancer Efficacy of Intravenously Injected CAR-NK Cells in Mice with Peritoneal Xenografts. Mol. Ther. Oncolytics 2020, 16, 75–85. [Google Scholar] [CrossRef]
- Costa, E.C.; Moreira, A.F.; De Melo-Diogo, D.; Gaspar, V.M.; Carvalho, M.P.; Correia, I. 3D tumor spheroids: An overview on the tools and techniques used for their analysis. Biotechnol. Adv. 2016, 34, 1427–1441. [Google Scholar] [CrossRef]
- Wang, M.; Yao, L.; Cheng, M.; Cai, D.; Martinek, J.; Pan, C.; Shi, W.; Ma, A.; White, R.W.D.V.; Airhart, S.; et al. Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J. 2018, 32, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Shuen, T.W.H.; Toh, T.B.; Chan, X.Y.; Liu, M.; Tan, S.Y.; Fan, Y.; Yang, H.; Lyer, S.G.; Bonney, G.K.; et al. Development of a new patient-derived xenograft humanised mouse model to study human-specific tumour microenvironment and immunotherapy. Gut 2018, 67, 1845–1854. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Huang, G.; Cheng, L.; Li, Z.; Xiao, Y.; Deng, Q.; Jiang, Y.; Li, B.; Lin, S.; Wang, S.; et al. Establishment of peripheral blood mononuclear cell-derived humanized lung cancer mouse models for studying efficacy of PD-L1/PD-1 targeted immunotherapy. mAbs 2018, 10, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Martins, F.; Sofiya, L.; Sykiotis, G.P.; Lamine, F.; Maillard, M.; Fraga, M.; Shabafrouz, K.; Ribi, C.; Cairoli, A.; Guex-Crosier, Y.; et al. Adverse effects of immune-checkpoint inhibitors: Epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019, 16, 563–580. [Google Scholar] [CrossRef]
- Oliva, A.; Kinter, A.L.; Vaccarezza, M.; Rubbert, A.; Catanzaro, A.; Moir, S.; Monaco, J.; Ehler, L.; Mizell, S.; Jackson, R.; et al. Natural killer cells from human immunodeficiency virus (HIV)-infected individuals are an important source of CC-chemokines and suppress HIV-1 entry and replication in vitro. J. Clin. Investig. 1998, 102, 223–231. [Google Scholar] [CrossRef]
- A Fehniger, T.; Shah, M.H.; Turner, M.J.; VanDeusen, J.B.; Whitman, S.P.; A Cooper, M.; Suzuki, K.; Wechser, M.; Goodsaid, F.; Caligiuri, M.A. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12: Implications for the innate immune response. J. Immunol. 1999, 162, 4511–4520. [Google Scholar]
- Roda, J.M.; Parihar, R.; Magro, C.; Nuovo, G.J.; Tridandapani, S.; Carson, W.E. Natural Killer Cells Produce T Cell–Recruiting Chemokines in Response to Antibody-Coated Tumor Cells. Cancer Res. 2006, 66, 517–526. [Google Scholar] [CrossRef]
- Wang, J.; Matosevic, S. Adenosinergic signaling as a target for natural killer cell immunotherapy. J. Mol. Med. 2018, 96, 903–913. [Google Scholar] [CrossRef]
- Akhiani, A.A.; Hallner, A.; Kiffin, R.; Aydin, E.; Werlenius, O.; Aurelius, J.; Martner, A.; Thorén, F.B.; Hellstrand, K. Idelalisib Rescues Natural Killer Cells from Monocyte-Induced Immunosuppression by Inhibiting NOX2-Derived Reactive Oxygen Species. Cancer Immunol. Res. 2020, 8, 1532–1541. [Google Scholar] [CrossRef]
- Cekic, C.; Day, Y.-J.; Sag, D.; Linden, J. Myeloid Expression of Adenosine A2A Receptor Suppresses T and NK Cell Responses in the Solid Tumor Microenvironment. Cancer Res. 2014, 74, 7250–7259. [Google Scholar] [CrossRef]
- Ghalamfarsa, G.; Kazemi, M.H.; Mohseni, S.R.; Masjedi, A.; Hojjat-Farsangi, M.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. CD73 as a potential opportunity for cancer immunotherapy. Expert Opin. Ther. Targets 2018, 23, 127–142. [Google Scholar] [CrossRef]
- Neo, S.Y.; Yang, Y.; Record, J.; Ma, R.; Chen, X.; Chen, Z.; Tobin, N.P.; Blake, E.; Seitz, C.; Thomas, R.; et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J. Clin. Investig. 2020, 130, 1185–1198. [Google Scholar] [CrossRef] [PubMed]
- Michelet, X.; Dyck, L.; Hogan, A.; Loftus, R.M.; Duquette, D.; Wei, K.; Beyaz, S.; Tavakkoli, A.; Foley, C.; Donnelly, R.; et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat. Immunol. 2018, 19, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Tobin, L.M.; Mavinkurve, M.; Carolan, E.; Kinlen, D.; O’Brien, E.C.; Little, M.A.; Finlay, D.K.; Cody, D.; Hogan, A.E.; O’Shea, D. NK cells in childhood obesity are activated, metabolically stressed, and functionally deficient. JCI Insight 2017, 2, 2. [Google Scholar] [CrossRef] [PubMed]
- Harmon, C.; Robinson, M.W.; Hand, F.; AlMuaili, D.; Mentor, K.; Houlihan, D.D.; Hoti, E.; Lynch, L.; Geoghegan, J.; O’Farrelly, C. Lactate-Mediated Acidification of Tumor Microenvironment Induces Apoptosis of Liver-Resident NK Cells in Colorectal Liver Metastasis. Cancer Immunol. Res. 2018, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Pouyssegur, J. Tumor Microenvironment: A Metabolic Player that Shapes the Immune Response. Int. J. Mol. Sci. 2019, 21, 157. [Google Scholar] [CrossRef] [PubMed]
- Pająk, B.; Siwiak, E.; Sołtyka, M.; Priebe, A.; Zieliński, R.; Fokt, I.; Ziemniak, M.; Jaśkiewicz, A.; Borowski, R.; Domoradzki, T.; et al. 2-Deoxy-d-Glucose and Its Analogs: From Diagnostic to Therapeutic Agents. Int. J. Mol. Sci. 2019, 21, 234. [Google Scholar] [CrossRef]
- Xu, X.; Meng, Y.; Li, L.; Xu, P.; Wang, J.; Li, Z.; Bian, J. Overview of the Development of Glutaminase Inhibitors: Achievements and Future Directions. J. Med. Chem. 2019, 62, 1096–1115. [Google Scholar] [CrossRef]
- Dhankhar, R.; Gupta, V.; Kumar, S.; Kapoor, R.K.; Gulati, P. Microbial Enzymes for Deprivation of Amino Acid Metabolism in Malignant Cells: Biological Strategy for Cancer Treatment. Available online: https://pubmed.ncbi.nlm.nih.gov/32037468/ (accessed on 1 December 2020).
- Mah, A.Y.; Rashidi, A.; Keppel, M.P.; Saucier, N.; Moore, E.K.; Alinger, J.B.; Tripathy, S.K.; Agarwal, S.K.; Jeng, E.K.; Wong, H.C.; et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight 2017, 2, 2. [Google Scholar] [CrossRef]
- Ando, M.; Sugimoto, K.; Itoh, Y.; Sasaki, M.; Mukai, K.; Ando, J.; Egashira, M.; Schuster, S.M.; Oshimi, K. Selective apoptosis of natural killer-cell tumours by l-asparaginase. Br. J. Haematol. 2005, 130, 860–868. [Google Scholar] [CrossRef]


{kind=link}
| Design Strategy | Target | Tumor Model | Origin of NK Cell | Reference |
|---|---|---|---|---|
| NK-Centric CAR | Mesothelin | Ovarian carcinoma | iPSC-derived NK cells | [44] |
| PSCA | Kidney cell transplant | YTS | [45] | |
| Armored CAR | GD2 | Neuroblastoma | NK-T cells | [48] |
| Inducible CAR | CD19 | Lymphoma | T cells | [49] |
| Dominant Negative Receptors | TGF-β | Glioblastoma | Cord blood-derived NK cells | [61] |
| TGF-β | Lung carcinoma | NK-92 | [62] | |
| Switch Receptors | TGF-β | Neuroblastoma Hepatocellular carcinoma, Prostate carcinoma | Cord blood-derived NK cells | [63] |
| TGF-β | NK-92 | [64] | ||
| Chimeric NK cell receptors | NKG2DL | Neuroblastoma | Primary NK cells | [73] |
| NKG2DL | Osteosarcoma | Primary NK cells | [74] | |
| NKG2DL | Colorectal carcinoma | Primary NK cells | [75] | |
| Enhanced Fcγ receptors | CD64/CD16A | Ovarian carcinoma | iPSC-derived NK cells, NK-92 | [84] |
| CD16A | Ovarian carcinoma | iPSC-derived NK cells | [85] | |
| Genetic Deletions | PD-1 | Ovarian carcinoma | Primary NK cells | [86] |
| Checkpoint Blockade | NKG2A | Head and neck carcinoma | - | [92] |
| TIM-3 | Lung adenocarcinoma, Melanoma | - | [100,101] | |
| TIGIT | Ovarian carcinoma, Ewing’s sarcoma, leiomyosarcoma | - | [105,106] | |
| BiKEs and TriKEs | CD33 | Myelodysplastic syndrome | - | [107,108] |
| CD33 | Acute myeloid leukemia | - | [109] | |
| B7-H3 | Ovarian carcinoma | - | [110] | |
| EpCAM | Colorectal carcinoma | - | [111] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navin, I.; Lam, M.T.; Parihar, R. Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment. Cancers 2020, 12, 3871. https://doi.org/10.3390/cancers12123871
Navin I, Lam MT, Parihar R. Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment. Cancers. 2020; 12(12):3871. https://doi.org/10.3390/cancers12123871
Chicago/Turabian StyleNavin, Ishwar, Michael T. Lam, and Robin Parihar. 2020. "Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment" Cancers 12, no. 12: 3871. https://doi.org/10.3390/cancers12123871
APA StyleNavin, I., Lam, M. T., & Parihar, R. (2020). Design and Implementation of NK Cell-Based Immunotherapy to Overcome the Solid Tumor Microenvironment. Cancers, 12(12), 3871. https://doi.org/10.3390/cancers12123871