Proteomic Resistance Biomarkers for PI3K Inhibitor in Triple Negative Breast Cancer Patient-Derived Xenograft Models

 , , , , ,
, , , , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
2.1. Generation and Characterization of TNBC PDX Models
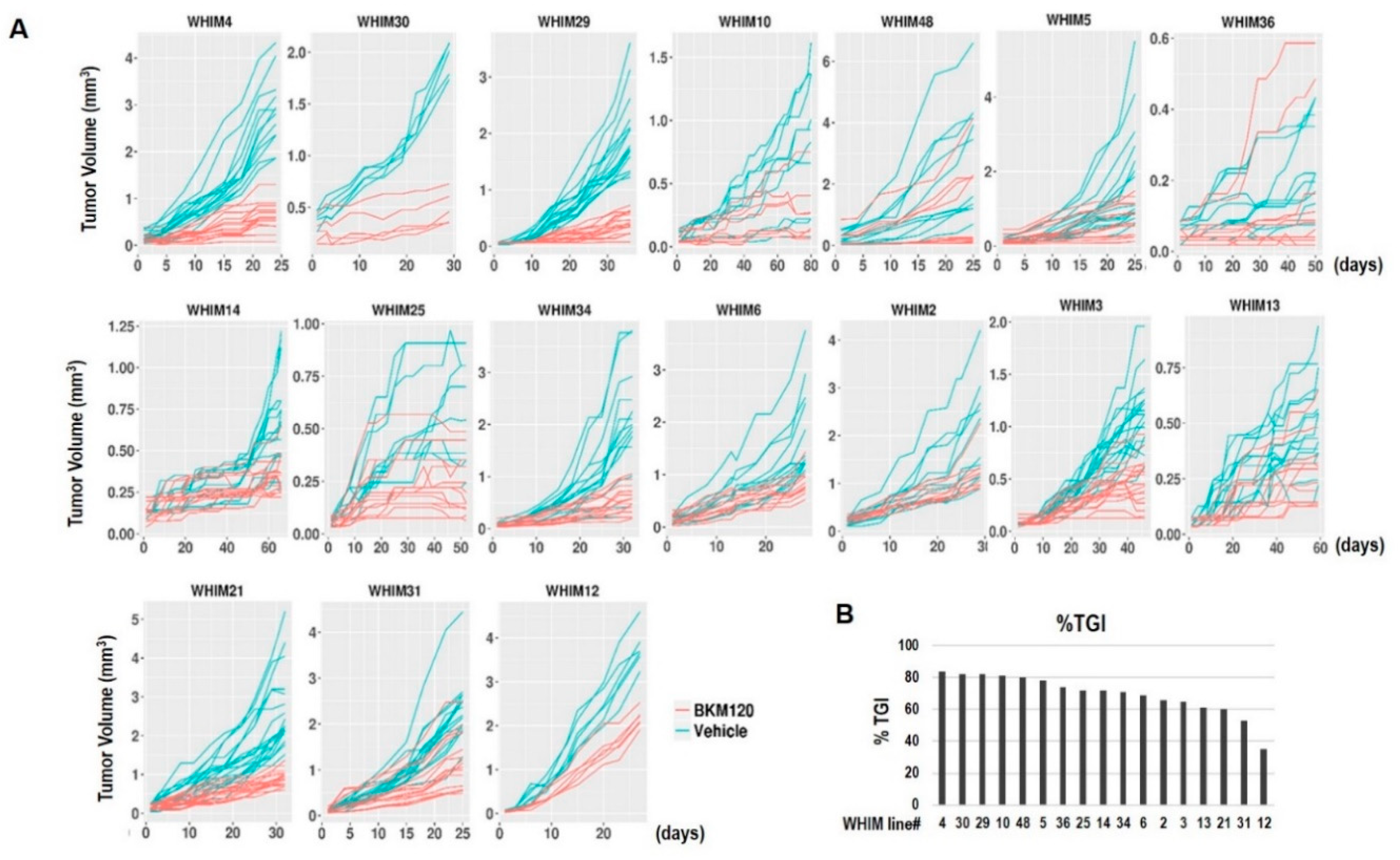
2.2. Tumor Growth Response to BKM120 in TNBC PDX Models
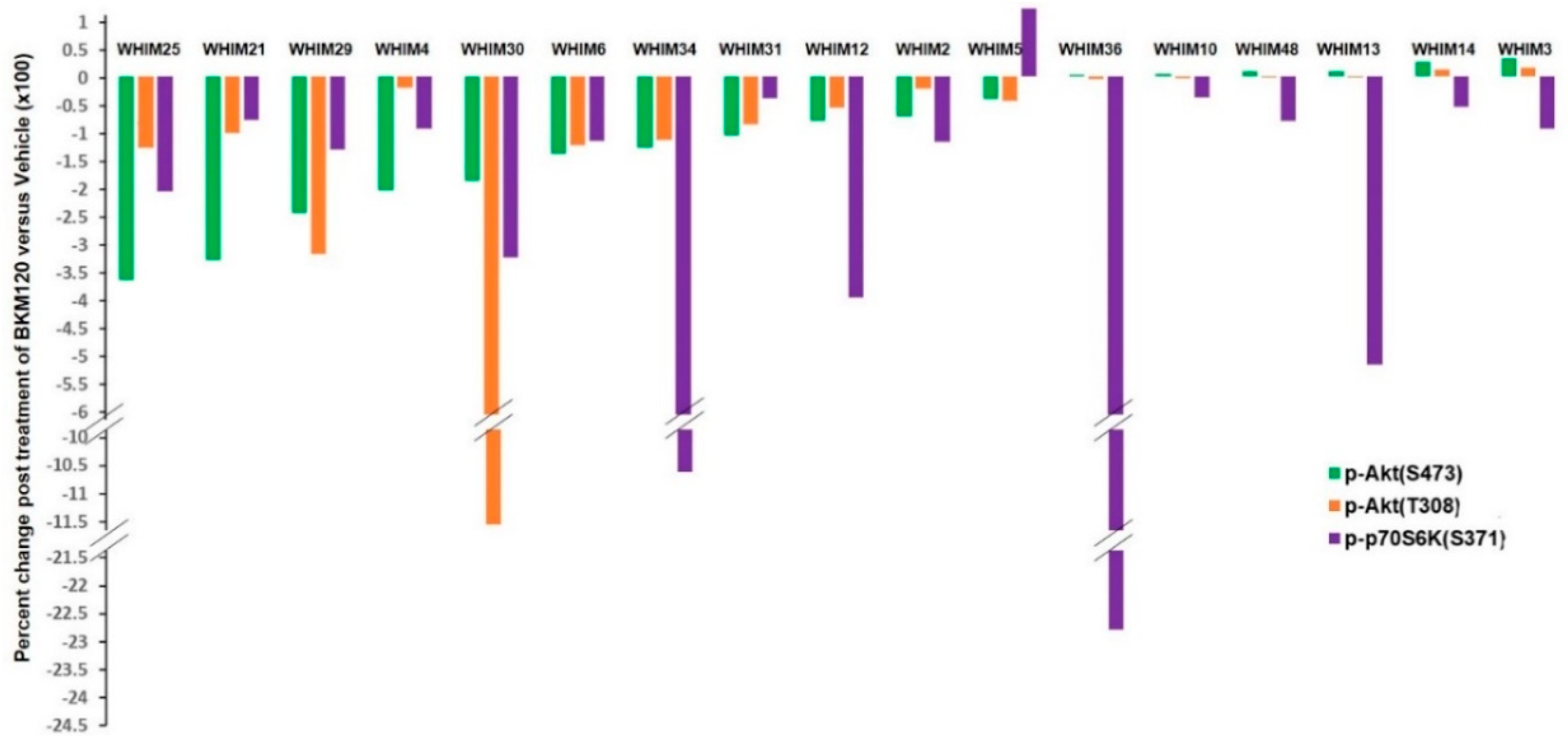
2.3. RPPA Analysis Indicating PI3K Pathway Inhibition by BKM120
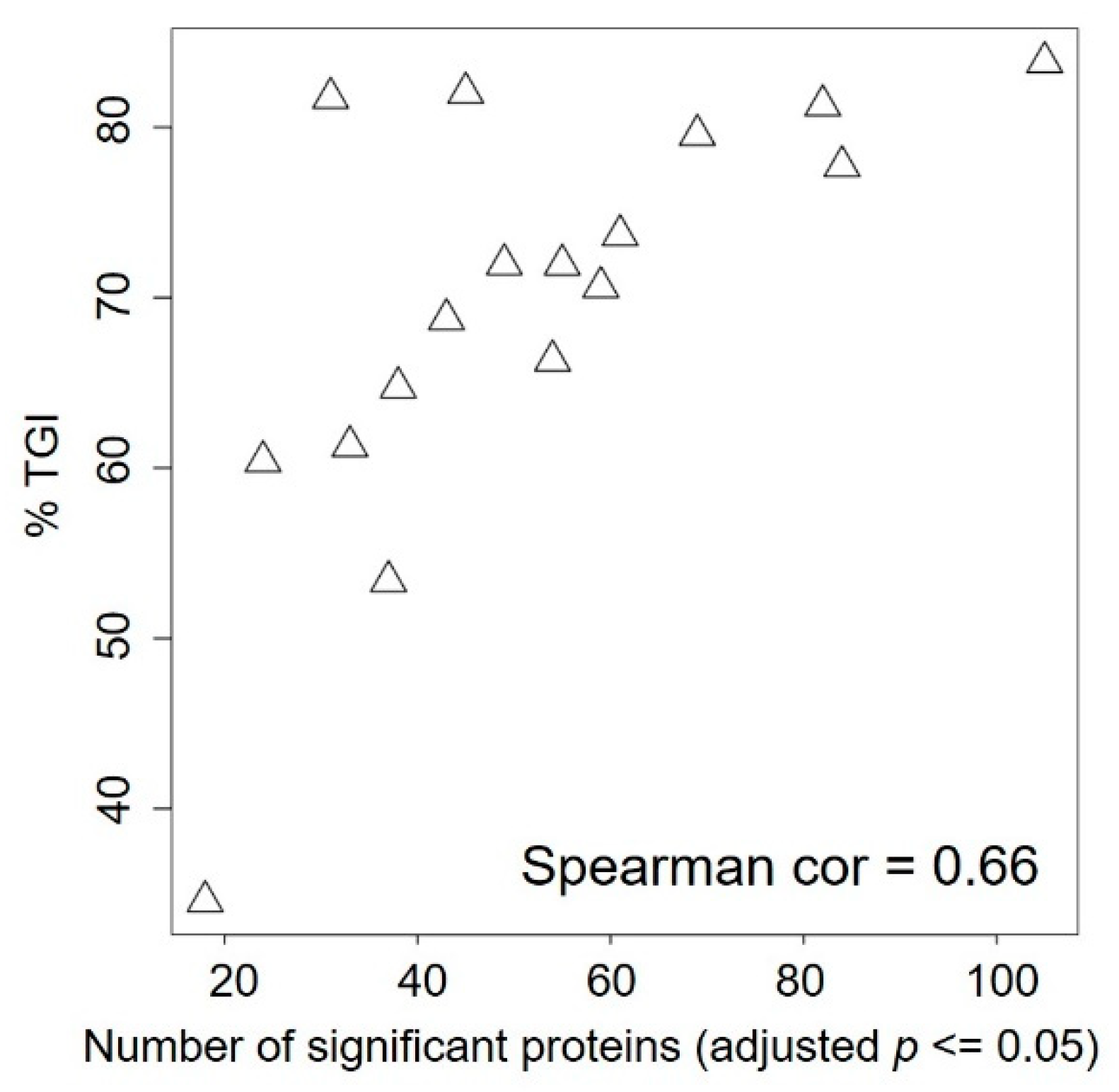
2.4. Baseline Proteomic Analysis Reveals Potential Resistance Biomarkers to BKM120
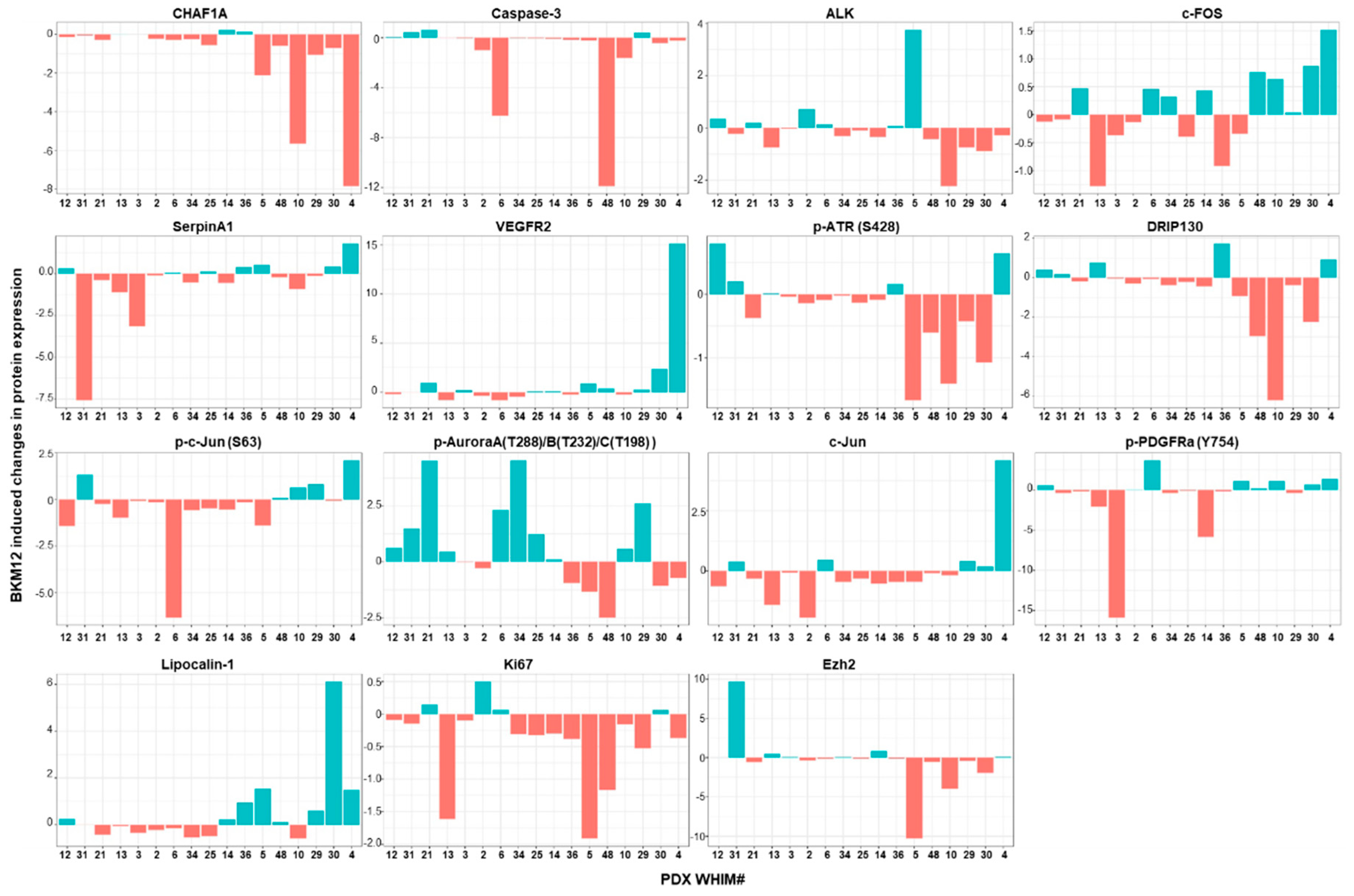
2.5. Treatment-Induced Proteomic Changes Reveal Potential Resistance Biomarkers to BKM120
3. Discussion
4. Materials and Methods
4.1. Chemicals and Antibodies
4.2. Generation of PDX Models
4.3. Immunohistochemistry
4.4. mRNA Gene Expression Analysis Using Agilent 4X44K Arrays
4.5. Whole Exome Sequencing Analysis
4.6. Reverse Phase Protein Array (RPPA) and PI3K Signature Score
4.7. Treatments of Patient-Derived Triple Negative Breast Cancer Xenograft Models
4.8. In Vitro Cytotoxic Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hudis, C.A.; Gianni, L. Triple-Negative Breast Cancer: An Unmet Medical Need. Oncologist 2011, 16, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crystal, A.S.; Shaw, A.T.; Sequist, L.V.; Friboulet, L.; Niederst, M.J.; Lockerman, E.L.; Frias, R.L.; Gainor, J.F.; Amzallag, A.; Greninger, P.; et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science 2014, 346, 1480–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, N.M.; Coon, J.J. Phosphoproteomics in the Age of Rapid and Deep Proteome Profiling. Anal. Chem. 2016, 88, 74–94. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosfjord, E.; Lucas, J.; Li, G.; Gerber, H.P. Advances in patient-derived tumor xenografts: From target identification to predicting clinical response rates in oncology. Biochem. Pharm. 2014, 91, 135–143. [Google Scholar] [CrossRef]
- Li, S.; Shen, D.; Shao, J.; Crowder, R.; Liu, W.; Prat, A.; He, X.; Liu, S.; Hoog, J.; Lu, C.; et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013, 4, 1116–1130. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ellis, M.J.; Li, S.; Larson, D.E.; Chen, K.; Wallis, J.W.; Harris, C.C.; McLellan, M.D.; Fulton, R.S.; Fulton, L.L.; et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 2010, 464, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-L.; Li, S.; Mertins, P.; Cao, S.; Gunawardena, H.P.; Ruggles, K.V.; Mani, D.R.; Clauser, K.R.; Tanioka, M.; Usary, J.; et al. Proteogenomic integration reveals therapeutic targets in breast cancer xenografts. Nat. Commun. 2017, 8, 14864. [Google Scholar] [CrossRef]
- Gao, H.; Korn, J.M.; Ferretti, S.; Monahan, J.E.; Wang, Y.; Singh, M.; Zhang, C.; Schnell, C.; Yang, G.; Zhang, Y.; et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat. Med. 2015, 21, 1318–1325. [Google Scholar] [CrossRef]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Marty, B.; Maire, V.; Gravier, E.; Rigaill, G.; Vincent-Salomon, A.; Kappler, M.; Lebigot, I.; Djelti, F.; Tourdes, A.; Gestraud, P.; et al. Frequent PTEN genomic alterations and activated phosphatidylinositol 3-kinase pathway in basal-like breast cancer cells. Breast Cancer Res. 2008, 10, R101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Knowles, E.; O’Toole, S.A.; McNeil, C.M.; Millar, E.K.A.; Qiu, M.R.; Crea, P.; Daly, R.J.; Musgrove, E.A.; Sutherland, R.L. PI3K pathway activation in breast cancer is associated with the basal-like phenotype and cancer-specific mortality. Int. J. Cancer 2010, 126, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.P.; Roth, A.; Goya, R.; Oloumi, A.; Ha, G.; Zhao, Y.; Turashvili, G.; Ding, J.; Tse, K.; Haffari, G.; et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 2012, 486, 395–399. [Google Scholar] [CrossRef]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [Green Version]
- Dourdin, N.; Schade, B.; Lesurf, R.; Hallett, M.; Munn, R.J.; Cardiff, R.D.; Muller, W.J. Phosphatase and tensin homologue deleted on chromosome 10 deficiency accelerates tumor induction in a mouse model of ErbB-2 mammary tumorigenesis. Cancer Res. 2008, 68, 2122–2131. [Google Scholar] [CrossRef] [Green Version]
- Saal, L.H.; Gruvberger-Saal, S.K.; Persson, C.; Lovgren, K.; Jumppanen, M.; Staaf, J.; Jonsson, G.; Pires, M.M.; Maurer, M.; Holm, K.; et al. Recurrent gross mutations of the PTEN tumor suppressor gene in breast cancers with deficient DSB repair. Nat. Genet. 2008, 40, 102–107. [Google Scholar] [CrossRef]
- Garrido-Castro, A.C.; Saura, C.; Barroso-Sousa, R.; Guo, H.; Ciruelos, E.; Bermejo, B.; Gavila, J.; Serra, V.; Prat, A.; Pare, L.; et al. Phase 2 study of buparlisib (BKM120), a pan-class I PI3K inhibitor, in patients with metastatic triple-negative breast cancer. Breast Cancer Res. 2020, 22, 120. [Google Scholar] [CrossRef]
- Rodon, J.; Brana, I.; Siu, L.L.; De Jonge, M.J.; Homji, N.; Mills, D.; Di Tomaso, E.; Sarr, C.; Trandafir, L.; Massacesi, C.; et al. Phase I dose-escalation and -expansion study of buparlisib (BKM120), an oral pan-Class I PI3K inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2014, 32, 670–681. [Google Scholar] [CrossRef]
- Brachmann, S.M.; Kleylein-Sohn, J.; Gaulis, S.; Kauffmann, A.; Blommers, M.J.; Kazic-Legueux, M.; Laborde, L.; Hattenberger, M.; Stauffer, F.; Vaxelaire, J.; et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol. Cancer 2012, 11, 1747–1757. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Mooradian, A.D.; Erdmann-Gilmore, P.; Zhang, Q.; Viner, R.; Davies, S.R.; Huang, K.-L.; Bomgarden, R.; Van Tine, B.A.; Shao, J.; et al. Breast tumors educate the proteome of stromal tissue in an individualized but coordinated manner. Sci. Signal 2017, 10, eaam8065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mundt, F.; Rajput, S.; Li, S.; Ruggles, K.V.; Mooradian, A.D.; Mertins, P.; Gillette, M.A.; Krug, K.; Guo, Z.; Hoog, J.; et al. Mass Spectrometry-Based Proteomics Reveals Potential Roles of NEK9 and MAP2K4 in Resistance to PI3K Inhibition in Triple-Negative Breast Cancers. Cancer Res. 2018, 78, 2732–2746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, K.W.; Yuca, E.; Akcakanat, A.; Scott, S.M.; Arango, N.P.; Zheng, X.; Chen, K.; Tapia, C.; Tarco, E.; Eterovic, A.K.; et al. A Population of Heterogeneous Breast Cancer Patient-Derived Xenografts Demonstrate Broad Activity of PARP Inhibitor in BRCA1/2 Wild-Type Tumors. Clin. Cancer Res. 2017, 23, 6468–6477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanker, A.B.; Kaklamani, V.; Arteaga, C.L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discov. 2019, 9, 482–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, E.M.; Kuba, M.G.; Miller, T.W.; Davies, B.R.; Arteaga, C.L. Autocrine IGF-I/insulin receptor axis compensates for inhibition of AKT in ER-positive breast cancer cells with resistance to estrogen deprivation. Breast Cancer Res. 2013, 15, R55. [Google Scholar] [CrossRef] [Green Version]
- Garrett, J.T.; Sutton, C.R.; Kurupi, R.; Bialucha, C.U.; Ettenberg, S.A.; Collins, S.D.; Sheng, Q.; Wallweber, J.; DeFazio-Eli, L.; Arteaga, C.L. Combination of Antibody That Inhibits Ligand-Independent HER3 Dimerization and a p110α Inhibitor Potently Blocks PI3K Signaling and Growth of HER2+ Breast Cancers. Cancer Res. 2013, 73, 6013–6023. [Google Scholar] [CrossRef] [Green Version]
- Rajput, S.; Guo, Z.; Li, S.; Ma, C.X. PI3K inhibition enhances the anti-tumor effect of eribulin in triple negative breast cancer. Oncotarget 2019, 10, 3667–3680. [Google Scholar] [CrossRef]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013, 19, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, M.; Aust, M.M.; Attkisson, E.; Williams, D.C.; Ferreira-Gonzalez, A.; Grant, S. Dual Inhibition of Bcl-2 and Bcl-xL Strikingly Enhances PI3K Inhibition-Induced Apoptosis in Human Myeloid Leukemia Cells through a GSK3- and Bim-Dependent Mechanism. Cancer Res. 2013, 73, 1340–1351. [Google Scholar] [CrossRef] [Green Version]
- Donnella, H.J.; Webber, J.T.; Levin, R.S.; Camarda, R.; Momcilovic, O.; Bayani, N.; Shah, K.N.; Korkola, J.E.; Shokat, K.M.; Goga, A.; et al. Kinome rewiring reveals AURKA limits PI3K-pathway inhibitor efficacy in breast cancer. Nat. Chem. Biol. 2018, 14, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Human and Mouse Linked Evaluation of Tumor (HAMLET) Core. Available online: http://digitalcommons.wustl.edu/hamlet/ (accessed on 14 December 2020).
- Xu, S.; Li, S.; Guo, Z.; Luo, J.; Ellis, M.J.; Ma, C.X. Combined targeting of mTOR and AKT is an effective strategy for basal-like breast cancer in patient-derived xenograft models. Mol. Cancer 2013, 12, 1665–1675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, M.J.; Suman, V.J.; Hoog, J.; Lin, L.; Snider, J.; Prat, A.; Parker, J.S.; Luo, J.; DeSchryver, K.; Allred, D.C.; et al. Randomized phase II neoadjuvant comparison between letrozole, anastrozole, and exemestane for postmenopausal women with estrogen receptor-rich stage 2 to 3 breast cancer: Clinical and biomarker outcomes and predictive value of the baseline PAM50-based intrinsic subtype—ACOSOG Z1031. J. Clin. Oncol. 2011, 29, 2342–2349. [Google Scholar] [PubMed] [Green Version]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Gray, W.H.; Lehmann, B.D.; Bauer, J.A.; Shyr, Y.; Pietenpol, J.A. TNBCtype: A Subtyping Tool for Triple-Negative Breast Cancer. Cancer Inform. 2012, 11, CIN.S9983. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [Green Version]
- TNBCtype. Available online: https://cbc.app.vumc.org/tnbc/index.php (accessed on 14 December 2020).
- Ahdesmäki, M.J.; Gray, S.R.; Johnson, J.H.; Lai, Z. Disambiguate: An open-source application for disambiguating two species in next generation sequencing data from grafted samples. F1000Research 2016, 5, 2741. [Google Scholar] [CrossRef]
- Somaticwrapper. Available online: https://github.com/ding-lab/somaticwrapper (accessed on 14 December 2020).
- Kim, S.; Scheffler, K.; Halpern, A.L.; Bekritsky, M.A.; Noh, E.; Kallberg, M.; Chen, X.; Kim, Y.; Beyter, D.; Krusche, P.; et al. Strelka2: Fast and accurate calling of germline and somatic variants. Nat. Methods 2018, 15, 591–594. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, K.; Schulz, M.H.; Long, Q.; Apweiler, R.; Ning, Z. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 2009, 25, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Genome Analysis Toolkit. Available online: https://gatk.broadinstitute.org (accessed on 14 December 2020).
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Fu, X.; Hennessy, B.T.; Casa, A.J.; Zhang, Y.; Gonzalez-Angulo, A.M.; Lluch, A.; Gray, J.W.; Brown, P.H.; Hilsenbeck, S.G.; et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen-receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res. 2010, 12, R40. [Google Scholar] [CrossRef] [Green Version]
- Hather, G.; Liu, R.; Bandi, S.; Mettetal, J.; Manfredi, M.; Shyu, W.-C.; Donelan, J.; Chakravarty, A. Growth rate analysis and efficient experimental design for tumor xenograft studies. Cancer Inform. 2014, 13, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDX ID | Selected Actionable Mutations and Significantly Mutated Genes | PI3K Pathway Alterations |
|---|---|---|
| WHIM2, 5 | PTCH2 p.W293S; JAK2 p.I166T; MAP3K8 p.P461L, TP53 p.Q167Tfs | AKT3 Amp |
| WHIM3 | KRAS p.G13D; NF1 p.Q1775L | |
| WHIM4 | TP53 p.E326Q, p.E285*, p.R175H; ALK p.R395H | PIK3CA amp |
| WHIM6 | ||
| WHIM10 | ||
| WHIM12 | TP53 p.R248Q; PTPRJ p.K1017N; HRAS p.G12D; PIK3CA p.E109fs; PTEN p.T319* | PIK3CA p.V105_E109delinsT; PTEN p.T319* |
| WHIM13 | TP53 p.C238Y; KIT p.D419H | |
| WHIM14 | TP53 p.I195T, CSF1R p.N241K; NTRK3 p.T332M | |
| WHIM21 | FGFR3 p.E135K, PTPRS p.P1506T; TP53 p.P151H | AKT1 Amp |
| WHIM25 | TP53 p.R273H | |
| WHIM29 | PTEN p.E284*, TP53 p.V216M | PTEN p.E284* |
| WHIM30 | BRCA1 p.E1410* (germline); TP53 p.X125_splice site; ATM p.D126E; ATR p.M211T | |
| WHIM31 | BRCA1 p.3604delA (germline); TP53 p.R342*, KRAS p.A134V; PIK3CG p.I287M | |
| WHIM34 | TP53 p.R248W; PTEN p.D310G, GATA3 p.T421Rfs*55 | PTEN p.D310G |
| WHIM36 | TP53 p.F134C; | |
| WHIM48 | TP53 p.Y205H; MET p.R988C |
| Protein Marker | SpearmanCorr2TGI | p Value |
|---|---|---|
| MMP9 | −0.70098 | 0.002347 |
| EGFR | −0.64461 | 0.006396 |
| Integrina5 | −0.57843 | 0.016803 |
| Caspase 3 | 0.541667 | 0.026735 |
| p-IkappaB (S32/36) | −0.5098 | 0.038642 |
| RANKL | −0.48284 | 0.051624 |
| p-HER3(Y1197) | −0.47304 | 0.057094 |
| Caveolin | −0.46569 | 0.061477 |
| CBP | −0.46324 | 0.062994 |
| PI3Kp85 | −0.45588 | 0.067714 |
| Bak | 0.443627 | 0.076168 |
| Bcl.xL | −0.43873 | 0.079763 |
| Vimentin | −0.43137 | 0.085389 |
| KLF4 | −0.42892 | 0.087329 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Z.; Primeau, T.; Luo, J.; Zhang, C.; Sun, H.; Hoog, J.; Gao, F.; Huang, S.; Edwards, D.P.; Davies, S.R.; et al. Proteomic Resistance Biomarkers for PI3K Inhibitor in Triple Negative Breast Cancer Patient-Derived Xenograft Models. Cancers 2020, 12, 3857. https://doi.org/10.3390/cancers12123857
Guo Z, Primeau T, Luo J, Zhang C, Sun H, Hoog J, Gao F, Huang S, Edwards DP, Davies SR, et al. Proteomic Resistance Biomarkers for PI3K Inhibitor in Triple Negative Breast Cancer Patient-Derived Xenograft Models. Cancers. 2020; 12(12):3857. https://doi.org/10.3390/cancers12123857
Chicago/Turabian StyleGuo, Zhanfang, Tina Primeau, Jingqin Luo, Cynthia Zhang, Hua Sun, Jeremy Hoog, Feng Gao, Shixia Huang, Dean P. Edwards, Sherri R. Davies, and et al. 2020. "Proteomic Resistance Biomarkers for PI3K Inhibitor in Triple Negative Breast Cancer Patient-Derived Xenograft Models" Cancers 12, no. 12: 3857. https://doi.org/10.3390/cancers12123857
APA StyleGuo, Z., Primeau, T., Luo, J., Zhang, C., Sun, H., Hoog, J., Gao, F., Huang, S., Edwards, D. P., Davies, S. R., Aft, R., Ding, L., Ellis, M. J., Li, S., & Ma, C. X. (2020). Proteomic Resistance Biomarkers for PI3K Inhibitor in Triple Negative Breast Cancer Patient-Derived Xenograft Models. Cancers, 12(12), 3857. https://doi.org/10.3390/cancers12123857