Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide

 ,
, 


Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
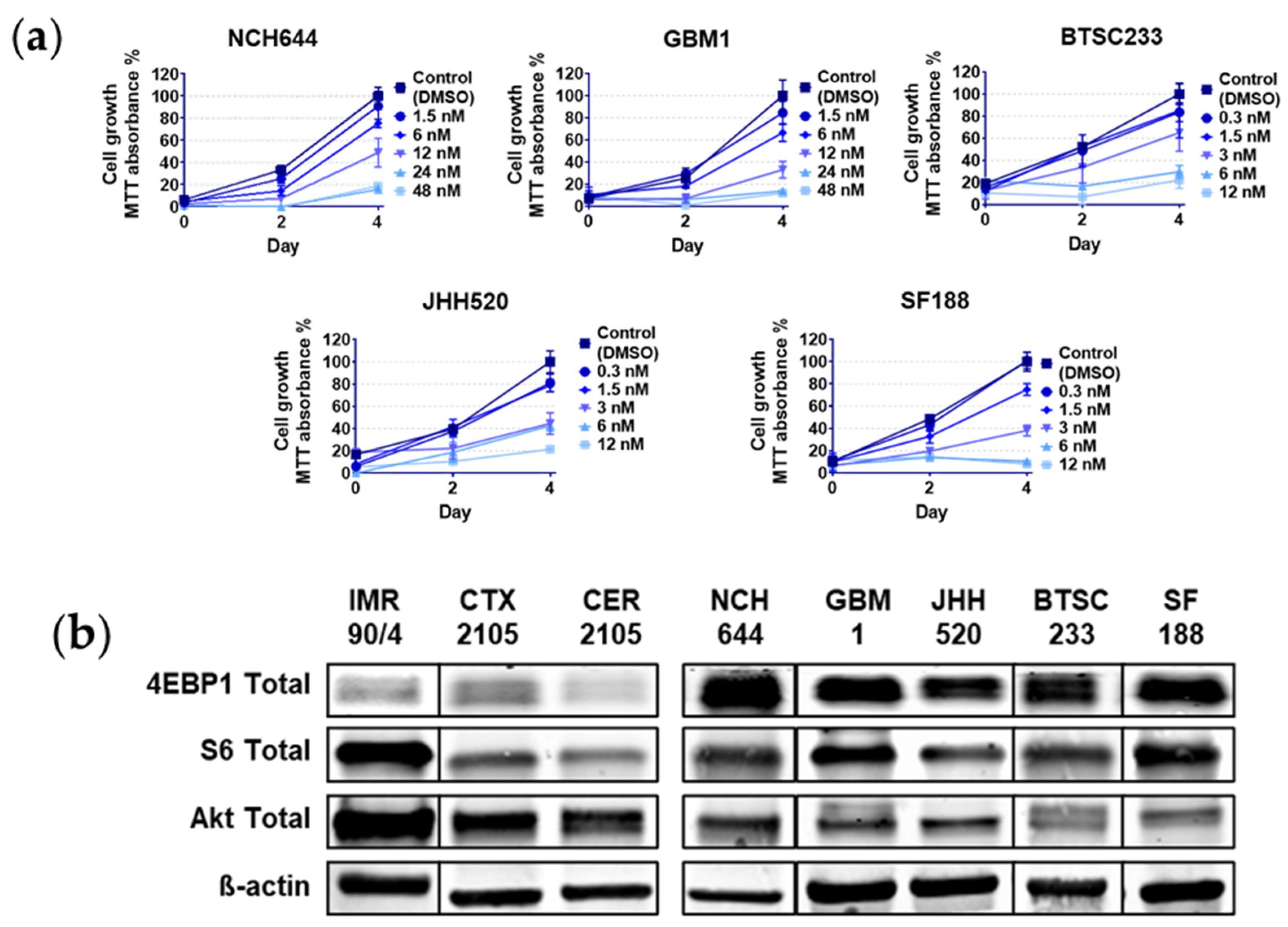
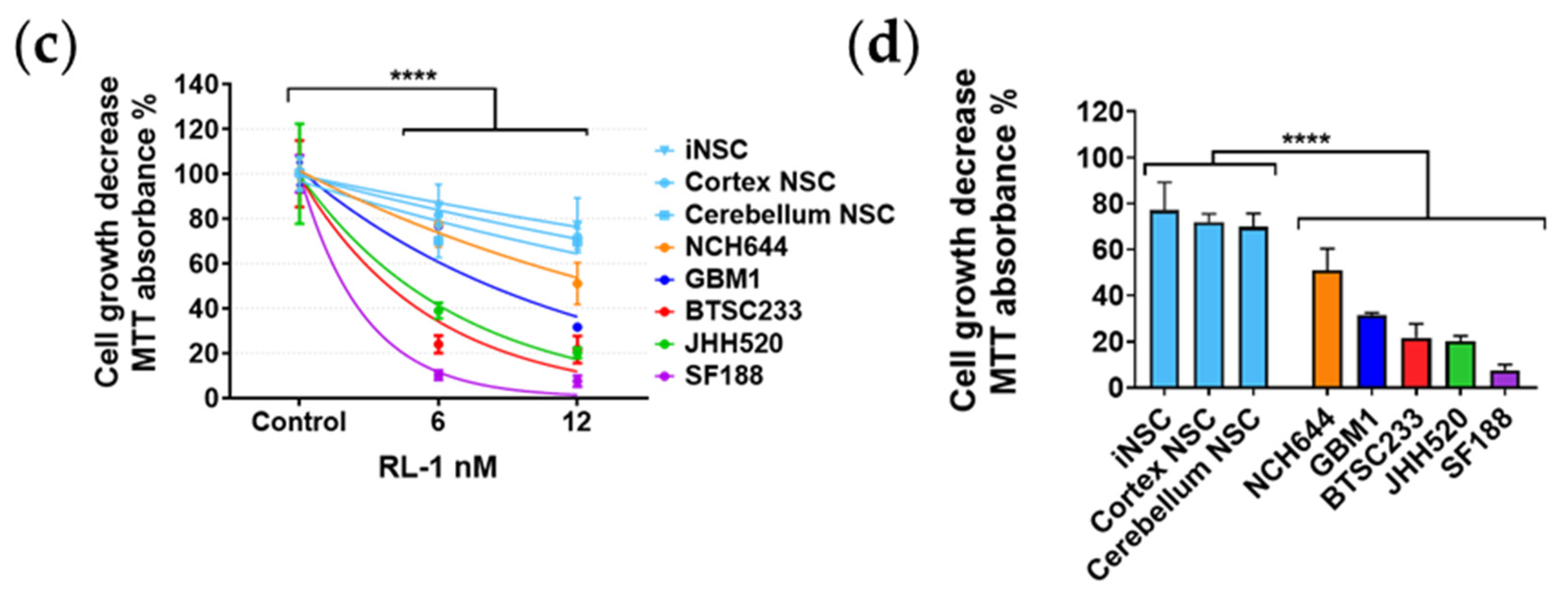
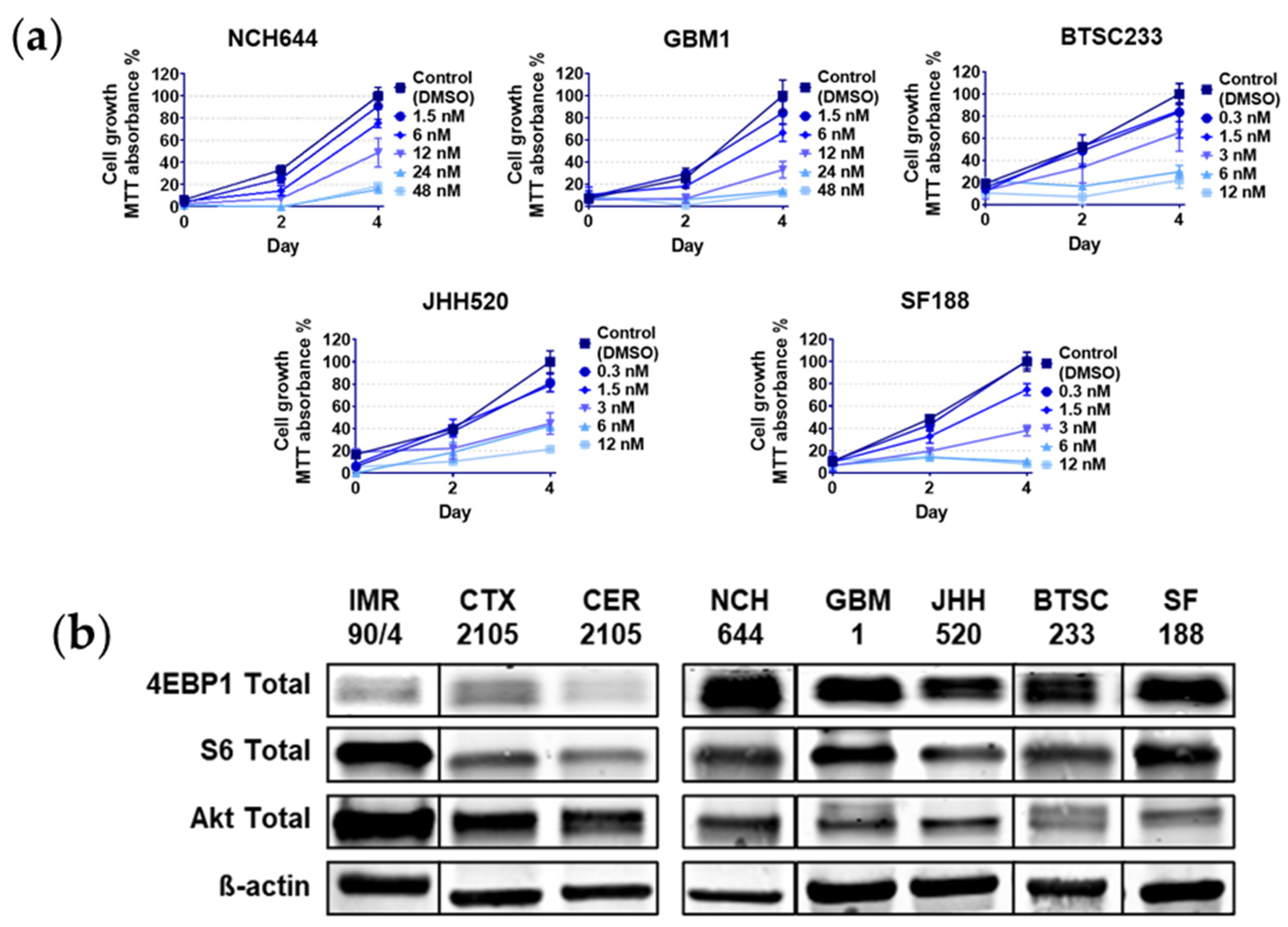
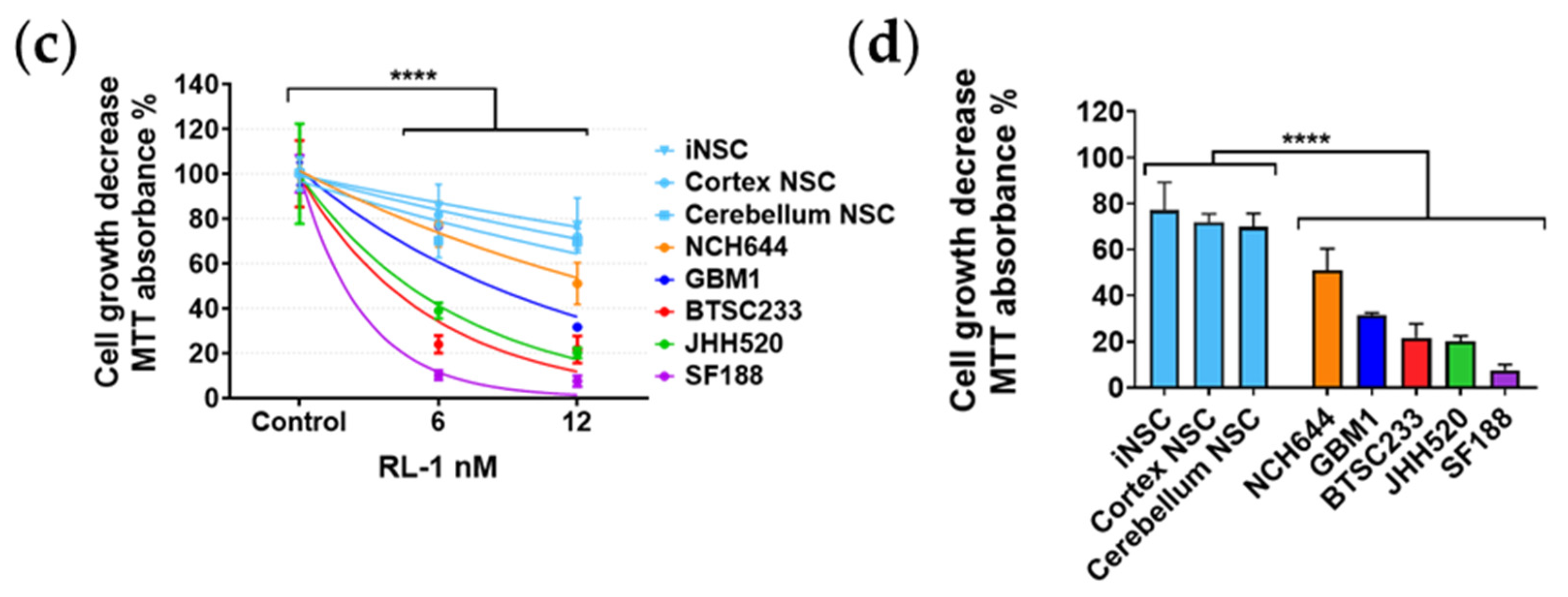
2.1. Rapalink-1 Inhibits Cell Growth in Glioblastoma Stem Cells (GSCs) Compared to Non-Cancerous Cells
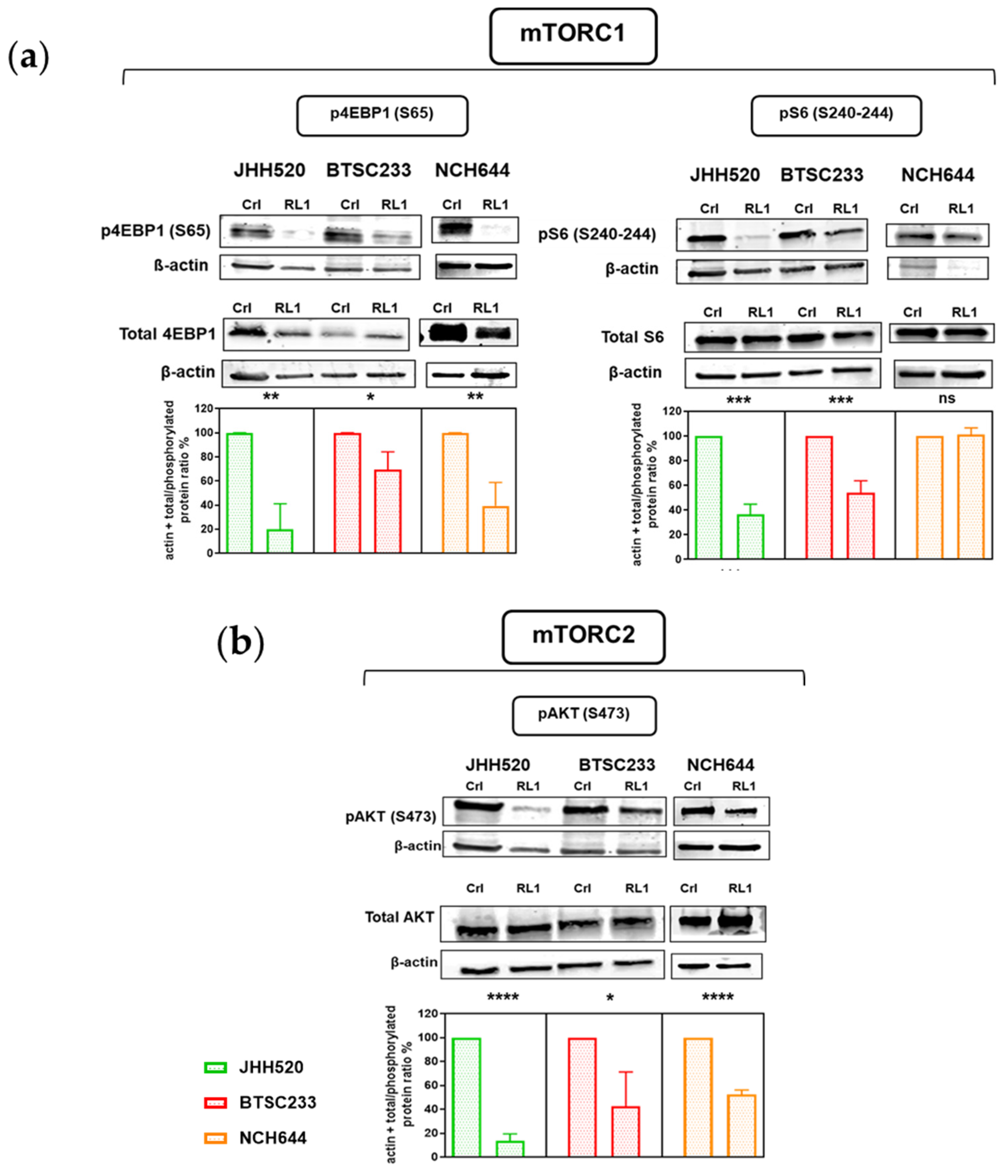
2.2. RL1 Inhibits mTOR Pathway Signaling Activity
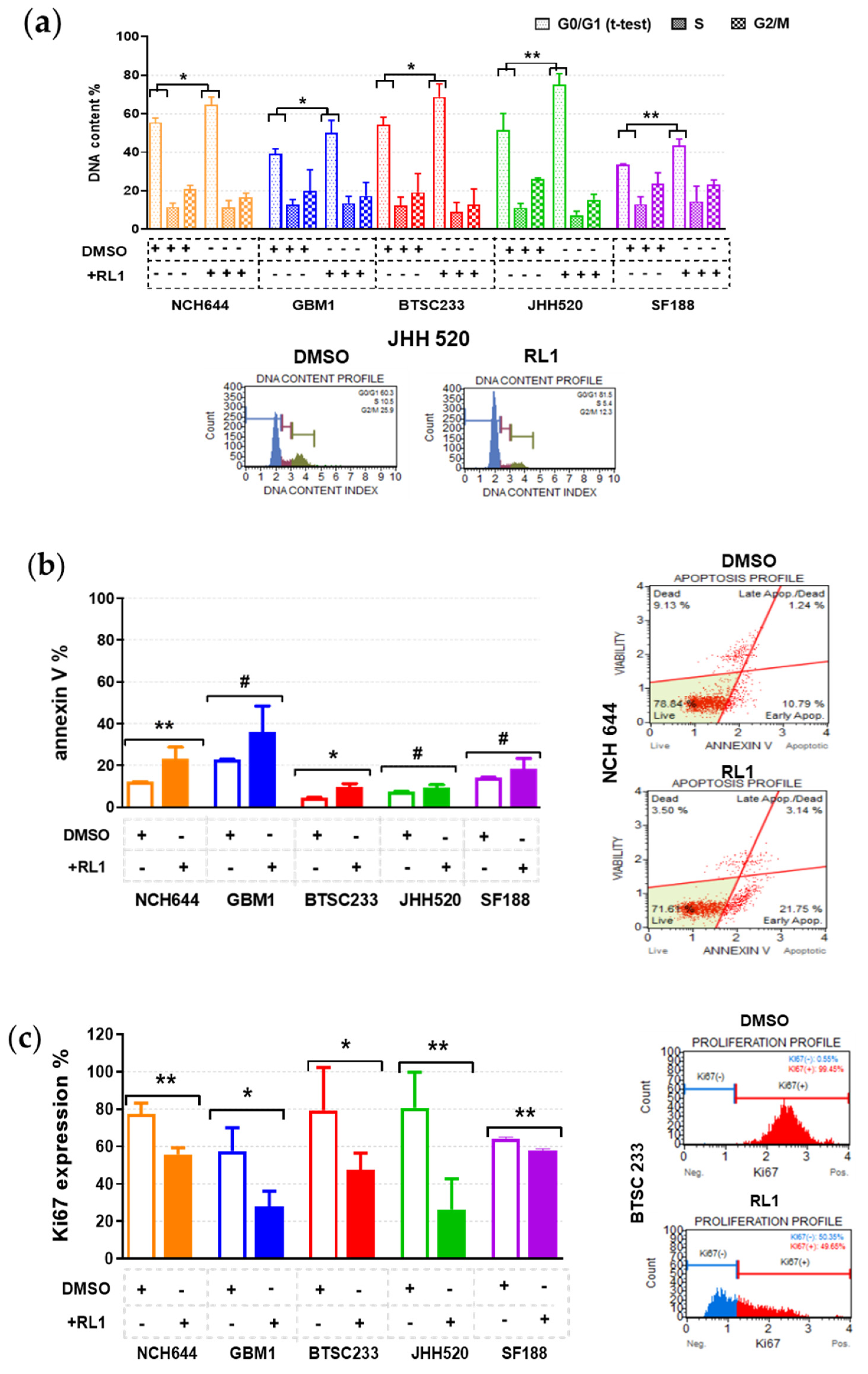
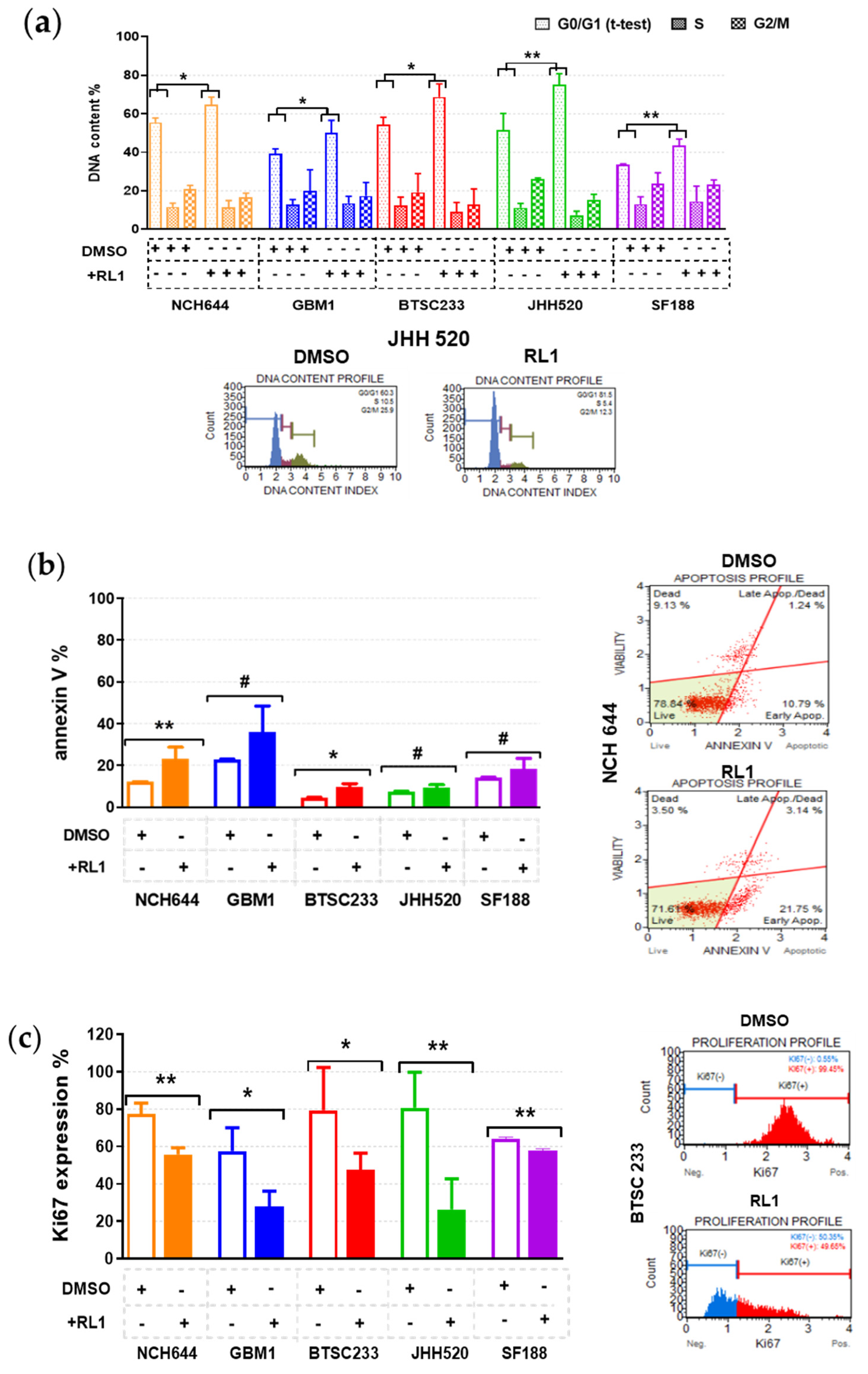
2.3. RL1 Induces Cell Cycle Arrest, Apoptosis, and Proliferation Inhibition
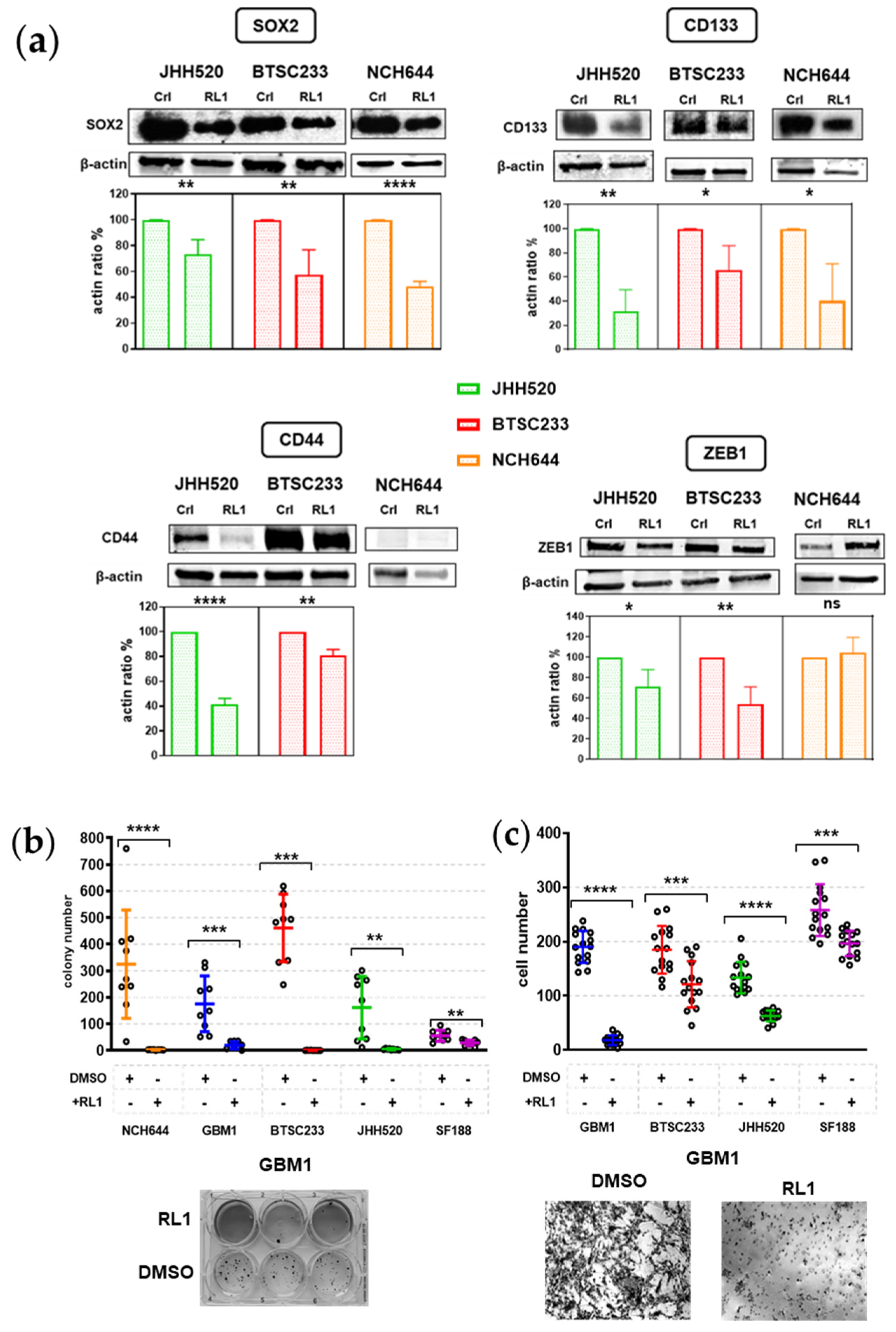
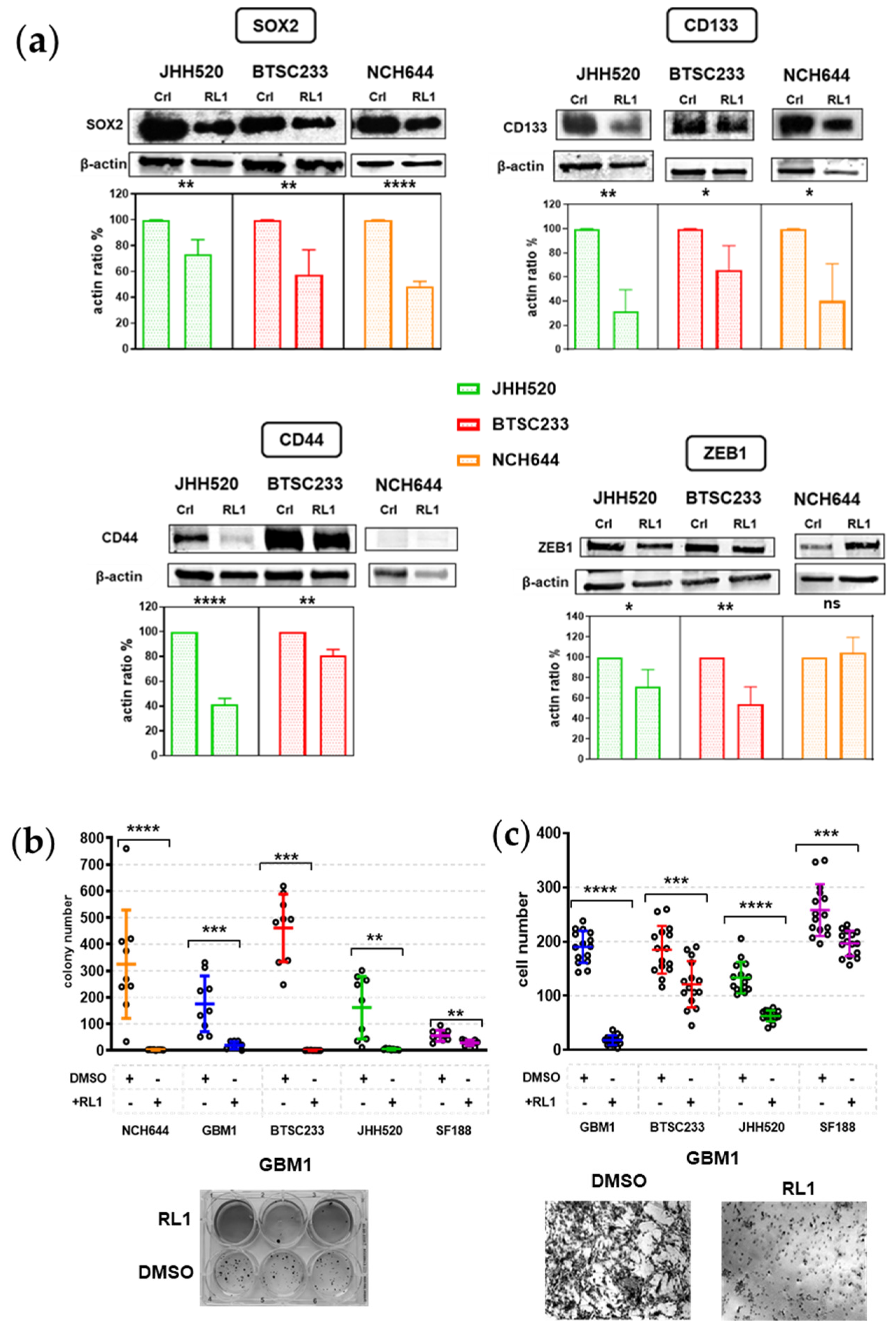
2.4. RL1 Inhibits Stemness and EMT
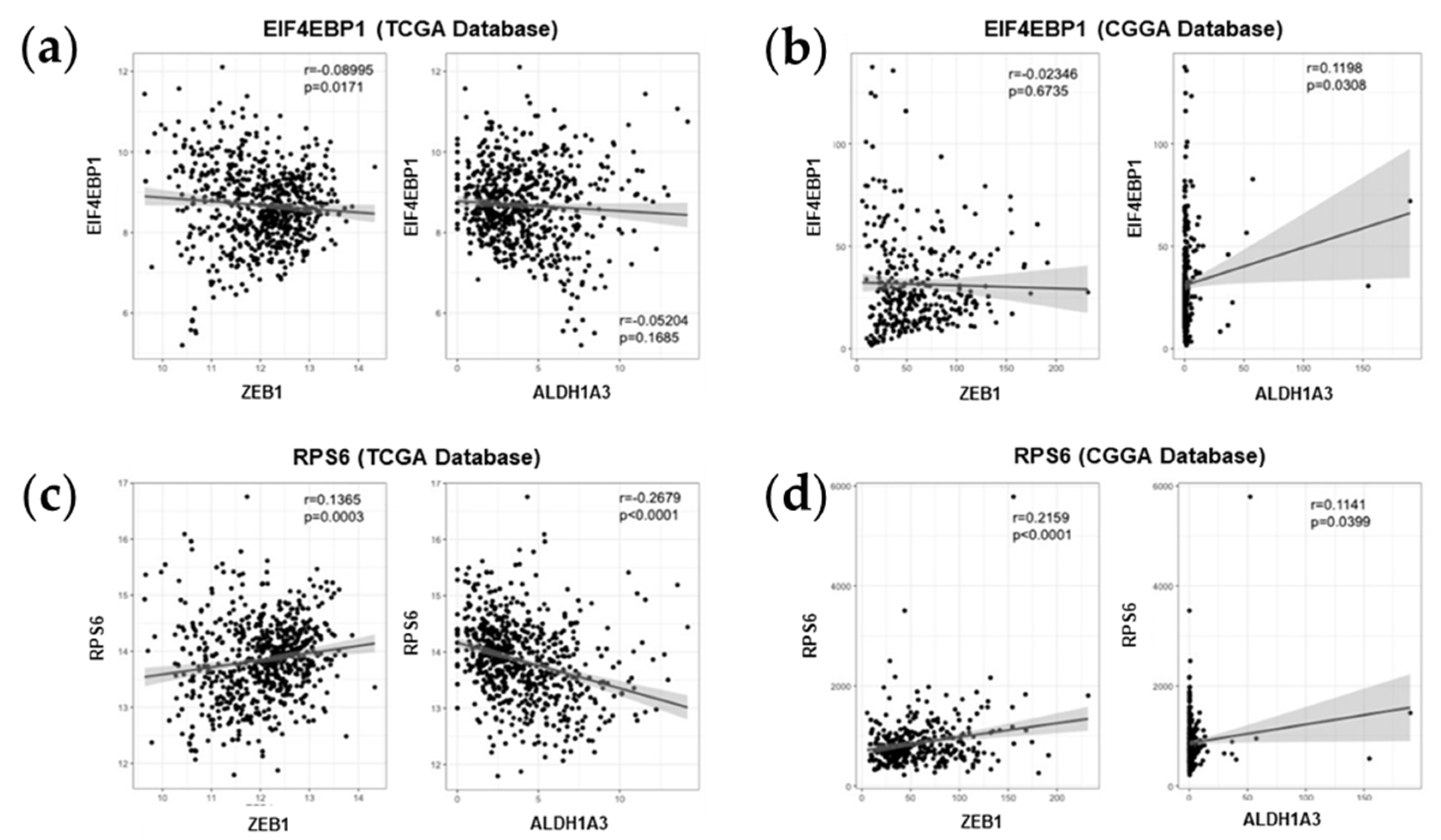
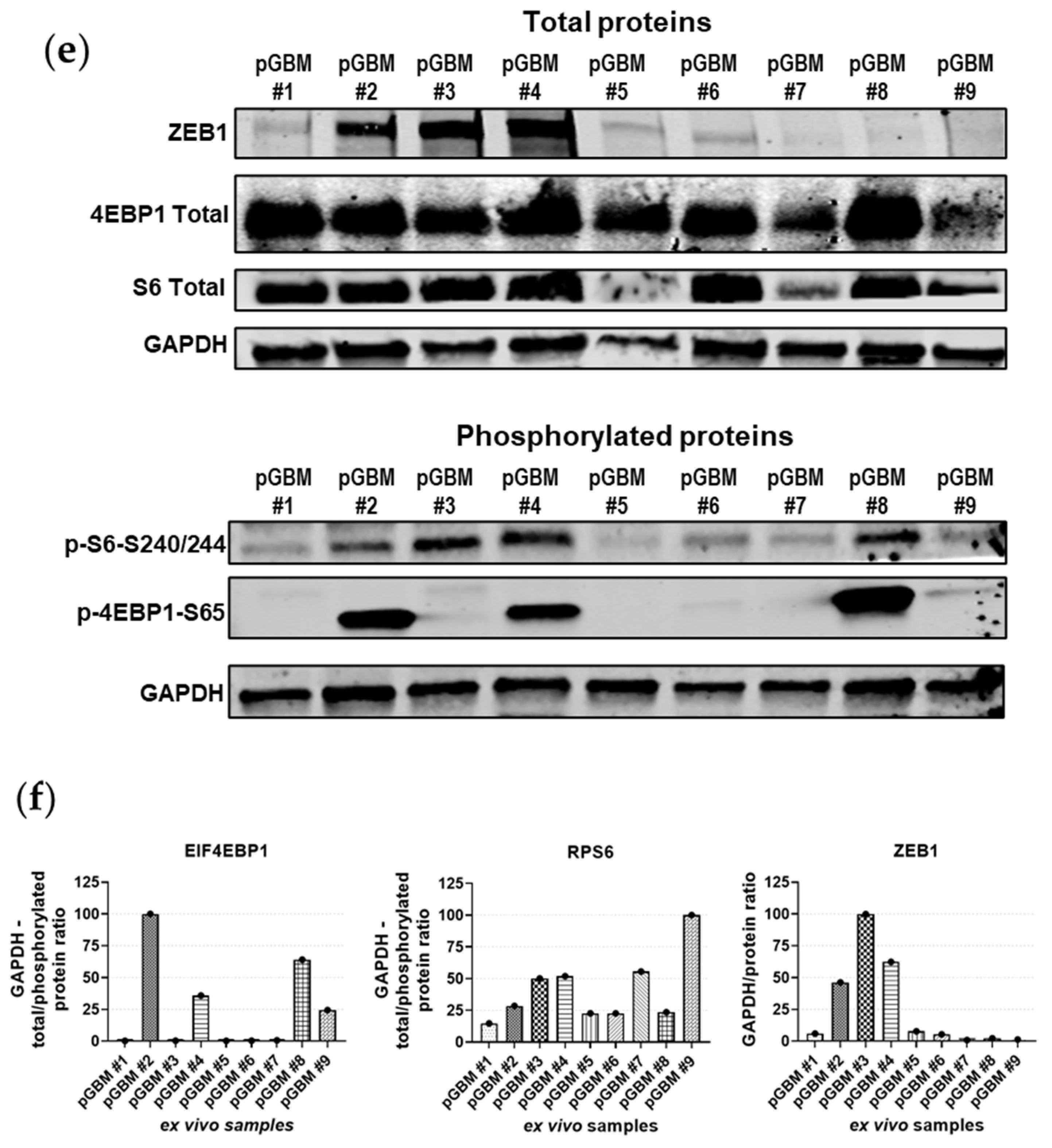
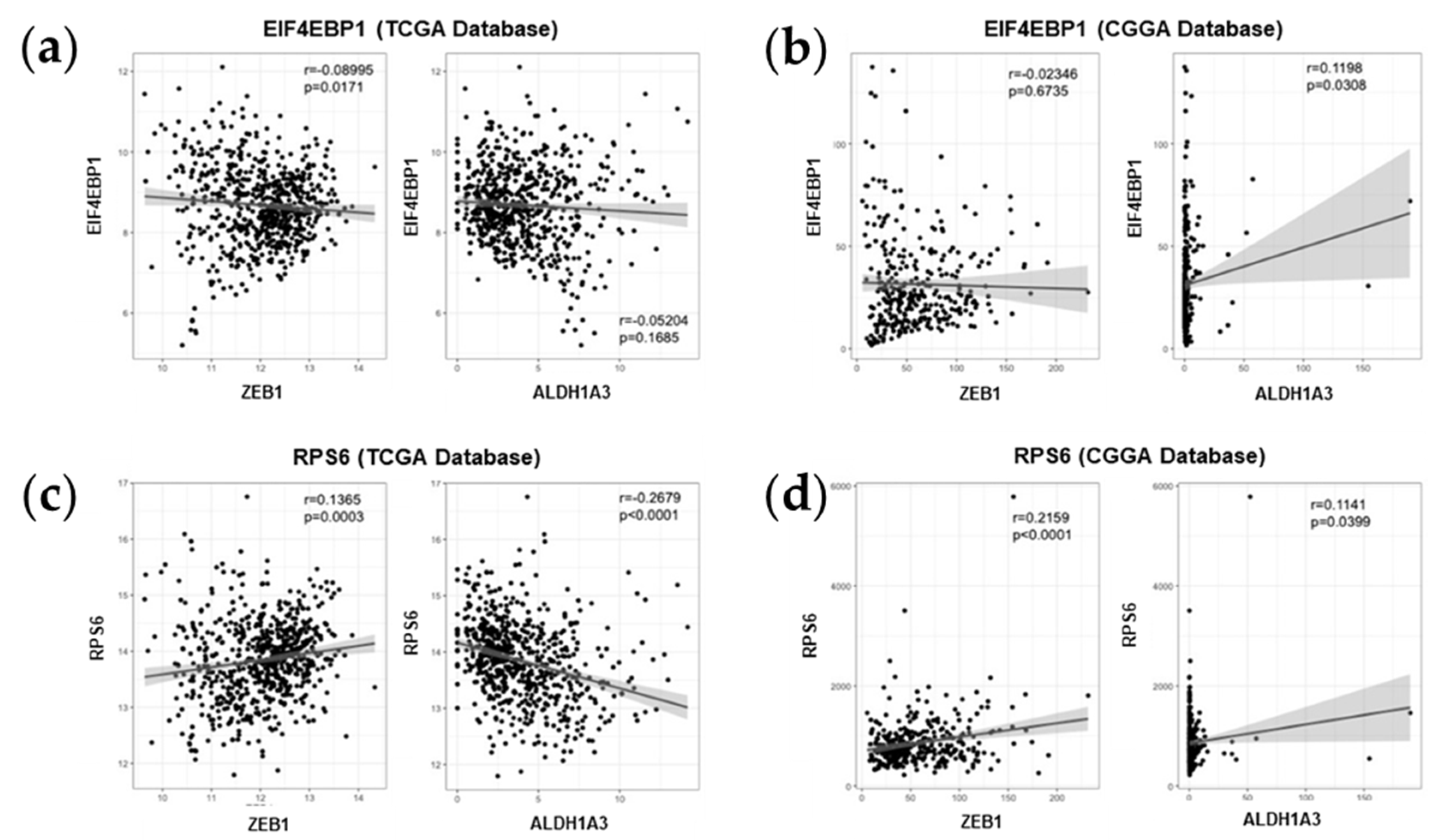
2.5. Association between mTOR Biomarkers and EMT/Mesenchymal Markers Biosamples of GBM Patients of Western and Eastern Ethnicity
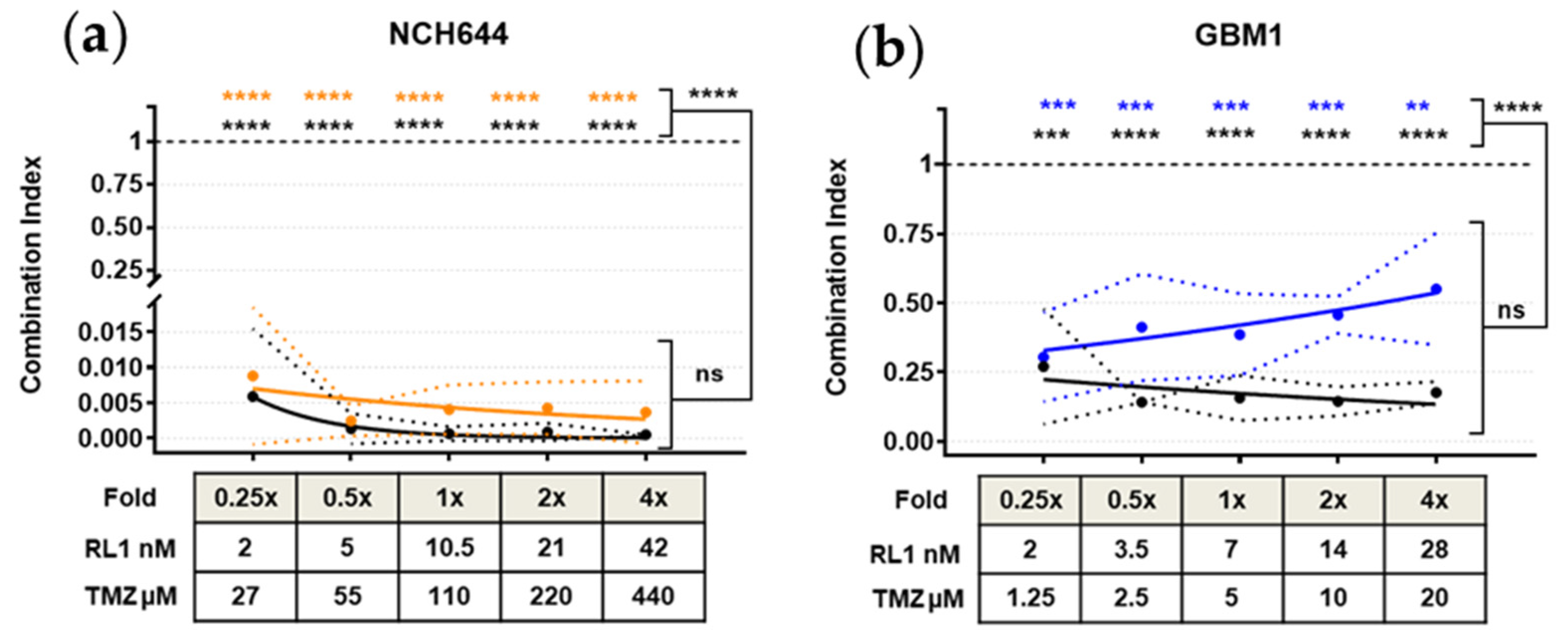
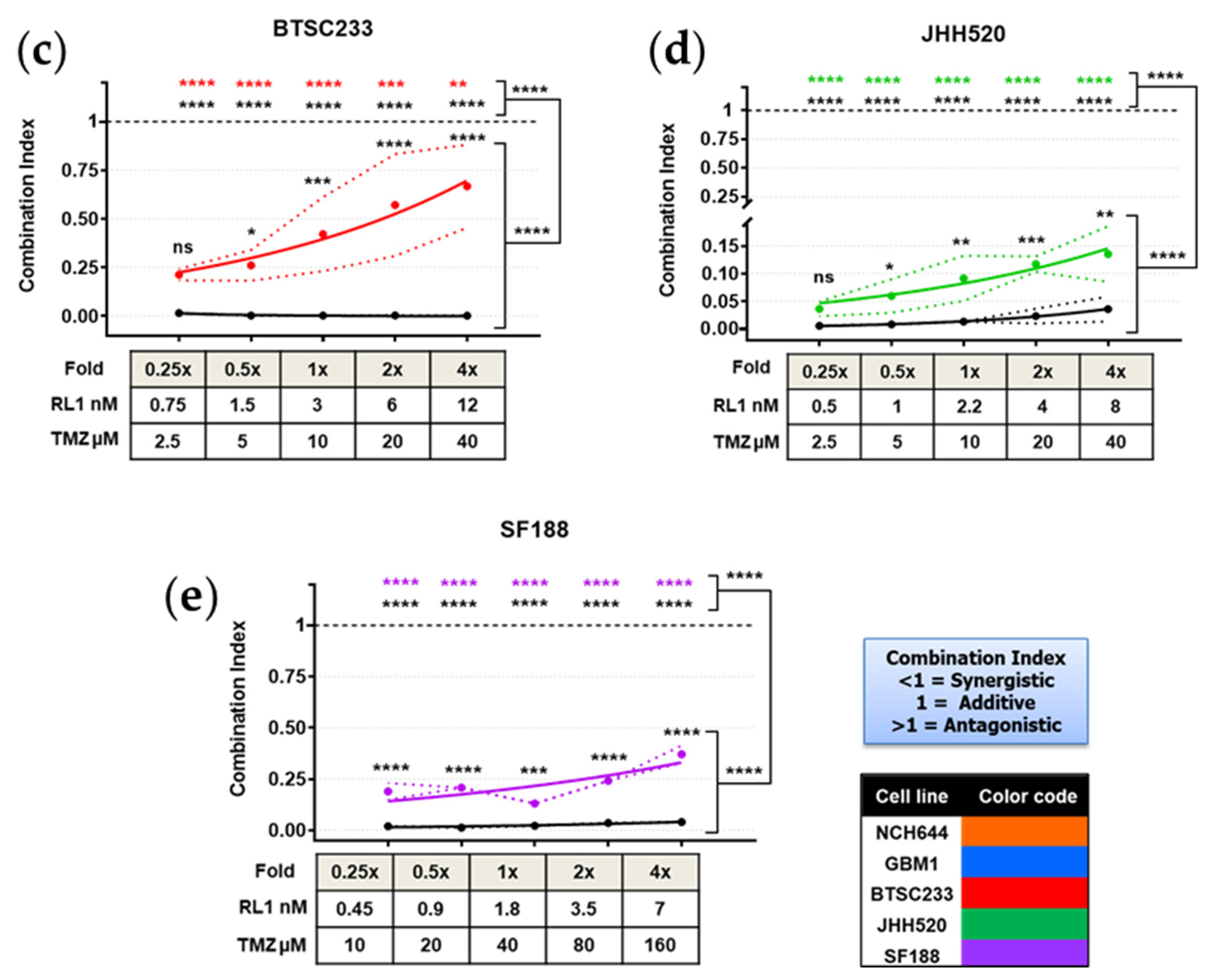
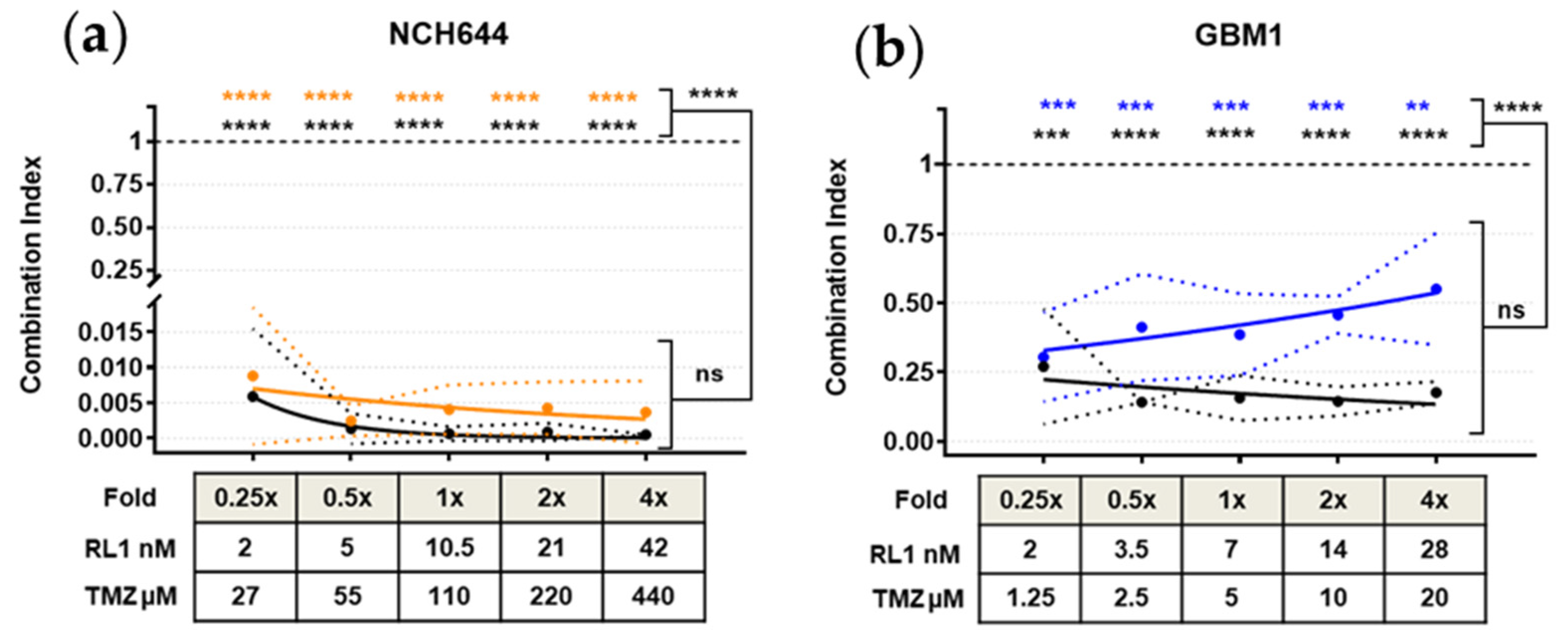
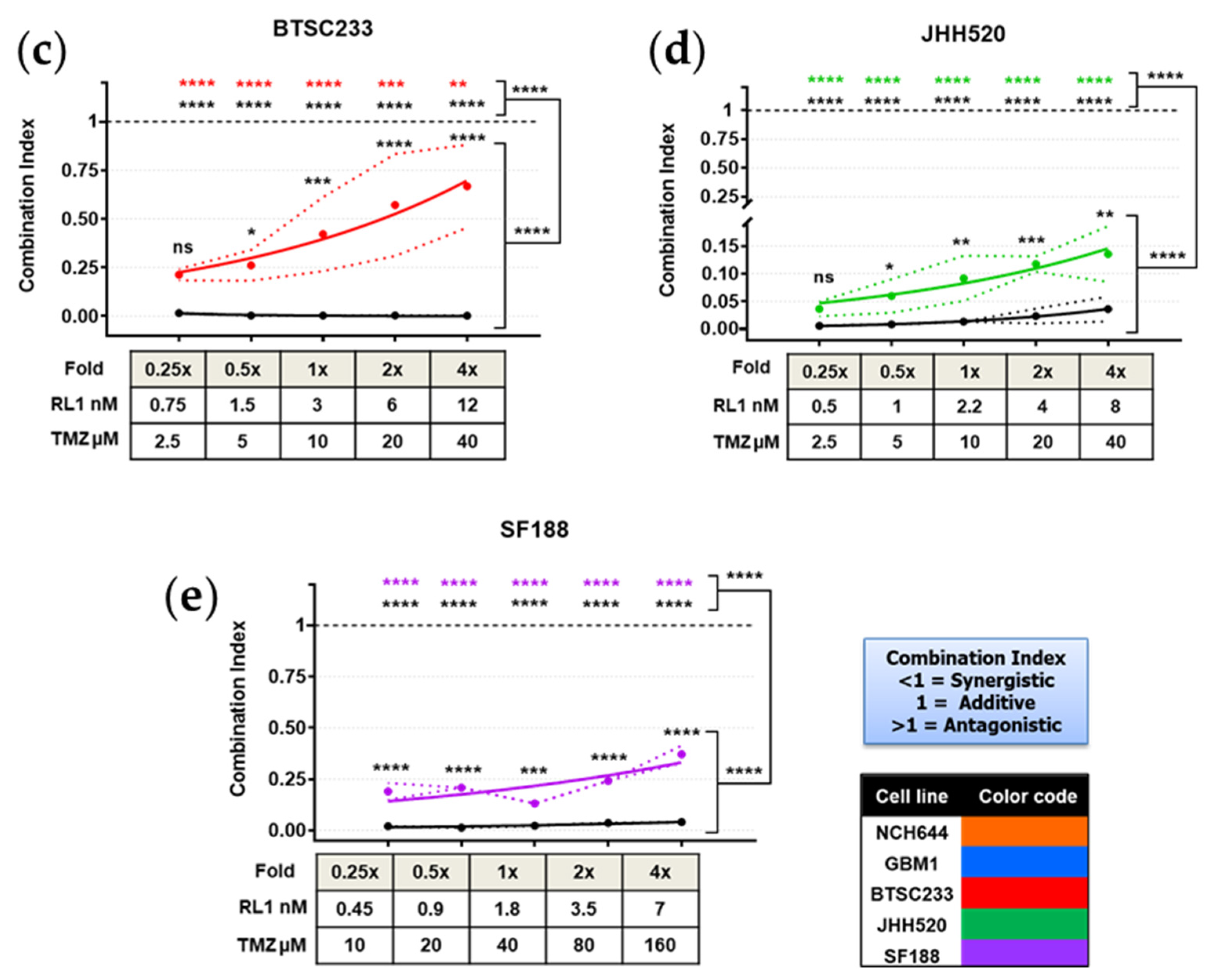
2.6. TTFields, RL1, and TMZ Synergistically Reduced Cell Growth
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Fresh Patient Samples, and Pharmacologic Substances
4.2. Cell Growth (MTT Assay)
4.3. Tumor Treating Fields
- IC50 fold of RL1 (0.25×, 0.5×, 1×, 2×, 4×) plus DMSO control.
- IC50 fold of TMZ (0.25, 0.5, 1, 2, 4) plus DMSO control.
- Combined IC50 folds of TMZ and RL1 in ascending order (0.25 + 0.25, 0.5 + 0.5, 1 + 1, 2 + 2, 4 + 4) plus DMSO control.
4.4. Synergy Assays
4.5. Flow Cytometry–Muse© Assays
4.6. Migration–Boyden Chamber Assay
4.7. Clonogenicity–Colony Formation Assay
4.8. Protein Expression–Western Blot
4.9. Bio-Informatic Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tonn, J.-C.; Reardon, D.A.; Rutka, J.T.; Westphal, M. (Eds.) Oncology of CNS Tumors; Springer: Cham, Switzerland, 2019; ISBN 978-3-030-04151-9. [Google Scholar]
- Stupp, R.; Hegi, M.E.; Mason, W.P. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 8. [Google Scholar] [CrossRef]
- Tabatabai, G.; Stupp, R.; van den Bent, M.J.; Hegi, M.E.; Tonn, J.C.; Wick, W.; Weller, M. Molecular diagnostics of gliomas: The clinical perspective. Acta Neuropathol. 2010, 120, 585–592. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Guardia, G.D.A.; Correa, B.R.; Araujo, P.R.; Qiao, M.; Burns, S.; Penalva, L.O.F.; Galante, P.A.F. Proneural and mesenchymal glioma stem cells display major differences in splicing and lncRNA profiles. NPJ Genomic Med. 2020, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, U.D.; Nikkhah, G.; Maciaczyk, J. Epithelial-to-mesenchymal (-like) transition as a relevant molecular event in malignant gliomas. Cancer Lett. 2013, 331, 131–138. [Google Scholar] [CrossRef]
- Stupp, R.; Weller, M.; Belanger, K.; Bogdahn, U.; Ludwin, S.K.; Lacombe, D.; Mirimanoff, R.O. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The Somatic Genomic Landscape of Glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Machado, L.E.; Alvarenga, A.W.; da Silva, F.F.; Roffé, M.; Begnami, M.D.; Torres, L.F.B.; da Cunha, I.W.; Martins, V.R.; Hajj, G.N.M. Overexpression of mTOR and p(240–244)S6 in IDH1 Wild-Type Human Glioblastomas Is Predictive of Low Survival. J. Histochem. Cytochem. 2018, 66, 403–414. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; deCarvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Wong, E.T.; Kanner, A.A.; Steinberg, D.; Engelhard, H.; Heidecke, V.; Kirson, E.D.; Taillibert, S.; Liebermann, F.; Dbalý, V.; et al. NovoTTF-100A versus physician’s choice chemotherapy in recurrent glioblastoma: A randomised phase III trial of a novel treatment modality. Eur. J. Cancer 2012, 48, 2192–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.; Read, W.; Steinberg, D.M.; Lhermitte, B.; Toms, S.; Idbaih, A.; Ahluwalia, M.S.; Fink, K.; et al. Effect of Tumor-Treating Fields Plus Maintenance Temozolomide vs. Maintenance Temozolomide Alone on Survival in Patients with Glioblastoma: A Randomized Clinical Trial. JAMA 2017, 318, 2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Pandey, M.; Ballo, M.T. Integration of Tumor-Treating Fields into the Multidisciplinary Management of Patients with Solid Malignancies. Oncologist 2019, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Dettmer, S.; Berberich, A.; Kessler, T.; Karapanagiotou-Schenkel, I.; Wick, A.; Winkler, F.; Pfaff, E.; Brors, B.; Debus, J.; et al. N2M2 (NOA-20) phase I/II trial of molecularly matched targeted therapies plus radiotherapy in patients with newly diagnosed non-MGMT hypermethylated glioblastoma. Neuro Oncol. 2019, 21, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wick, W.; Gorlia, T.; Bady, P.; Platten, M.; Taphoorn, M.J.B.; Steuve, J.; Brandes, A.A.; Hamou, M.-F.; Wick, A.; Kosch, M.; et al. Phase II Study of Radiotherapy and Temsirolimus versus Radiochemotherapy with Temozolomide in Patients with Newly Diagnosed Glioblastoma without MGMT Promoter Hypermethylation. Clin. Cancer Res. 2016, 22, 4797–4806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamouille, S.; Connolly, E.; Smyth, J.W.; Akhurst, R.J.; Derynck, R. TGF-induced activation of mTOR complex 2 drives epithelial-mesenchymal transition and cell invasion. J. Cell Sci. 2012, 125, 1259–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsuno, Y.; Meyer, D.S.; Zhang, Z.; Shokat, K.M.; Akhurst, R.J.; Miyazono, K.; Derynck, R. Chronic TGF-b exposure drives stabilized EMT, tumor stemness, and cancer drug resistance with vulnerability to bitopic mTOR inhibition. Sci. Signal. 2019, 12, eaau8544. [Google Scholar] [CrossRef]
- Karimi Roshan, M.; Soltani, A.; Soleimani, A.; Rezaie Kahkhaie, K.; Afshari, A.R.; Soukhtanloo, M. Role of AKT and mTOR signaling pathways in the induction of epithelial-mesenchymal transition (EMT) process. Biochimie 2019, 165, 229–234. [Google Scholar] [CrossRef]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F.; et al. Phase II Trial of Temsirolimus (CCI-779) in Recurrent Glioblastoma Multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in Glioblastoma: Rationale and Preclinical/Clinical Evidence. Dis. Markers 2018, 2018, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.; Aksoy, O.; Wong, R.A.; Ilkhanizadeh, S.; Novotny, C.J.; Gustafson, W.C.; Truong, A.Y.-Q.; Cayanan, G.; Simonds, E.F.; Haas-Kogan, D.; et al. A Kinase Inhibitor Targeted to mTORC1 Drives Regression in Glioblastoma. Cancer Cell 2017, 31, 424–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Podergajs, N.; Brekka, N.; Radlwimmer, B.; Herold-Mende, C.; Talasila, K.M.; Tiemann, K.; Rajcevic, U.; Lah, T.T.; Bjerkvig, R.; Miletic, H. Expansive growth of two glioblastoma stem-like cell lines is mediated by bFGF and not by EGF. Radiol. Oncol. 2013, 47, 330–337. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedele, V.; Dai, F.; Masilamani, A.P.; Heiland, D.H.; Kling, E.; Gätjens-Sanchez, A.M.; Ferrarese, R.; Platania, L.; Soroush, D.; Kim, H.; et al. Epigenetic Regulation of ZBTB18 Promotes Glioblastoma Progression. Mol. Cancer Res. MCR 2017, 15, 998–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, Z.A.; Wilson, K.M.; Salmasi, V.; Orr, B.A.; Eberhart, C.G.; Siu, I.-M.; Lim, M.; Weingart, J.D.; Quinones-Hinojosa, A.; Bettegowda, C.; et al. Establishment and Biological Characterization of a Panel of Glioblastoma Multiforme (GBM) and GBM Variant Oncosphere Cell Lines. PLoS ONE 2016, 11, e0150271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.M.; Mathews-Griner, L.A.; Williamson, T.; Guha, R.; Chen, L.; Shinn, P.; McKnight, C.; Michael, S.; Klumpp-Thomas, C.; Binder, Z.A.; et al. Mutation Profiles in Glioblastoma 3D Oncospheres Modulate Drug Efficacy. SLAS Technol. Transl. Life Sci. Innov. 2019, 24, 28–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network. Central Nervous System Cancers (Version 1.2016). Available online: https://www.nccn.org/patients/guidelines/content/PDF/brain-gliomas-patient.pdf (accessed on 16 October 2020).
- Chou, T.-C. Drug Combination Studies and Their Synergy Quantification Using the Chou-Talalay Method. Cancer Res. 2010, 70, 440–446. [Google Scholar] [CrossRef] [Green Version]
- Meng, D.; Frank, A.R.; Jewell, J.L. mTOR signaling in stem and progenitor cells. Development 2018, 145, dev152595. [Google Scholar] [CrossRef] [Green Version]
- Duzgun, Z.; Eroglu, Z.; Biray Avci, C. Role of mTOR in glioblastoma. Gene 2016, 575, 187–190. [Google Scholar] [CrossRef]
- Magaway, C.; Kim, E.; Jacinto, E. Targeting mTOR and Metabolism in Cancer: Lessons and Innovations. Cells 2019, 8, 1584. [Google Scholar] [CrossRef] [Green Version]
- Munster, P.; Mita, M.; Mahipal, A.; Nemunaitis, J.; Massard, C.; Mikkelsen, T.; Cruz, C.; Paz-Ares, L.; Hidalgo, M.; Rathkopf, D.; et al. First-In-Human Phase I Study of a Dual mTOR Kinase and DNA-PK Inhibitor (CC-115) in Advanced Malignancy. Cancer Manag. Res. 2019, 11, 10463–10476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarini, F.; Evangelisti, C.; Lattanzi, G.; McCubrey, J.A.; Martelli, A.M. Advances in understanding the mechanisms of evasive and innate resistance to mTOR inhibition in cancer cells. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 1322–1337. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.W.; Nicolaides, T.P.; Weiss, W.A. Inhibiting 4EBP1 in Glioblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.; Hartmann, R.; Schröter, F.; Suwala, A.K.; Maciaczyk, D.; Krüger, A.C.; Willbold, D.; Kahlert, U.D.; Maciaczyk, J. Reciprocal regulation of the cholinic phenotype and epithelial-mesenchymal transition in glioblastoma cells. Oncotarget 2016, 7, 73414–73431. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, U.D.; Suwala, A.K.; Koch, K.; Natsumeda, M.; Orr, B.A.; Hayashi, M.; Maciaczyk, J.; Eberhart, C.G. Pharmacologic Wnt Inhibition Reduces Proliferation, Survival, and Clonogenicity of Glioblastoma Cells. J. Neuropathol. Exp. Neurol. 2015, 74, 889–900. [Google Scholar] [CrossRef]
- Fedele, M.; Cerchia, L.; Pegoraro, S.; Sgarra, R.; Manfioletti, G. Proneural-Mesenchymal Transition: Phenotypic Plasticity to Acquire Multitherapy Resistance in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 2746. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.C.; Richardson, C.J.; Tee, A.R.; Cheatham, L.; Tsou, C.; Blenis, J. mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol. Cell. Biol. 2004, 24, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487. [Google Scholar] [CrossRef] [Green Version]
- Kwasnicki, A.; Jeevan, D.; Braun, A.; Murali, R.; Jhanwar-Uniyal, M. Involvement of mTOR signaling pathways in regulating growth and dissemination of metastatic brain tumors via EMT. Anticancer Res. 2015, 35, 689–696. [Google Scholar]
- Hung, C.-M.; Garcia-Haro, L.; Sparks, C.A.; Guertin, D.A. mTOR-dependent cell survival mechanisms. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Park, A.K.; Kim, P.; Ballester, L.Y.; Esquenazi, Y.; Zhao, Z. Subtype-specific signaling pathways and genomic aberrations associated with prognosis of glioblastoma. Neuro Oncol. 2019, 21, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Kong, K.-K.; Xu, X.-K.; Chen, C.; Li, H.; Wang, F.-Y.; Peng, X.-F.; Zhang, Z.; Li, P.; Li, J.-L.; et al. Downregulation of miR-205 is associated with glioblastoma cell migration, invasion, and the epithelial-mesenchymal transition, by targeting ZEB1 via the Akt/mTOR signaling pathway. Int. J. Oncol. 2018, 52, 485–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Zhang, W.; Li, Y.; Alvarez, A.; Li, Z.; Wang, Y.; Song, L.; Lv, D.; Nakano, I.; Hu, B.; et al. SHP-2-upregulated ZEB1 is important for PDGFRα-driven glioma epithelial-mesenchymal transition and invasion in mice and humans. Oncogene 2016, 35, 5641–5652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Chen, Y.; Li, Y.; Lyu, X.; Cui, J.; Cheng, Y.; Zhao, L.; Zhao, G. Metformin inhibits TGF-β1-induced epithelial-to-mesenchymal transition-like process and stem-like properties in GBM via AKT/mTOR/ZEB1 pathway. Oncotarget 2018, 9, 7023–7035. [Google Scholar] [CrossRef] [Green Version]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, E.; Tigges, J.; Hartmann, J.; Kapr, J.; Serafini, M.M.; Viviani, B. Neural In Vitro Models for Studying Substances Acting on the Central Nervous System. Handb. Exp. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Kahlert, U.D.; Suwala, A.K.; Raabe, E.H.; Siebzehnrubl, F.A.; Suarez, M.J.; Orr, B.A.; Bar, E.E.; Maciaczyk, J.; Eberhart, C.G. ZEB1 Promotes Invasion in Human Fetal Neural Stem Cells and Hypoxic Glioma Neurospheres: ZEB1 Regulates Motility in Hypoxic Gliomas. Brain Pathol. 2015, 25, 724–732. [Google Scholar] [CrossRef] [Green Version]
- Kahlert, U.D.; Cheng, M.; Koch, K.; Marchionni, L.; Fan, X.; Raabe, E.H.; Maciaczyk, J.; Glunde, K.; Eberhart, C.G. Alterations in cellular metabolome after pharmacological inhibition of Notch in glioblastoma cells. Int. J. Cancer 2016, 138, 1246–1255. [Google Scholar] [CrossRef] [Green Version]
- Suwala, A.K.; Koch, K.; Rios, D.H.; Aretz, P.; Uhlmann, C.; Ogorek, I.; Felsberg, J.; Reifenberger, G.; Köhrer, K.; Deenen, R.; et al. Inhibition of Wnt/beta-catenin signaling downregulates expression of aldehyde dehydrogenase isoform 3A1 (ALDH3A1) to reduce resistance against temozolomide in glioblastoma in vitro. Oncotarget 2018, 9, 22703–22716. [Google Scholar] [CrossRef] [Green Version]
- Vargas-Toscano, A.; Khan, D.; Nickel, A.-C.; Hewera, M.; Kamp, M.A.; Fischer, I.; Steiger, H.-J.; Zhang, W.; Muhammad, S.; Hänggi, D.; et al. Robot technology identifies a Parkinsonian therapeutics repurpose to target stem cells of glioblastoma. CNS Oncol. 2020, 9, CNS58. [Google Scholar] [CrossRef] [PubMed]
- Niyazi, M.; Niyazi, I.; Belka, C. Counting colonies of clonogenic assays by using densitometric software. Radiat. Oncol. 2007, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, K.; Hartmann, R.; Tsiampali, J.; Uhlmann, C.; Nickel, A.-C.; He, X.; Kamp, M.A.; Sabel, M.; Barker, R.A.; Steiger, H.-J.; et al. A comparative pharmaco-metabolomic study of glutaminase inhibitors in glioma stem-like cells confirms biological effectiveness but reveals differences in target-specificity. Cell Death Discov. 2020, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Cell Line | Color Code 1 | RL-1 IC50 nM | TMZ IC50 µM | Gender | Age Group | Molecular Subtype (Verhaak) | MGMT Status | IDH Status | ALDH1A3 Expression |
|---|---|---|---|---|---|---|---|---|---|---|
| Glioblastoma stem cells | NCH644 | 10.5 | 110 | Female | Adult | Proneural | Methylated | Wildtype | Negative | |
| GBM1 | 7 | 5 | Male | Adult | Classical | Methylated | Wildtype | Positive | ||
| BTSC233 | 3 | 10 | Female | Adult | Mesenchymal | Methylated | Wildtype | Positive | ||
| JHH520 | 2.2 | 10 | Female | Adult | Mesenchymal | Methylated | Wildtype | Positive | ||
| SF188 | 1.8 | 40 | Male | Pediatric | - | Unmethylated | Wildtype | Positive | ||
| Induced Neural Stem Cell | IMR 90/4 | 15.5 | - | - | - | - | - | - | - | |
| Neural Stem Cells | Cortex | 12 | - | - | - | - | - | - | - | |
| Cerebellum | 11 | - | - | - | - | - | - | - |
| Dish Configuration | Value | Drug | Concentration |
|---|---|---|---|
| Frequency | 200 kHz | IC50 folds of RL1 | 0.25×, 0.5×, 1×, 2×, 4× Plus DMSO control. |
| Temperature | 37 °C | ||
| Current | 12 mA | IC50 folds of TMZ | 0.25×, 0.5×, 1×, 2×, 4× Plus DMSO control. |
| Voltage | 1.7 V/cm RMS | Combined IC50 folds of TMZ and RL1 in ascending order | 0.25 + 0.25, 0.5 + 0.5, 1 + 1, 2 + 2, 4 + 4 Plus DMSO control. |
| Incubation Time | All 48 h BTSC233 96 h |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vargas-Toscano, A.; Nickel, A.-C.; Li, G.; Kamp, M.A.; Muhammad, S.; Leprivier, G.; Fritsche, E.; Barker, R.A.; Sabel, M.; Steiger, H.-J.; et al. Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide. Cancers 2020, 12, 3859. https://doi.org/10.3390/cancers12123859
Vargas-Toscano A, Nickel A-C, Li G, Kamp MA, Muhammad S, Leprivier G, Fritsche E, Barker RA, Sabel M, Steiger H-J, et al. Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide. Cancers. 2020; 12(12):3859. https://doi.org/10.3390/cancers12123859
Chicago/Turabian StyleVargas-Toscano, Andres, Ann-Christin Nickel, Guanzhang Li, Marcel Alexander Kamp, Sajjad Muhammad, Gabriel Leprivier, Ellen Fritsche, Roger A. Barker, Michael Sabel, Hans-Jakob Steiger, and et al. 2020. "Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide" Cancers 12, no. 12: 3859. https://doi.org/10.3390/cancers12123859
APA StyleVargas-Toscano, A., Nickel, A.-C., Li, G., Kamp, M. A., Muhammad, S., Leprivier, G., Fritsche, E., Barker, R. A., Sabel, M., Steiger, H.-J., Zhang, W., Hänggi, D., & Kahlert, U. D. (2020). Rapalink-1 Targets Glioblastoma Stem Cells and Acts Synergistically with Tumor Treating Fields to Reduce Resistance against Temozolomide. Cancers, 12(12), 3859. https://doi.org/10.3390/cancers12123859