The Role of cis- and trans-Acting RNA Regulatory Elements in Leukemia



Abstract
:Simple Summary
Abstract
1. Introduction
2. cis-Acting RNA Regulatory Motifs
2.1. Aberrant Pre-mRNA Splicing
2.2. Alterations in Untranslated Regions (UTR) of mRNA
2.2.1. 5′ UTR Alterations in Leukemogenesis
2.2.2. 3′ UTR Alterations in Leukemogenesis
3. Prospective Therapeutic Value of Targeting Non-Coding Pre-mRNA and mRNA Sequences
4. Regulatory Non-Coding RNA Molecules
4.1. Long Non-Coding RNA
4.2. Circular RNA
4.3. Short Non-Coding RNAs
miRNA
5. Therapeutic Approaches for Targeting RNA Molecules
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, J.; Nimer, S. Recent advances in the understanding and treatment of acute myeloid leukemia. F1000Research 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artz, A.; Ridgeway, J.A. Managing the Continuum of Myeloid Malignancies. J. Adv. Pract. Oncol. 2018, 9, 345–349. [Google Scholar]
- Ghia, P.; Hallek, M. Management of chronic lymphocytic leukemia. Haematologica 2014, 99, 965–972. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L.; et al. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211–1218. [Google Scholar] [CrossRef] [Green Version]
- Medinger, M.; Passweg, J.R. Acute myeloid leukaemia genomics. Br. J. Haematol. 2017, 179, 530–542. [Google Scholar] [CrossRef]
- Andersson, A.K.; Ma, J.; Wang, J.; Chen, X.; Gedman, A.L.; Dang, J.; Nakitandwe, J.; Holmfeldt, L.; Parker, M.; Easton, J.; et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat. Genet. 2015, 47, 330–337. [Google Scholar] [CrossRef]
- Mullighan, C.G.; Goorha, S.; Radtke, I.; Miller, C.B.; Coustan-Smith, E.; Dalton, J.D.; Girtman, K.; Mathew, S.; Ma, J.; Pounds, S.B.; et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446, 758–764. [Google Scholar] [CrossRef]
- Liu, Y.F.; Wang, B.Y.; Zhang, W.N.; Huang, J.Y.; Li, B.S.; Zhang, M.; Jiang, L.; Li, J.F.; Wang, M.J.; Dai, Y.J.; et al. Genomic Profiling of Adult and Pediatric B-cell Acute Lymphoblastic Leukemia. EBioMedicine 2016, 8, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Brown, F.C.; Cifani, P.; Drill, E.; He, J.; Still, E.; Zhong, S.; Balasubramanian, S.; Pavlick, D.; Yilmazel, B.; Knapp, K.M.; et al. Genomics of primary chemoresistance and remission induction failure in paediatric and adult acute myeloid leukaemia. Br. J. Haematol. 2017, 176, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Chalandon, Y.; Thomas, X.; Hayette, S.; Cayuela, J.M.; Abbal, C.; Huguet, F.; Raffoux, E.; Leguay, T.; Rousselot, P.; Lepretre, S.; et al. Randomized study of reduced-intensity chemotherapy combined with imatinib in adults with Ph-positive acute lymphoblastic leukemia. Blood 2015, 125, 3711–3719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sherman, R.M.; Salzberg, S.L. Pan-genomics in the human genome era. Nat. Rev. Genet. 2020, 21, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Pennisi, E. ENCODE Project Writes Eulogy for Junk DNA. Science 2012, 337, 1159–1161. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Adams, J.C. Physiological roles of non-coding RNAs. Am. J. Physiol. Cell Physiol. 2019, 317, C1–C2. [Google Scholar] [CrossRef]
- Salzberg, S.L. Open questions: How many genes do we have? BMC Biol. 2018, 16, 94. [Google Scholar] [CrossRef]
- Pertea, M. The human transcriptome: An unfinished story. Genes (Basel) 2012, 3, 344–360. [Google Scholar] [CrossRef] [Green Version]
- Kehr, B.; Helgadottir, A.; Melsted, P.; Jonsson, H.; Helgason, H.; Jonasdottir, A.; Jonasdottir, A.; Sigurdsson, A.; Gylfason, A.; Halldorsson, G.H.; et al. Diversity in non-repetitive human sequences not found in the reference genome. Nat. Genet. 2017, 49, 588–593. [Google Scholar] [CrossRef]
- Ballouz, S.; Dobin, A.; Gillis, J.A. Is it time to change the reference genome? Genome Biol. 2019, 20, 159. [Google Scholar] [CrossRef] [Green Version]
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-wide association studies of cancer: Current insights and future perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef]
- Gutierrez-Camino, A.; Martin-Guerrero, I.; Garcia de Andoin, N.; Sastre, A.; Carbone Baneres, A.; Astigarraga, I.; Navajas, A.; Garcia-Orad, A. Confirmation of involvement of new variants at CDKN2A/B in pediatric acute lymphoblastic leukemia susceptibility in the Spanish population. PLoS ONE 2017, 12, e0177421. [Google Scholar] [CrossRef] [PubMed]
- Vijayakrishnan, J.; Qian, M.; Studd, J.B.; Yang, W.; Kinnersley, B.; Law, P.J.; Broderick, P.; Raetz, E.A.; Allan, J.; Pui, C.H.; et al. Identification of four novel associations for B-cell acute lymphoblastic leukaemia risk. Nat. Commun. 2019, 10, 5348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederichs, S.; Bartsch, L.; Berkmann, J.C.; Frose, K.; Heitmann, J.; Hoppe, C.; Iggena, D.; Jazmati, D.; Karschnia, P.; Linsenmeier, M.; et al. The dark matter of the cancer genome: Aberrations in regulatory elements, untranslated regions, splice sites, non-coding RNA and synonymous mutations. EMBO Mol. Med. 2016, 8, 442–457. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, C.; Zhan, X.; Li, L.; Lei, J.; Shi, Y. Structure of the human activated spliceosome in three conformational states. Cell Res. 2018, 28, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Maquat, L.E.; Kinniburgh, A.J.; Beach, L.R.; Honig, G.R.; Lazerson, J.; Ershler, W.B.; Ross, J. Processing of human beta-globin mRNA precursor to mRNA is defective in three patients with beta+-thalassemia. Proc. Natl. Acad. Sci. USA 1980, 77, 4287–4291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thein, S.L. The molecular basis of β-thalassemia. Cold Spring Harb. Perspect. Med. 2013, 3, a011700. [Google Scholar] [CrossRef] [Green Version]
- Sotillo, E.; Barrett, D.M.; Black, K.L.; Bagashev, A.; Oldridge, D.; Wu, G.; Sussman, R.; Lanauze, C.; Ruella, M.; Gazzara, M.R.; et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015, 5, 1282–1295. [Google Scholar] [CrossRef] [Green Version]
- Fischer, J.; Paret, C.; El Malki, K.; Alt, F.; Wingerter, A.; Neu, M.A.; Kron, B.; Russo, A.; Lehmann, N.; Roth, L.; et al. CD19 Isoforms Enabling Resistance to CART-19 Immunotherapy Are Expressed in B-ALL Patients at Initial Diagnosis. J. Immunother. 2017, 40, 187–195. [Google Scholar] [CrossRef] [Green Version]
- Orlando, E.J.; Han, X.; Tribouley, C.; Wood, P.A.; Leary, R.J.; Riester, M.; Levine, J.E.; Qayed, M.; Grupp, S.A.; Boyer, M.; et al. Genetic mechanisms of target antigen loss in CAR19 therapy of acute lymphoblastic leukemia. Nat. Med. 2018, 24, 1504–1506. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, A.E.; Geron, I.; Gotlib, J.; Dao, K.-H.T.; Barroga, C.F.; Newton, I.G.; Giles, F.J.; Durocher, J.; Creusot, R.S.; Karimi, M.; et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc. Natl. Acad. Sci. USA 2009, 106, 3925–3929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, X.S.; Bea, S.; Valdes-Mas, R.; Villamor, N.; Gutierrez-Abril, J.; Martin-Subero, J.I.; Munar, M.; Rubio-Perez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.J.; Cooper, T.A. The pathobiology of splicing. J. Pathol. 2010, 220, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Shuai, S.; Suzuki, H.; Diaz-Navarro, A.; Nadeu, F.; Kumar, S.A.; Gutierrez-Fernandez, A.; Delgado, J.; Pinyol, M.; López-Otín, C.; Puente, X.S.; et al. The U1 spliceosomal RNA is recurrently mutated in multiple cancers. Nature 2019, 574, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.; Lee, S.C. Mutations in spliceosome genes and therapeutic opportunities in myeloid malignancies. Genes Chromosomes Cancer 2019, 58, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Black, K.L.; Naqvi, A.S.; Asnani, M.; Hayer, K.E.; Yang, S.Y.; Gillespie, E.; Bagashev, A.; Pillai, V.; Tasian, S.K.; Gazzara, M.R.; et al. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2018, 46, 11357–11369. [Google Scholar] [CrossRef] [Green Version]
- Crews, L.A.; Balaian, L.; Delos Santos, N.P.; Leu, H.S.; Court, A.C.; Lazzari, E.; Sadarangani, A.; Zipeto, M.A.; La Clair, J.J.; Villa, R.; et al. RNA Splicing Modulation Selectively Impairs Leukemia Stem Cell Maintenance in Secondary Human AML. Cell Stem Cell 2016, 19, 599–612. [Google Scholar] [CrossRef] [Green Version]
- Rojas, E.A.; Corchete, L.A.; Mateos, M.V.; García-Sanz, R.; Misiewicz-Krzeminska, I.; Gutiérrez, N.C. Transcriptome analysis reveals significant differences between primary plasma cell leukemia and multiple myeloma even when sharing a similar genetic background. Blood Cancer J. 2019, 9, 90. [Google Scholar] [CrossRef]
- Li, J.; Liu, C. Coding or Noncoding, the Converging Concepts of RNAs. Front. Genet. 2019, 10, 496. [Google Scholar] [CrossRef]
- Schuster, S.L.; Hsieh, A.C. The Untranslated Regions of mRNAs in Cancer. Trends Cancer 2019, 5, 245–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pippucci, T.; Savoia, A.; Perrotta, S.; Pujol-Moix, N.; Noris, P.; Castegnaro, G.; Pecci, A.; Gnan, C.; Punzo, F.; Marconi, C.; et al. Mutations in the 5' UTR of ANKRD26, the ankirin repeat domain 26 gene, cause an autosomal-dominant form of inherited thrombocytopenia, THC2. Am. J. Hum. Genet. 2011, 88, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marconi, C.; Canobbio, I.; Bozzi, V.; Pippucci, T.; Simonetti, G.; Melazzini, F.; Angori, S.; Martinelli, G.; Saglio, G.; Torti, M.; et al. 5’UTR point substitutions and N-terminal truncating mutations of ANKRD26 in acute myeloid leukemia. J. Hematol. Oncol. 2017, 10, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Seraihi, A.F.; Rio-Machin, A.; Tawana, K.; Bödör, C.; Wang, J.; Nagano, A.; Heward, J.A.; Iqbal, S.; Best, S.; Lea, N.; et al. GATA2 monoallelic expression underlies reduced penetrance in inherited GATA2-mutated MDS/AML. Leukemia 2018, 32, 2502–2507. [Google Scholar] [CrossRef]
- Diaz de Arce, A.J.; Noderer, W.L.; Wang, C.L. Complete motif analysis of sequence requirements for translation initiation at non-AUG start codons. Nucleic Acids Res. 2017, 46, 985–994. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Ranum, L.P. Repeat associated non-ATG (RAN) translation: New starts in microsatellite expansion disorders. Curr. Opin. Genet. Dev. 2014, 26, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Green, K.M.; Linsalata, A.E.; Todd, P.K. RAN translation-What makes it run? Brain Res. 2016, 1647, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Benzow, K.A.; Koob, M.D.; Condie, A.; Catovsky, D.; Matutes, E.; Yuille, M.R.; Houlston, R.S. Instability of CAG-trinucleotide repeats in chronic lymphocytic leukemia. Leuk. Lymphoma 2002, 43, 1987–1990. [Google Scholar] [CrossRef]
- Dunna, N.R.; Naushad, S.M.; Vuree, S.; Anuradha, C.; Sailaja, K.; Surekha, D.; Rao, D.R.; Vishnupriya, S. Association of thymidylate synthase 5'-UTR 28bp tandem repeat and serine hydroxymethyltransfarase C1420T polymorphisms with susceptibility to acute leukemia. Asian Pac. J. Cancer Prev. 2014, 15, 1719–1723. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, C.; Peixeiro, I.; Romao, L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet 2013, 9, e1003529. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.G.; Wilusz, J.E. Non-AUG translation: A new start for protein synthesis in eukaryotes. Genes Dev. 2017, 31, 1717–1731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sendoel, A.; Dunn, J.G.; Rodriguez, E.H.; Naik, S.; Gomez, N.C.; Hurwitz, B.; Levorse, J.; Dill, B.D.; Schramek, D.; Molina, H.; et al. Translation from unconventional 5' start sites drives tumour initiation. Nature 2017, 541, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat. Rev. Mol. Cell Biol. 2017, 18, 18–30. [Google Scholar] [CrossRef]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3' untranslated regions and fewer microRNA target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3' untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 7028–7033. [Google Scholar] [CrossRef] [Green Version]
- Singh, P.; Alley, T.L.; Wright, S.M.; Kamdar, S.; Schott, W.; Wilpan, R.Y.; Mills, K.D.; Graber, J.H. Global changes in processing of mRNA 3' untranslated regions characterize clinically distinct cancer subtypes. Cancer Res. 2009, 69, 9422–9430. [Google Scholar] [CrossRef] [Green Version]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Mayr, C.; Bartel, D.P. Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [Green Version]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3'-UTR landscape across seven tumour types. Nat. Commun. 2014, 5, 5274. [Google Scholar] [CrossRef] [Green Version]
- Ramsingh, G.; Koboldt, D.C.; Trissal, M.; Chiappinelli, K.B.; Wylie, T.; Koul, S.; Chang, L.W.; Nagarajan, R.; Fehniger, T.A.; Goodfellow, P.; et al. Complete characterization of the microRNAome in a patient with acute myeloid leukemia. Blood 2010, 116, 5316–5326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Benito, M.; Loayza-Puch, F.; Oude Vrielink, J.A.; Odero, M.D.; Agami, R. 3'UTR-mediated gene silencing of the Mixed Lineage Leukemia (MLL) gene. PLoS ONE 2011, 6, e25449. [Google Scholar] [CrossRef]
- Wiestner, A.; Tehrani, M.; Chiorazzi, M.; Wright, G.; Gibellini, F.; Nakayama, K.; Liu, H.; Rosenwald, A.; Muller-Hermelink, H.K.; Ott, G.; et al. Point mutations and genomic deletions in CCND1 create stable truncated cyclin D1 mRNAs that are associated with increased proliferation rate and shorter survival. Blood 2007, 109, 4599–4606. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Singh, I.; Tisdale, S.; Abdel-Wahab, O.; Leslie, C.S.; Mayr, C. Widespread intronic polyadenylation inactivates tumour suppressor genes in leukaemia. Nature 2018, 561, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Asnani, M.; Thomas-Tikhonenko, A. Exons of Leukemia Suppressor Genes: Creative Assembly Required. Trends Cancer 2018, 4, 796–798. [Google Scholar] [CrossRef]
- Climente-Gonzalez, H.; Porta-Pardo, E.; Godzik, A.; Eyras, E. The Functional Impact of Alternative Splicing in Cancer. Cell Rep. 2017, 20, 2215–2226. [Google Scholar] [CrossRef] [Green Version]
- Asnani, M.; Hayer, K.E.; Naqvi, A.S.; Zheng, S.; Yang, S.Y.; Oldridge, D.; Ibrahim, F.; Maragkakis, M.; Gazzara, M.R.; Black, K.L.; et al. Retention of CD19 intron 2 contributes to CART-19 resistance in leukemias with subclonal frameshift mutations in CD19. Leukemia 2020, 34, 1202–1207. [Google Scholar] [CrossRef]
- de Necochea-Campion, R.; Shouse, G.P.; Zhou, Q.; Mirshahidi, S.; Chen, C.-S. Aberrant splicing and drug resistance in AML. J. Hematol. Oncol. 2016, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Böttcher, S.; et al. Mutations driving CLL and their evolution in progression and relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Mullighan, C.G.; Phillips, L.A.; Su, X.; Ma, J.; Miller, C.B.; Shurtleff, S.A.; Downing, J.R. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science 2008, 322, 1377–1380. [Google Scholar] [CrossRef] [Green Version]
- Anguille, S.; Van Tendeloo, V.F.; Berneman, Z.N. Leukemia-associated antigens and their relevance to the immunotherapy of acute myeloid leukemia. Leukemia 2012, 26, 2186–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acheampong, D.O.; Adokoh, C.K.; Asante, D.B.; Asiamah, E.A.; Barnie, P.A.; Bonsu, D.O.M.; Kyei, F. Immunotherapy for acute myeloid leukemia (AML): A potent alternative therapy. Biomed Pharm. 2018, 97, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Ocadlikova, D.; Lecciso, M.; Isidori, A.; Loscocco, F.; Visani, G.; Amadori, S.; Cavo, M.; Curti, A. Chemotherapy-Induced Tumor Cell Death at the Crossroads Between Immunogenicity and Immunotolerance: Focus on Acute Myeloid Leukemia. Front. Oncol. 2019, 9, 1004. [Google Scholar] [CrossRef] [PubMed]
- Rech, A.J.; Balli, D.; Mantero, A.; Ishwaran, H.; Nathanson, K.L.; Stanger, B.Z.; Vonderheide, R.H. Tumor Immunity and Survival as a Function of Alternative Neopeptides in Human Cancer. Cancer Immunol. Res. 2018, 6, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasagi, M.; Shukla, S.A.; Fritsch, E.F.; Keskin, D.B.; DeLuca, D.; Carmona, E.; Zhang, W.; Sougnez, C.; Cibulskis, K.; Sidney, J.; et al. Systematic identification of personal tumor-specific neoantigens in chronic lymphocytic leukemia. Blood 2014, 124, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Cancer Genome Atlas Research, N.; et al. Comprehensive Analysis of Alternative Splicing Across Tumors from 8705 Patients. Cancer Cell 2018, 34, 211–224.e216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasinghe, R.G.; Cao, S.; Gao, Q.; Wendl, M.C.; Vo, N.S.; Reynolds, S.M.; Zhao, Y.; Climente-Gonzalez, H.; Chai, S.; Wang, F.; et al. Systematic Analysis of Splice-Site-Creating Mutations in Cancer. Cell Rep. 2018, 23, 270–281.e273. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.C.; Wu, Q.; Chen, J.; Xuan, Z.; Jung, Y.-C.; Zhang, M.Q.; Rowley, J.D.; Wang, S.M. The transcriptome of human CD34+ hematopoietic stem-progenitor cells. Proc. Natl. Acad. Sci. USA 2009, 106, 8278–8283. [Google Scholar] [CrossRef] [Green Version]
- Schwarzer, A.; Emmrich, S.; Schmidt, F.; Beck, D.; Ng, M.; Reimer, C.; Adams, F.F.; Grasedieck, S.; Witte, D.; Käbler, S.; et al. The non-coding RNA landscape of human hematopoiesis and leukemia. Nat. Commun. 2017, 8, 218. [Google Scholar] [CrossRef]
- Wilson, N.K.; Göttgens, B. Single-Cell Sequencing in Normal and Malignant Hematopoiesis. HemaSphere 2018, 2, e34. [Google Scholar] [CrossRef]
- Van Galen, P.; Hovestadt, V.; Wadsworth Ii, M.H.; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Lombardi Story, J.; et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell 2019, 176, 1265–1281.e1224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Xiao, Y.; Sun, J.; Sun, H.; Chen, H.; Zhu, Y.; Fu, H.; Yu, C.; Weigao, E.; Lai, S.; et al. A single-cell survey of cellular hierarchy in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 128. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Bajic, V.B.; Zhang, Z. On the classification of long non-coding RNAs. RNA Biol. 2013, 10, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, J.D.; Wei, Y.; Khavari, P.A. The functions and unique features of long intergenic non-coding RNA. Nat. Rev. Mol. Cell Biol. 2018, 19, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Orera, J.; Albà, M.M. Conserved regions in long non-coding RNAs contain abundant translation and protein–RNA interaction signatures. NAR Genom. Bioinform. 2019, 1, e2. [Google Scholar] [CrossRef] [Green Version]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palazzo, A.F.; Lee, E.S. Sequence Determinants for Nuclear Retention and Cytoplasmic Export of mRNAs and lncRNAs. Front. Genet. 2018, 9, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trimarchi, T.; Bilal, E.; Ntziachristos, P.; Fabbri, G.; Dalla-Favera, R.; Tsirigos, A.; Aifantis, I. Genome-wide mapping and characterization of Notch-regulated long noncoding RNAs in acute leukemia. Cell 2014, 158, 593–606. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zhou, N.; Watabe, K.; Lu, Z.; Wu, F.; Xu, M.; Mo, Y.Y. Long non-coding RNA UCA1 promotes breast tumor growth by suppression of p27 (Kip1). Cell Death Dis. 2014, 5, e1008. [Google Scholar] [CrossRef]
- Belver, L.; Ferrando, A. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat. Rev. Cancer 2016, 16, 494–507. [Google Scholar] [CrossRef]
- Luo, H.; Zhu, G.; Xu, J.; Lai, Q.; Yan, B.; Guo, Y.; Fung, T.K.; Zeisig, B.B.; Cui, Y.; Zha, J.; et al. HOTTIP lncRNA Promotes Hematopoietic Stem Cell Self-Renewal Leading to AML-like Disease in Mice. Cancer Cell 2019, 36, 645–659.e648. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Leong, W.Z.; Ngoc, P.C.T.; Tan, T.K.; Bertulfo, F.C.; Lim, M.C.; An, O.; Li, Z.; Yeoh, A.E.J.; Fullwood, M.J.; et al. The enhancer RNA ARIEL activates the oncogenic transcriptional program in T-cell acute lymphoblastic leukemia. Blood 2019, 134, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Amodio, N.; Raimondi, L.; Juli, G.; Stamato, M.A.; Caracciolo, D.; Tagliaferri, P.; Tassone, P. MALAT1: A druggable long non-coding RNA for targeted anti-cancer approaches. J. Hematol. Oncol. 2018, 11, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradeepa, M.M.; McKenna, F.; Taylor, G.C.; Bengani, H.; Grimes, G.R.; Wood, A.J.; Bhatia, S.; Bickmore, W.A. Psip1/p52 regulates posterior Hoxa genes through activation of lncRNA Hottip. PLoS Genet 2017, 13, e1006677. [Google Scholar] [CrossRef] [Green Version]
- Tran, N.T.; Su, H.; Khodadadi-Jamayran, A.; Lin, S.; Zhang, L.; Zhou, D.; Pawlik, K.M.; Townes, T.M.; Chen, Y.; Mulloy, J.C.; et al. The AS-RBM15 lncRNA enhances RBM15 protein translation during megakaryocyte differentiation. EMBO Rep. 2016, 17, 887–900. [Google Scholar] [CrossRef]
- Fernando, T.R.; Contreras, J.R.; Zampini, M.; Rodriguez-Malave, N.I.; Alberti, M.O.; Anguiano, J.; Tran, T.M.; Palanichamy, J.K.; Gajeton, J.; Ung, N.M.; et al. The lncRNA CASC15 regulates SOX4 expression in RUNX1-rearranged acute leukemia. Mol. Cancer 2017, 16, 126. [Google Scholar] [CrossRef]
- Jianyong, S.; Yanlu, X.; Kuo, J.; Bo, X.; Tongtong, J.; Renji, W.; Yuankang, Z.; Hong, T.; Tao, J.; Angang, Y.; et al. Hypoxia-sensitive Long Noncoding RNA CASC15 Promotes Lung Tumorigenesis by Regulating the SOX4/β-catenin Axis. J. Exp. Amp; Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Chen, L.; Fan, X.; Zhu, J.; Chen, X.; Liu, Y.; Zhou, H. LncRNA MAGI2-AS3 inhibits the self-renewal of leukaemic stem cells by promoting TET2-dependent DNA demethylation of the LRIG1 promoter in acute myeloid leukaemia. RNA Biol. 2020, 17, 784–793. [Google Scholar] [CrossRef]
- Hughes, J.M.; Legnini, I.; Salvatori, B.; Masciarelli, S.; Marchioni, M.; Fazi, F.; Morlando, M.; Bozzoni, I.; Fatica, A. C/EBPalpha-p30 protein induces expression of the oncogenic long non-coding RNA UCA1 in acute myeloid leukemia. Oncotarget 2015, 6, 18534–18544. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Li, W.; Sun, Y.; Yu, D.; Wen, X.; Wang, H.; Cui, J.; Wang, G.; Hoffman, A.R.; Hu, J.F. A novel antisense long noncoding RNA within the IGF1R gene locus is imprinted in hematopoietic malignancies. Nucleic Acids Res. 2014, 42, 9588–9601. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Wang, F.; Zha, J.; Li, H.; Yan, B.; Du, Q.; Yang, F.; Sobh, A.; Vulpe, C.; Drusbosky, L.; et al. CTCF boundary remodels chromatin domain and drives aberrant HOX gene transcription in acute myeloid leukemia. Blood 2018, 132, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Ebralidze, A.K.; Guibal, F.C.; Steidl, U.; Zhang, P.; Lee, S.; Bartholdy, B.; Jorda, M.A.; Petkova, V.; Rosenbauer, F.; Huang, G.; et al. PU.1 expression is modulated by the balance of functional sense and antisense RNAs regulated by a shared cis-regulatory element. Genes Dev. 2008, 22, 2085–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Kouwe, E.; Heller, G.; Czibere, A.; Castilla, L.H.; Delwel, R.; Di Ruscio, A.; Ebralidze, A.K.; Forte, M.; Kazianka, L.; Kornauth, C.; et al. Core binding factor leukemia hijacks T-cell prone PU.1 antisense promoter. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sun, M.D.; Zheng, Y.Q.; Wang, L.P.; Zhao, H.T.; Yang, S. Long noncoding RNA UCA1 promotes cell proliferation, migration and invasion of human leukemia cells via sponging miR-126. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, Y.; Xu, X. Knockdown of LncRNA-UCA1 suppresses chemoresistance of pediatric AML by inhibiting glycolysis through the microRNA-125a/hexokinase 2 pathway. J. Cell. Biochem. 2018, 119, 6296–6308. [Google Scholar] [CrossRef]
- Bousard, A.; Raposo, A.C.; Zylicz, J.J.; Picard, C.; Pires, V.B.; Qi, Y.; Gil, C.; Syx, L.; Chang, H.Y.; Heard, E.; et al. The role of Xist-mediated Polycomb recruitment in the initiation of X-chromosome inactivation. EMBO Rep. 2019, 20, e48019. [Google Scholar] [CrossRef]
- Yildirim, E.; Kirby, J.E.; Brown, D.E.; Mercier, F.E.; Sadreyev, R.I.; Scadden, D.T.; Lee, J.T. Xist RNA is a potent suppressor of hematologic cancer in mice. Cell 2013, 152, 727–742. [Google Scholar] [CrossRef] [Green Version]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; Zhou, B.; Li, H.; Huang, X.; Wu, Y.; Xing, C.; Yu, X.; Ji, Y. Long noncoding RNA HOTAIR promotes the self-renewal of leukemia stem cells through epigenetic silencing of p15. Exp. Hematol. 2018, 67, 32–40.e33. [Google Scholar] [CrossRef]
- Xing, C.Y.; Hu, X.Q.; Xie, F.Y.; Yu, Z.J.; Li, H.Y.; Bin, Z.; Wu, J.B.; Tang, L.Y.; Gao, S.M. Long non-coding RNA HOTAIR modulates c-KIT expression through sponging miR-193a in acute myeloid leukemia. FEBS Lett. 2015, 589, 1981–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, W.; Cao, L.; Li, Z.; Wang, X. Long Non-Coding RNA CCAT1 Acts as a Competing Endogenous RNA to Regulate Cell Growth and Differentiation in Acute Myeloid Leukemia. Mol. Cells 2016, 39, 330–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lian, Z.; Padden, C.; Gerstein, M.B.; Rozowsky, J.; Snyder, M.; Gingeras, T.R.; Kapranov, P.; Weissman, S.M.; Newburger, P.E. A myelopoiesis-associated regulatory intergenic noncoding RNA transcript within the human HOXA cluster. Blood 2009, 113, 2526–2534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Weissman, S.M.; Newburger, P.E. Long intergenic non-coding RNA HOTAIRM1 regulates cell cycle progression during myeloid maturation in NB4 human promyelocytic leukemia cells. RNA Biol. 2014, 11, 777–787. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.Q.D.; Dostie, J. Reciprocal regulation of chromatin state and architecture by HOTAIRM1 contributes to temporal collinear HOXA gene activation. Nucleic Acids Res. 2017, 45, 1091–1104. [Google Scholar] [CrossRef]
- Chen, Z.H.; Wang, W.T.; Huang, W.; Fang, K.; Sun, Y.M.; Liu, S.R.; Luo, X.Q.; Chen, Y.Q. The lncRNA HOTAIRM1 regulates the degradation of PML-RARA oncoprotein and myeloid cell differentiation by enhancing the autophagy pathway. Cell Death Differ. 2017, 24, 212–224. [Google Scholar] [CrossRef]
- Garzon, R.; Volinia, S.; Papaioannou, D.; Nicolet, D.; Kohlschmidt, J.; Yan, P.S.; Mrózek, K.; Bucci, D.; Carroll, A.J.; Baer, M.R.; et al. Expression and prognostic impact of lncRNAs in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, 18679–18684. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, D.; Nicolet, D.; Volinia, S.; Mrózek, K.; Yan, P.; Bundschuh, R.; Carroll, A.J.; Kohlschmidt, J.; Blum, W.; Powell, B.L.; et al. Prognostic and biologic significance of long non-coding RNA profiling in younger adults with cytogenetically normal acute myeloid leukemia. Haematologica 2017, 102, 1391–1400. [Google Scholar] [CrossRef] [Green Version]
- De Clara, E.; Gourvest, M.; Ma, H.; Vergez, F.; Tosolini, M.; Dejean, S.; Demur, C.; Delabesse, E.; Recher, C.; Touriol, C.; et al. Long non-coding RNA expression profile in cytogenetically normal acute myeloid leukemia identifies a distinct signature and a new biomarker in NPM1-mutated patients. Haematologica 2017, 102, 1718–1726. [Google Scholar] [CrossRef] [Green Version]
- Helsmoortel, H.H.; De Moerloose, B.; Pieters, T.; Ghazavi, F.; Bresolin, S.; Cave, H.; de Vries, A.; de Haas, V.; Flotho, C.; Labarque, V.; et al. LIN28B is over-expressed in specific subtypes of pediatric leukemia and regulates lncRNA H19. Haematologica 2016, 101, e240–e244. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wu, P.; Lin, R.; Rong, L.; Xue, Y.; Fang, Y. LncRNA NALT interaction with NOTCH1 promoted cell proliferation in pediatric T cell acute lymphoblastic leukemia. Sci. Rep. 2015, 5, 13749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yang, X.; Sun, X.; Rong, L.; Kang, M.; Wu, P.; Ji, X.; Lin, R.; Huang, J.; Xue, Y.; et al. Bone marrow infiltrated Lnc-INSR induced suppressive immune microenvironment in pediatric acute lymphoblastic leukemia. Cell Death Dis. 2018, 9, 1043. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Gong, C.; Yuan, K. LncRNA UCA1 promotes cell proliferation, invasion and migration of laryngeal squamous cell carcinoma cells by activating Wnt/beta-catenin signaling pathway. Exp. Ther. Med. 2019, 17, 1182–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, L.; Wang, Y.Y.; Jiang, P. LncRNA LINC00909 promotes cell proliferation and metastasis in pediatric acute myeloid leukemia via miR-625-mediated modulation of Wnt/beta-catenin signaling. Biochem. Biophys. Res. Commun. 2020, 527, 654–661. [Google Scholar] [CrossRef]
- Liang, Y.; Li, E.; Zhang, H.; Zhang, L.; Tang, Y.; Wanyan, Y. Silencing of lncRNA UCA1 curbs proliferation and accelerates apoptosis by repressing SIRT1 signals by targeting miR-204 in pediatric AML. J. Biochem. Mol. Toxicol. 2020, 34, e22435. [Google Scholar] [CrossRef]
- Li, X.; Song, F.; Sun, H. Long non-coding RNA AWPPH interacts with ROCK2 and regulates the proliferation and apoptosis of cancer cells in pediatric T-cell acute lymphoblastic leukemia. Oncol. Lett. 2020, 20, 239. [Google Scholar] [CrossRef]
- Chen, L.; Shi, Y.; Li, J.; Yang, X.; Li, R.; Zhou, X.; Zhu, L. LncRNA CDKN2B-AS1 contributes to tumorigenesis and chemoresistance in pediatric T-cell acute lymphoblastic leukemia through miR-335-3p/TRAF5 axis. Anti-Cancer Drugs 2020. [Google Scholar] [CrossRef]
- Su, W.; Hong, Y.; Jiang, H. Analysis of Relationship between Long Non-Coding RNA Small Nucleolar RNA Host Gene 1 and Acute Myeloid Leukemia Risk and Prognosis in Pediatric Patients. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2020, 28, 1127–1136. [Google Scholar] [CrossRef]
- Guan, X.; Wen, X.; Xiao, J.; An, X.; Yu, J.; Guo, Y. Lnc-SOX6-1 upregulation correlates with poor risk stratification and worse treatment outcomes, and promotes cell proliferation while inhibits apoptosis in pediatric acute myeloid leukemia. Int. J. Lab. Hematol. 2019, 41, 234–241. [Google Scholar] [CrossRef]
- Cuadros, M.; Andrades, A.; Coira, I.F.; Balinas, C.; Rodriguez, M.I.; Alvarez-Perez, J.C.; Peinado, P.; Arenas, A.M.; Garcia, D.J.; Jimenez, P.; et al. Expression of the long non-coding RNA TCL6 is associated with clinical outcome in pediatric B-cell acute lymphoblastic leukemia. Blood Cancer J. 2019, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Hofmans, M.; Lammens, T.; Helsmoortel, H.H.; Bresolin, S.; Cavé, H.; Flotho, C.; Hasle, H.; van den Heuvel-Eibrink, M.M.; Niemeyer, C.; Stary, J.; et al. The long non-coding RNA landscape in juvenile myelomonocytic leukemia. Haematologica 2018, 103, e501–e504. [Google Scholar] [CrossRef] [PubMed]
- Hofmans, M.; Depreter, B.; Lammens, T.; Cave, H.; Flotho, C.; Hasle, H.; de Haas, V.; Niemeyer, C.M.; Stary, J.; Van Vlierberghe, P.; et al. Long Non-Coding RNAs As Novel Therapeutic Targets in Juvenile Myelomonocytic Leukemia: Proof of Concept Study. Blood 2019, 134, 1701. [Google Scholar] [CrossRef]
- Cao, L.; Xiao, P.F.; Tao, Y.F.; Hu, S.Y.; Lu, J.; Zhao, W.L.; Li, Z.H.; Wang, N.N.; Wang, J.; Feng, X.; et al. Microarray profiling of bone marrow long non-coding RNA expression in Chinese pediatric acute myeloid leukemia patients. Oncol. Rep. 2016, 35, 757–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernando, T.R.; Rodriguez-Malave, N.I.; Waters, E.V.; Yan, W.; Casero, D.; Basso, G.; Pigazzi, M.; Rao, D.S. LncRNA Expression Discriminates Karyotype and Predicts Survival in B-Lymphoblastic Leukemia. Mol. Cancer Res. 2015, 13, 839–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Malave, N.I.; Fernando, T.R.; Patel, P.C.; Contreras, J.R.; Palanichamy, J.K.; Tran, T.M.; Anguiano, J.; Davoren, M.J.; Alberti, M.O.; Pioli, K.T.; et al. BALR-6 regulates cell growth and cell survival in B-lymphoblastic leukemia. Mol. Cancer 2015, 14, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diffner, E.; Beck, D.; Gudgin, E.; Thoms, J.A.I.; Knezevic, K.; Pridans, C.; Foster, S.; Goode, D.; Lim, W.K.; Boelen, L.; et al. Activity of a heptad of transcription factors is associated with stem cell programs and clinical outcome in acute myeloid leukemia. Blood 2013, 121, 2289–2300. [Google Scholar] [CrossRef] [PubMed]
- Krivtsov, A.V.; Twomey, D.; Feng, Z.; Stubbs, M.C.; Wang, Y.; Faber, J.; Levine, J.E.; Wang, J.; Hahn, W.C.; Gilliland, D.G.; et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL–AF9. Nature 2006, 442, 818–822. [Google Scholar] [CrossRef]
- Yan, X.; Hu, Z.; Feng, Y.; Hu, X.; Yuan, J.; Zhao, S.D.; Zhang, Y.; Yang, L.; Shan, W.; He, Q.; et al. Comprehensive Genomic Characterization of Long Non-coding RNAs across Human Cancers. Cancer Cell 2015, 28, 529–540. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Li, X.; Zhi, H.; Zhang, Y.; Wang, P.; Wang, Y.; Shang, S.; Fang, Y.; Shen, W.; Ning, S.; et al. Comprehensive Characterization of Somatic Mutations Impacting lncRNA Expression for Pan-Cancer. Mol. Ther. Nucleic Acids 2019, 18, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Minotti, L.; Agnoletto, C.; Baldassari, F.; Corra, F.; Volinia, S. SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies? High Throughput 2018, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Perez de Acha, O.; Rossi, M.; Gorospe, M. Circular RNAs in Blood Malignancies. Front. Mol. Biosci. 2020, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhong, C.; Jiao, J.; Li, P.; Cui, B.; Ji, C.; Ma, D. Characterization of hsa_circ_0004277 as a New Biomarker for Acute Myeloid Leukemia via Circular RNA Profile and Bioinformatics Analysis. Int. J. Mol. Sci. 2017, 18, 597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Liu, T.; Liu, J.; Feng, Y.; Wang, B.; Wang, J.; Bai, J.; Zhao, W.; Shen, Y.; Wang, X.; et al. Circ-ANAPC7 is Upregulated in Acute Myeloid Leukemia and Appears to Target the MiR-181 Family. Cell. Physiol. Biochem. 2018, 47, 1998–2007. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.Y.; Yi, J.; Zhu, X.; Zhang, J.; Zhou, J.; Tang, X.; Lin, J.; Wang, P.; Deng, Z.Q. Circular RNA of vimentin expression as a valuable predictor for acute myeloid leukemia development and prognosis. J. Cell. Physiol. 2019, 234, 3711–3719. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Du, Y.; Beckford, J.; Alachkar, H. Upregulation of the EMT marker vimentin is associated with poor clinical outcome in acute myeloid leukemia. J. Transl. Med. 2018, 16, 170. [Google Scholar] [CrossRef] [Green Version]
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 165, 289–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Chen, W.M.; Wang, Z.H.; Wei, T.N.; Chen, Z.Z.; Wu, W.B. CircPAN3 mediates drug resistance in acute myeloid leukemia through the miR-153-5p/miR-183-5p-XIAP axis. Exp. Hematol. 2019, 70, 42–54.e43. [Google Scholar] [CrossRef]
- L Abbate, A.; Tolomeo, D.; Cifola, I.; Severgnini, M.; Turchiano, A.; Augello, B.; Squeo, G.; D Addabbo, P.; Traversa, D.; Daniele, G.; et al. MYC-containing amplicons in acute myeloid leukemia: Genomic structures, evolution, and transcriptional consequences. Leukemia 2018, 32, 2152–2166. [Google Scholar] [CrossRef]
- Hu, J.; Han, Q.; Gu, Y.; Ma, J.; McGrath, M.; Qiao, F.; Chen, B.; Song, C.; Ge, Z. Circular RNA PVT1 expression and its roles in acute lymphoblastic leukemia. Epigenomics 2018, 10, 723–732. [Google Scholar] [CrossRef]
- Yuan, D.M.; Ma, J.; Fang, W.B. Identification of non-coding RNA regulatory networks in pediatric acute myeloid leukemia reveals circ-0004136 could promote cell proliferation by sponging miR-142. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 9251–9258. [Google Scholar] [CrossRef]
- Huang, W.; Fang, K.; Chen, T.-Q.; Zeng, Z.-C.; Sun, Y.-M.; Han, C.; Sun, L.-Y.; Chen, Z.-H.; Yang, Q.-Q.; Pan, Q.; et al. circRNA circAF4 functions as an oncogene to regulate MLL-AF4 fusion protein expression and inhibit MLL leukemia progression. J. Hematol. Oncol. 2019, 12, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaffo, E.; Boldrin, E.; Dal Molin, A.; Bresolin, S.; Bonizzato, A.; Trentin, L.; Frasson, C.; Debatin, K.-M.; Meyer, L.H.; Te Kronnie, G. Circular RNA differential expression in blood cell populations and exploration of circRNA deregulation in pediatric acute lymphoblastic leukemia. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, R.; Ma, X.-K.; Chen, L.-L.; Yang, L. Increased complexity of circRNA expression during species evolution. RNA Biol. 2017, 14, 1064–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Qu, H. circVAR database: Genome-wide archive of genetic variants for human circular RNAs. BMC Genom. 2020, 21, 750. [Google Scholar] [CrossRef]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [Green Version]
- Pekarsky, Y.; Croce, C.M. Role of miR-15/16 in CLL. Cell Death Differ. 2015, 22, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Starczynowski, D.T.; Kuchenbauer, F.; Argiropoulos, B.; Sung, S.; Morin, R.; Muranyi, A.; Hirst, M.; Hogge, D.; Marra, M.; Wells, R.A.; et al. Identification of miR-145 and miR-146a as mediators of the 5q–syndrome phenotype. Nat. Med. 2010, 16, 49–58. [Google Scholar] [CrossRef]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef]
- Pekarsky, Y.; Croce, C.M. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget 2010, 1, 224–227. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, A.A.; So, A.Y.; Mehta, A.; Minisandram, A.; Sinha, N.; Jonsson, V.D.; Rao, D.S.; O'Connell, R.M.; Baltimore, D. Oncomir miR-125b regulates hematopoiesis by targeting the gene Lin28A. Proc. Natl. Acad. Sci. USA 2012, 109, 4233–4238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- So, A.Y.-L.; Sookram, R.; Chaudhuri, A.A.; Minisandram, A.; Cheng, D.; Xie, C.; Lim, E.L.; Flores, Y.G.; Jiang, S.; Kim, J.T.; et al. Dual mechanisms by which miR-125b represses IRF4 to induce myeloid and B-cell leukemias. Blood 2014, 124, 1502–1512. [Google Scholar] [CrossRef] [PubMed]
- Narayan, N.; Morenos, L.; Phipson, B.; Willis, S.N.; Brumatti, G.; Eggers, S.; Lalaoui, N.; Brown, L.M.; Kosasih, H.J.; Bartolo, R.C.; et al. Functionally distinct roles for different miR-155 expression levels through contrasting effects on gene expression, in acute myeloid leukaemia. Leukemia 2017, 31, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Lechman, E.R.; Gentner, B.; Ng, S.W.K.; Schoof, E.M.; van Galen, P.; Kennedy, J.A.; Nucera, S.; Ciceri, F.; Kaufmann, K.B.; Takayama, N.; et al. miR-126 Regulates Distinct Self-Renewal Outcomes in Normal and Malignant Hematopoietic Stem Cells. Cancer Cell 2016, 29, 602–606. [Google Scholar] [CrossRef]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.L.; et al. Bone marrow niche trafficking of miR-126 controls the self-renewal of leukemia stem cells in chronic myelogenous leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Chen, P.; Su, R.; Li, Y.; Hu, C.; Wang, Y.; Arnovitz, S.; He, M.; Gurbuxani, S.; Zuo, Z.; et al. Overexpression and knockout of miR-126 both promote leukemogenesis. Blood 2015, 126, 2005–2015. [Google Scholar] [CrossRef] [Green Version]
- Luan, C.; Yang, Z.; Chen, B. The functional role of microRNA in acute lymphoblastic leukemia: Relevance for diagnosis, differential diagnosis, prognosis, and therapy. Onco Targets Ther. 2015, 8, 2903–2914. [Google Scholar] [CrossRef] [Green Version]
- Wallace, J.A.; O’Connell, R.M. MicroRNAs and acute myeloid leukemia: Therapeutic implications and emerging concepts. Blood 2017, 130, 1290–1301. [Google Scholar] [CrossRef] [Green Version]
- Trino, S.; Lamorte, D.; Caivano, A.; Laurenzana, I.; Tagliaferri, D.; Falco, G.; Del Vecchio, L.; Musto, P.; De Luca, L. MicroRNAs as New Biomarkers for Diagnosis and Prognosis, and as Potential Therapeutic Targets in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2018, 19, 460. [Google Scholar] [CrossRef] [Green Version]
- Grobbelaar, C.; Ford, A.M. The Role of MicroRNA in Paediatric Acute Lymphoblastic Leukaemia: Challenges for Diagnosis and Therapy. J. Oncol. 2019, 2019, 8941471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wu, H.; Qin, X. MicroRNA-206 serves as a tumor suppressor in pediatric acute myeloid leukemia by targeting Cyclin D1. Pathol. Res. Pract. 2019, 215, 152554. [Google Scholar] [CrossRef] [PubMed]
- Rzepiel, A.; Kutszegi, N.; Gézsi, A.; Sági, J.C.; Egyed, B.; Péter, G.; Butz, H.; Nyírő, G.; Müller, J.; Kovács, G.T.; et al. Circulating microRNAs as minimal residual disease biomarkers in childhood acute lymphoblastic leukemia. J. Transl. Med. 2019, 17, 372. [Google Scholar] [CrossRef] [PubMed]
- Stamatopoulos, B.; Van Damme, M.; Crompot, E.; Dessars, B.; Housni, H.E.; Mineur, P.; Meuleman, N.; Bron, D.; Lagneaux, L. Opposite Prognostic Significance of Cellular and Serum Circulating MicroRNA-150 in Patients with Chronic Lymphocytic Leukemia. Mol. Med. 2015, 21, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Ramsingh, G.; Jacoby, M.A.; Shao, J.; De Jesus Pizzaro, R.E.; Shen, D.; Trissal, M.; Getz, A.H.; Ley, T.J.; Walter, M.J.; Link, D.C. Acquired copy number alterations of miRNA genes in acute myeloid leukemia are uncommon. Blood 2013, 122, e44–e51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cammaerts, S.; Strazisar, M.; De Rijk, P.; Del Favero, J. Genetic variants in microRNA genes: Impact on microRNA expression, function, and disease. Front. Genet. 2015, 6, 186. [Google Scholar] [CrossRef] [Green Version]
- Kawahara, Y. Human diseases caused by germline and somatic abnormalities in microRNA and microRNA-related genes. Congenit. Anom. (Kyoto) 2014, 54, 12–21. [Google Scholar] [CrossRef]
- Saunders, M.A.; Liang, H.; Li, W.-H. Human polymorphism at microRNAs and microRNA target sites. Proc. Natl. Acad. Sci. USA 2007, 104, 3300–3305. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Tong, Y.; Zhang, H.M.; Wang, K.; Hu, T.; Shan, G.; Sun, J.; Guo, A.Y. Genome-wide identification of SNPs in microRNA genes and the SNP effects on microRNA target binding and biogenesis. Hum. Mutat. 2012, 33, 254–263. [Google Scholar] [CrossRef]
- Han, M.; Zheng, Y. Comprehensive analysis of single nucleotide polymorphisms in human microRNAs. PLoS ONE 2013, 8, e78028. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, A.; Ziebarth, J.D.; Cui, Y. SomamiR: A database for somatic mutations impacting microRNA function in cancer. Nucleic Acids Res. 2013, 41, D977–D982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, A.; Cui, Y. SomamiR 2.0: A database of cancer somatic mutations altering microRNA-ceRNA interactions. Nucleic Acids Res. 2016, 44, D1005–D1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, J.K.; Chow, M.Y.; Zhang, Y.; Leung, S.W. siRNA Versus miRNA as Therapeutics for Gene Silencing. Mol. Ther. Nucleic Acids 2015, 4, e252. [Google Scholar] [CrossRef] [Green Version]
- Dorrance, A.M.; Neviani, P.; Ferenchak, G.J.; Huang, X.; Nicolet, D.; Maharry, K.S.; Ozer, H.G.; Hoellarbauer, P.; Khalife, J.; Hill, E.B.; et al. Targeting leukemia stem cells in vivo with antagomiR-126 nanoparticles in acute myeloid leukemia. Leukemia 2015, 29, 2143–2153. [Google Scholar] [CrossRef] [Green Version]
- Bouchie, A. First microRNA mimic enters clinic. Nat. Biotechnol. 2013, 31, 577. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.L.; Kim, T.Y.; et al. Phase 1 study of MRX34, a liposomal miR-34a mimic, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- O'Brien, S.; Moore, J.O.; Boyd, T.E.; Larratt, L.M.; Skotnicki, A.B.; Koziner, B.; Chanan-Khan, A.A.; Seymour, J.F.; Gribben, J.; Itri, L.M.; et al. 5-year survival in patients with relapsed or refractory chronic lymphocytic leukemia in a randomized, phase III trial of fludarabine plus cyclophosphamide with or without oblimersen. J. Clin. Oncol. 2009, 27, 5208–5212. [Google Scholar] [CrossRef]
- Moreno, P.M.; Pego, A.P. Therapeutic antisense oligonucleotides against cancer: Hurdling to the clinic. Front. Chem. 2014, 2, 87. [Google Scholar] [CrossRef] [Green Version]
- Hoshiko, T.; Kubota, Y.; Akisawa, T.; Watanabe, T.; Tanigawara, K.; Yano, J.; Kimura, S. Naked antisense double-stranded DNA oligonucleotide efficiently suppresses BCR-ABL positive leukemic cells. Investig. New Drugs 2020, 38, 1012–1019. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E.; Peel, S.; Schantz, A.; England, R.; Beano, M.; Bates, S.; Desai, A.; Puri, S.; Ashford, M.; Jones, A. Endocytic profiling of Cancer cell models reveals critical factors influencing lipid nanoparticle mediated mRNA delivery and protein expression. Mol. Ther. 2019, 27, 1950–1962. [Google Scholar] [CrossRef] [PubMed]
- Kedmi, R.; Veiga, N.; Ramishetti, S.; Goldsmith, M.; Rosenblum, D.; Dammes, N.; Hazan-Halevy, I.; Nahary, L.; Leviatan-Ben-Arye, S.; Harlev, M.; et al. A modular platform for targeted RNAi therapeutics. Nat. Nanotechnol. 2018, 13, 214–219. [Google Scholar] [CrossRef]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Stein, C.A.; Castanotto, D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scotti, M.M.; Swanson, M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016, 17, 19–32. [Google Scholar] [CrossRef]
- Kendall, G.C.; Mokhonova, E.I.; Moran, M.; Sejbuk, N.E.; Wang, D.W.; Silva, O.; Wang, R.T.; Martinez, L.; Lu, Q.L.; Damoiseaux, R.; et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci. Transl. Med. 2012, 4, 164ra160. [Google Scholar] [CrossRef]
- Naryshkin, N.A.; Weetall, M.; Dakka, A.; Narasimhan, J.; Zhao, X.; Feng, Z.; Ling, K.K.; Karp, G.M.; Qi, H.; Woll, M.G.; et al. Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345, 688–693. [Google Scholar] [CrossRef]
- Liu, S.; Huang, Z.; Tang, A.; Wu, X.; Aube, J.; Xu, L.; Xing, C.; Huang, Y. Inhibition of RNA-binding protein HuR reduces glomerulosclerosis in experimental nephritis. Clin. Sci. (Lond) 2020, 134, 1433–1448. [Google Scholar] [CrossRef]
- Zapp, M.L.; Stern, S.; Green, M.R. Small molecules that selectively block RNA binding of HIV-1 Rev protein inhibit Rev function and viral production. Cell 1993, 74, 969–978. [Google Scholar] [CrossRef]
- Velagapudi, S.P.; Costales, M.G.; Vummidi, B.R.; Nakai, Y.; Angelbello, A.J.; Tran, T.; Haniff, H.S.; Matsumoto, Y.; Wang, Z.F.; Chatterjee, A.K.; et al. Approved Anti-cancer Drugs Target Oncogenic Non-coding RNAs. Cell Chem. Biol. 2018, 25, 1086–1094.e1087. [Google Scholar] [CrossRef] [PubMed]
- Donlic, A.; Hargrove, A.E. Targeting RNA in mammalian systems with small molecules. Wiley Interdiscip. Rev. RNA 2018, 9, e1477. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Rooney, S.; Michlewski, G. RNA-Targeted Therapies and High-Throughput Screening Methods. Int. J. Mol. Sci. 2020, 21, 2996. [Google Scholar] [CrossRef] [PubMed]
- Wu, P. Inhibition of RNA-binding proteins with small molecules. Nat. Rev. Chem. 2020, 4, 441–458. [Google Scholar] [CrossRef]
- Mattick, J.S. Non-coding RNAs: The architects of eukaryotic complexity. EMBO Rep. 2001, 2, 986–991. [Google Scholar] [CrossRef]
- Guo, L.; Du, Y.; Chang, S.; Zhang, K.; Wang, J. rSNPBase: A database for curated regulatory SNPs. Nucleic Acids Res. 2013, 42, D1033–D1039. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef]
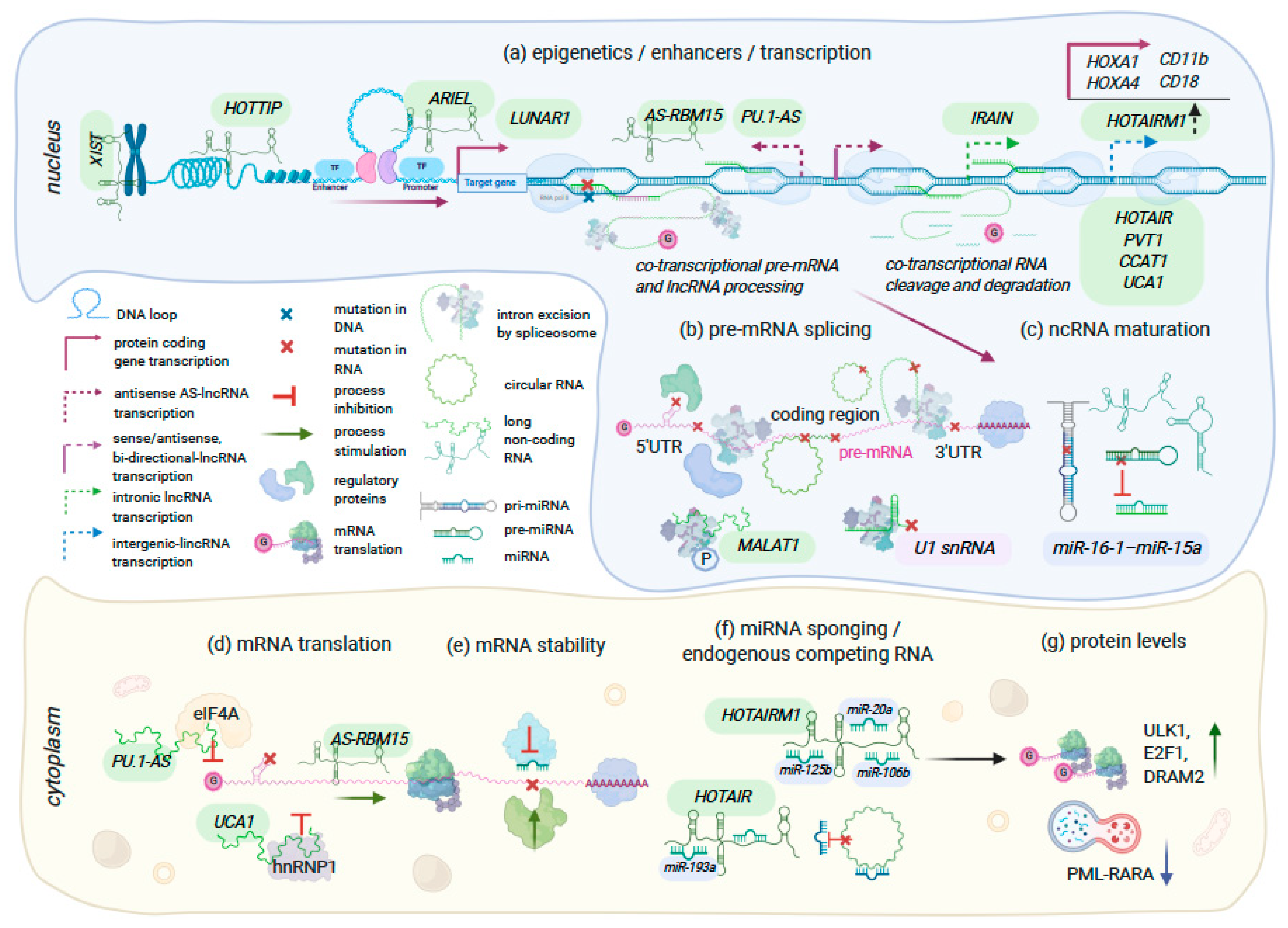

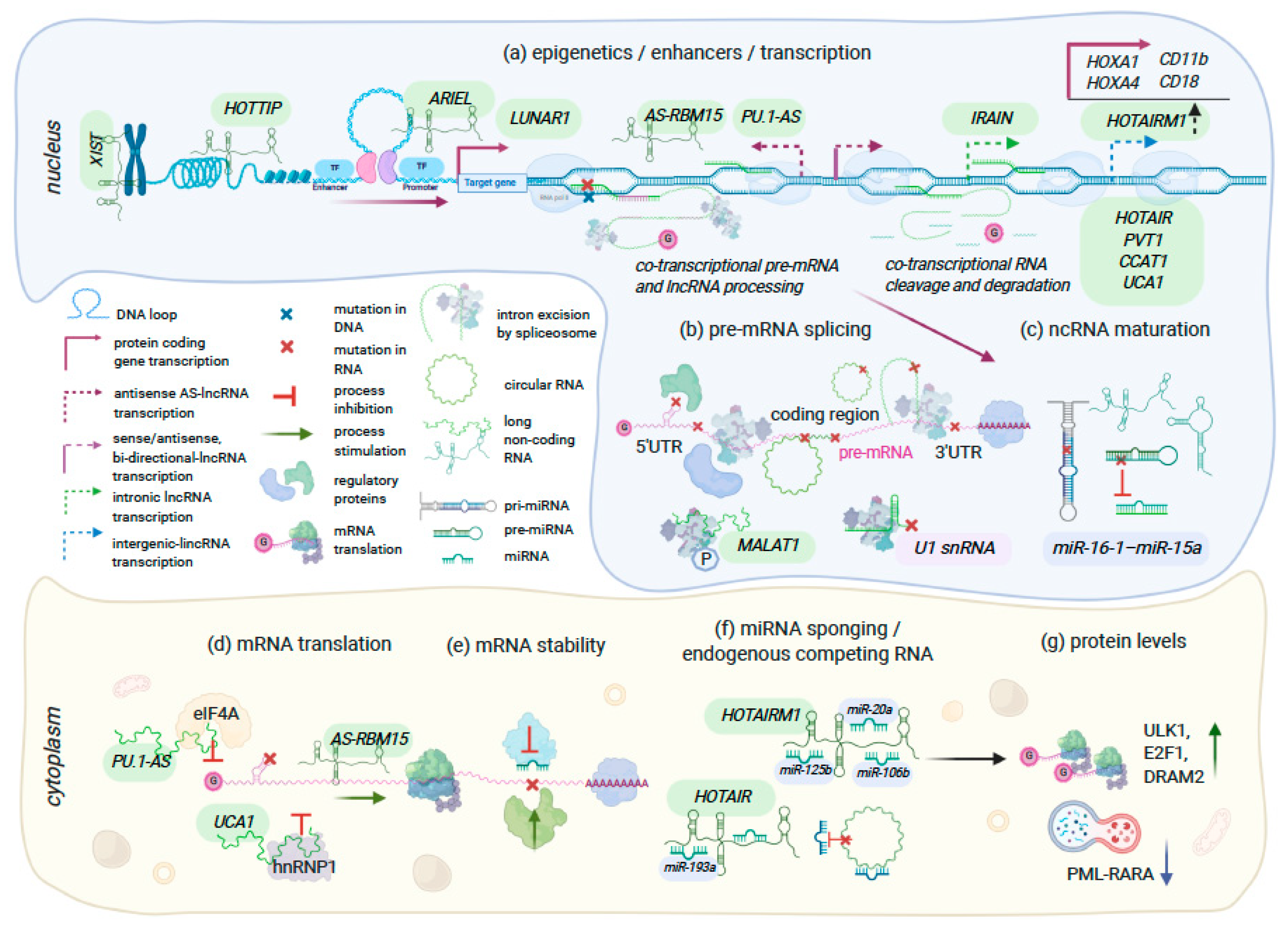{kind=link}
| lncRNA Gene Name | Type of Cancer | Expression in Cancer | Mechanisms | Gene Expression Regulators | Ref |
|---|---|---|---|---|---|
| Nuclear Function: Chromatin Folding and Transcription | |||||
| HOTTIP | MLLr+ NPM1C+ AML | Upregulated | Remodels chromatin accessibility and alters hematopoietic transcription programs affecting multiple pathways (cell cycle, apoptosis, myeloid/leukocyte cell differentiation, JAK-STAT signaling, and regulation of cell development); promotes HSCs self-renewal leading to AML-like disease in mice; lower survival in AML patients | CCCTC-binding factor (CTCF) active at a binding site located between HOXA7 and HOXA9 genes (CBS7/9); Psip1/p52 | [91,94,101] |
| MAGI2-AS3 | AML | Downregulated | Inhibits self-renewal in leukemic stem cells by promoting TET2-dependent DNA demethylation of the LRIG1 promoter in acute myeloid leukemia; a better survival with overexpression | Unknown | [98] |
| IRAIN | AML | Downregulated | Intrachromosomal interactions, enhancer-promoter loop within IGF1R gene | Unknown | [100] |
| MALAT1 | AML, CLL, CMML, MM, HCC, other cancers | Upregulated | Regulates the phosphorylation status of serin-rich splicing factors (SRSF), their subcellular localization in HeLa cells; interacts with PCR2, transcription factors and sequesters miRNA in the cytoplasm. Aberrant expression in Del 13q14 CLL; MALAT1 depletion increases cytarabine sensitivity in AML and response to ATRA-treatment in CMML | Multiple transcription factors e.g., SP1, SP3, HIF1-alpha, c-MYC | Reviewed in [93] |
| CASC15 | RUNX1r+ B-ALL, AML | Upregulated | CASC15 regulates expression of SOX4 (B cell reg.) and YY1; overexpression opposes cellular proliferation and promotes myeloid bias in vivo; associated with a better prognosis | HIF1-alpha hypoxia sensitive elements within CASC15 promoter | [96,97] |
| ARIEL | TAL1+ T-ALL | Upregulated | Enhancer RNA: recruits mediator proteins to the ARID5B enhancer, promotes enhancer-promoter interactions, activates ARID5B expression, thereby positively regulating the TAL1-induced transcriptional program and MYC oncogene | ARIEL transcription is activated by TAL1 complex | [92] |
| LUNAR1 | NOTCH-regulated T-ALL | Downregulated | enhancer lncRNA: activates IGF1R expression, T cell proliferation | Regulated by NOTCH1 | [88,90] |
| Cytoplasmic Function: Protein Translation, mRNA Stability | |||||
| AS-RBM15 | AMKL | Downregulated | AS-RBM15 promotes terminal differentiation by enhancing RBM15 translation in a 5′ cap-dependent manner. The overlapping region between AS-RBM15 RNA and 5′ UTR of RBM15 mRNA function as enhancer of RBM15 protein synthesis | AS-RBM15 transcription is activated by RUNX1 and repressed by RUNX1-ETO | [95] |
| PU.1-AS | AML | Upregulated | The simultaneous expression of both sense mRNA and anti-sense RNA (PU.1-AS) transcripts; PU.1-AS RNAs consist ~12–15% of PU.1 mRNA level but are more stable than PU.1 mRNA; PU.1-AS RNA forms complex with eIF4A and stalls PU.1 mRNA translation between initiation and elongation steps | Upstream regulatory element (URE) which physically interacts with both sense and anti-sense promoters; CBF fusions (RUNX1-ETO and CBFβ-MYH11) in AML | [102,103] |
| UCA1 | AML, breast cancer, other types | Upregulated | hnRNP1 is a splicing factor that also promotes cap-independent translation through binding with IRES and recruiting ribosomes to p53 and p27 (Kip1) mRNAs. lncRNA UCA1 binding with phosphorylated cytosolic form of hnRNP1 has anti-apoptotic effect in breast cancer. In leukemia, UCA1 sponges for miR-126, miR-125a, miR-16, and activates PI3K/AKT and JAK/STAT signaling | Regulated by CCAAT/enhancer-binding protein-alpha | [89,99,104,105] |
| lncRNA Gene Name | Type of Cancer | Study Design | Expression in Cancer | Prognostic Significance or Function in Cancer | Ref |
|---|---|---|---|---|---|
| SNHG1 | AML pediatric | newly diagnosed AML (n = 209), healthy controls(n = 67), BM, qRT-PCR | upregulated | shorter event-free and overall survival (p < 0.001) | [128] |
| SOX6-1 | AML pediatric | de novo AML (n = 146), nonhematologic cancer controls(n = 73), BM, proliferation, qRT-PCR apoptosis CCK-8 and AV/PI assay | upregulated | poor-risk stratification, overall survival (p < 0.001), | [129] |
| LINC00909 | AML pediatric | untreated AML (n = 93), healthy controls (n = 31), BM, RT-qPCR analysis, RNA-pull down; luciferase reporter assay; cell viability, migration | upregulated | sponge miR-625, activate WNT-signaling, poor prognosis, AML progression | [124] |
| UCA1 | AML pediatric | UCA1 expression in AML (n = 27) before and after adriamycin (ADR)-based chemotherapy, cell lines, qRT-PCR, luciferase reporter assay, RIP | upregulated | chemoresistance, inhibits glycolysis through the microRNA-125a/hexokinase 2 pathway | [105] |
| UCA1 | AML pediatric | untreated AML (n = 27), PB healthy donor controls, cell lines | upregulated | sustains AML proliferation similar to adult AML | [125] |
| H19 | AML pediatric | gene expression profiles from 1361 childhood leukemia patients in 14 independent studies using available Affymetrix data | upregulated | LIN28B and LIN28B-driven H19 expression present in aggressive subsets of pediatric leukemia | [120] |
| ENST00000435695 ENST00000415964 | AML pediatric | Arraystar Human IncRNA Array V3.0 in three AML vs. controls followed by qRT-PCR in AML BM (n = 22) | 372 dysregulated IncRNAs (difference ≥ 10-fold) | ENST00000435695 (most upregulated) ENST00000415964 (most downregulated) | [133] |
| lnc-THADA4-1 lnc-SUPT3H-1 | JMML pediatric | lncRNA landscapes in untreated JMML (n = 44, n = 19) and healthy BM donors, clinical and molecular characteristics, lncRNA-mRNA interaction network, LNA™ GapmeRs inhibition, cell viability | lnc-THADA4-1 (highest) lnc-SUPT3H-1 (lowest) lncRNA specific for granulocytic lineage– lnc-ACSL1-1,lnc-BASP1-3 | Defined lncRNA associated with favorable and unfavorable prognosis, JMML lncRNA score: difference in the event-free survival from HSCT is significant, p < 0.0001 | [131,132] |
| TCL6 CCDC26 | B-ALL pediatric | ETV6-RUNX1-positive (n = 24) versus ETV6-RUNX1-negative (n = 18) B-ALL, RNA seq, clustering analysis | TCL6(highest) CCDC26(lowest) | TCL6 levels may be associated with poor disease-free survival, even within ETV6-RUNX1-positive B-ALL (p < 0.05) | [130] |
| CDKN2B-AS (ANRIL) | B-ALL pediatric | genotype association study 217 B-ALL patients and 338 controls in CDKN2A/B (9p21.3) locus containing lnc-ANRIL | SNP | Six SNP inducing most strongly associated with B-ALL susceptibility rs2811712 located in the intron 1 on lnc-ANRIL | [21] |
| BALR-2, (BALR-6, LIN00958) | B-ALL pediatric | pediatric B-ALL MLLr+, TEL-AML1, E2A-PBX1, BCR-ABL1 (n = 160) | Upregulated | Poor overall survival (p = 0.005) | [134] |
| AWPPH | T-ALL pediatric | de novo, untreated T-ALL (n = 32) healthy controls, BM, cell proliferation, apoptosis | upregulated | supports proliferation and inhibits apoptosis | [126] |
| CDKN2B-AS1 | T-ALL pediatric | de novo untreated T-ALL (n = 21) and ADR-based therapies treated (n = 21), total T-ALL patients (n = 42), IP, RIP, Luc assay; | upregulated | ADR resistance, positive regulation of TRAF5 through miR-335-3p sponging | [127] |
| INSR | T-ALL pediatric | de novo, untreated T-ALL (n = 3) and healthy BM controls, anti-CD3 sorting, MNC RNAseq, lncRNA cellular localization | upregulated | lnc-INSR promotes tumor progression by promoting an immunosuppressive microenvironment in vivo | [122] |
| NALT | T-ALL pediatric | T-ALL (n = 20), BM, proliferation assay in vitro and in vivo PDX | upregulated | Co-expressed and supports NOTCH1 signaling, nuclear localization, novel cis-acting element regulating NOTCH1 | [121] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elcheva, I.A.; Spiegelman, V.S. The Role of cis- and trans-Acting RNA Regulatory Elements in Leukemia. Cancers 2020, 12, 3854. https://doi.org/10.3390/cancers12123854
Elcheva IA, Spiegelman VS. The Role of cis- and trans-Acting RNA Regulatory Elements in Leukemia. Cancers. 2020; 12(12):3854. https://doi.org/10.3390/cancers12123854
Chicago/Turabian StyleElcheva, Irina A., and Vladimir S. Spiegelman. 2020. "The Role of cis- and trans-Acting RNA Regulatory Elements in Leukemia" Cancers 12, no. 12: 3854. https://doi.org/10.3390/cancers12123854
APA StyleElcheva, I. A., & Spiegelman, V. S. (2020). The Role of cis- and trans-Acting RNA Regulatory Elements in Leukemia. Cancers, 12(12), 3854. https://doi.org/10.3390/cancers12123854