Mechanisms Generating Cancer Genome Complexity: Back to the Future

Abstract
Simple Summary
Abstract
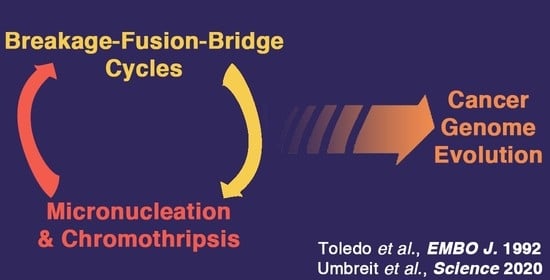
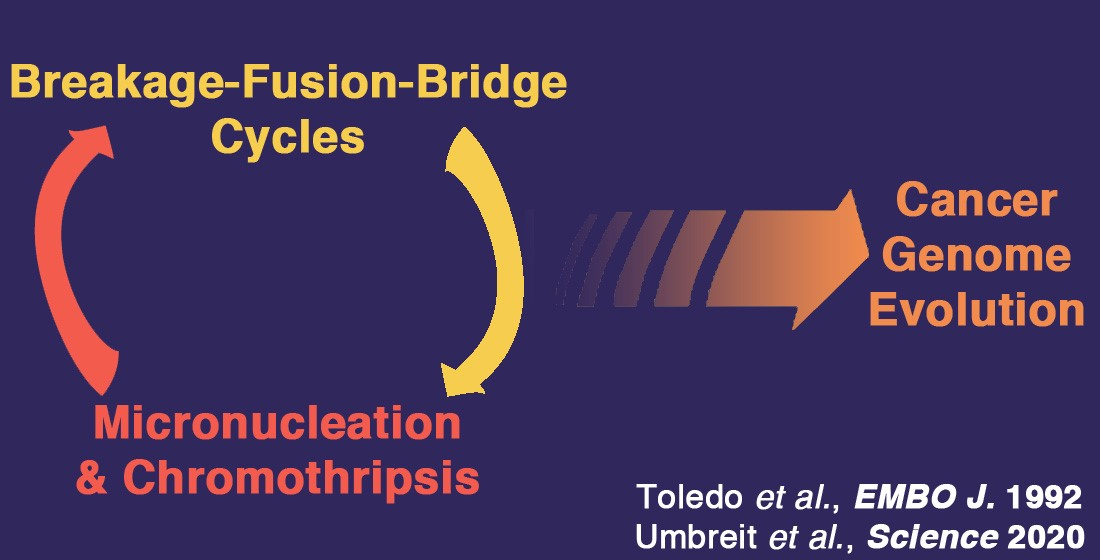{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Early Stages of Gene Amplification are Characterized by Extreme Genomic Instability
3. A Major Role for Breakage-Fusion-Bridge Cycles in Gene Amplification in Mammalian Cells
4. Further Rearrangements of the Amplified DNA: The Interphase Breakage Model
5. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Umbreit, N.T.; Zhang, C.-Z.; Lynch, L.D.; Blaine, L.J.; Cheng, A.M.; Tourdot, R.; Sun, L.; Almubarak, H.F.; Judge, K.; Mitchell, T.J.; et al. Mechanisms generating cancer genome complexity from a single cell division error. Science 2020, 368, eaba0712. [Google Scholar] [CrossRef] [PubMed]
- McClintock, B. Chromosome organization and genic expression. Cold Spring Harb. Symp. Quant. Biol. 1951, 16, 13–47. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Toledo, F.; Le Roscouet, D.; Buttin, G.; Debatisse, M. Co-amplified markers alternate in megabase long chromosomal inverted repeats and cluster independently in interphase nuclei at early steps of mammalian gene amplification. EMBO J 1992, 11, 2665–2673. [Google Scholar] [CrossRef]
- Schwab, M.; Amler, L.C. Amplification of cellular oncogenes: A predictor of clinical outcome in human cancer. Genes Chrom. Cancer 1990, 1, 181–193. [Google Scholar] [CrossRef]
- Toledo, F.; Smith, K.A.; Buttin, G.; Debatisse, M. The evolution of the amplified adenylate deaminase 2 domains in Chinese hamster cells suggests the sequential operation of different mechanisms of DNA amplification. Mutat. Res. 1992, 276, 261–273. [Google Scholar] [CrossRef]
- Coquelle, A.; Toledo, F.; Stern, S.; Bieth, A.; Debatisse, M. A new role for hypoxia in tumor progression: Induction of fragile site triggering genomic rearrangements and formation of complex DMs and HSRs. Mol. Cell 1998, 2, 259–265. [Google Scholar] [CrossRef]
- Trask, B.J.; Hamlin, J.L. Early dihydrofolate reductase gene amplification events in CHO cells usually occur on the same chromosome arm as the original locus. Genes Dev. 1989, 3, 1913–1925. [Google Scholar] [CrossRef]
- Windle, B.; Draper, B.W.; Yin, Y.X.; O’Gorman, S.; Wahl, G.M. A central role for chromosome breakage in gene amplification, deletion formation, and amplicon integration. Genes Dev. 1991, 5, 160–174. [Google Scholar] [CrossRef]
- Ma, C.; Martin, S.; Trask, B.; Hamlin, J.L. Sister chromatid fusion initiates amplification of the dihydrofolate reductase gene in Chinese hamster cells. Genes Dev. 1993, 7, 605–620. [Google Scholar] [CrossRef]
- Coquelle, A.; Pipiras, E.; Toledo, F.; Buttin, G.; Debatisse, M. Expression of fragile sites triggers intrachromosomal mammalian gene amplification and sets boundaries to early amplicons. Cell 1997, 89, 215–225. [Google Scholar] [CrossRef]
- Smith, K.A.; Gorman, P.A.; Stark, M.B.; Groves, R.P.; Stark, G.R. Distinctive chromosomal structures are formed very early in the amplification of CAD genes in Syrian hamster cells. Cell 1990, 63, 1219–1227. [Google Scholar] [CrossRef]
- Smith, K.A.; Stark, M.B.; Gorman, P.A.; Stark, G.R. Fusions near telomeres occur very early in the amplification of CAD genes in Syrian hamster cells. Proc. Natl. Acad. Sci. USA 1992, 89, 5427–5431. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, L.; Attolini, C.; Tessera, L.; Mucciolo, E.; Giulotto, E. Telomeric and nontelomeric (TTAGGG)n sequences in gene amplification and chromosome stability. Genomics 1994, 24, 53–62. [Google Scholar] [CrossRef]
- Toledo, F.; Buttin, G.; Debatisse, M. The origin of chromosome rearrangements at early stages of AMPD2 gene amplification in Chinese hamster cells. Curr. Biol. 1993, 3, 255–264. [Google Scholar] [CrossRef]
- McClintock, B. The fusion of broken ends of sister half-chromatids following chromatid breakage at meiotic anaphases. MO Agric. Exp. Sta. Res. Bull. 1938, 290, 1–48. [Google Scholar]
- McClintock, B. Mutable loci in maize. Carnegie Inst. Wash. Year Book 1948, 47, 155–169. [Google Scholar]
- Sabatier, L.; Ricoul, M.; Pottier, G.; Murnane, J.P. The loss of a single telomere can result in instability of multiple chromosomes in a human tumor cell line. Mol. Cancer Res. 2005, 3, 139–150. [Google Scholar] [CrossRef]
- Lo, A.W.I.; Sabatier, L.; Fouladi, B.; Pottier, G.; Ricoul, M.; Murnane, J.P. DNA amplification by breakage/fusion/bridge cycles initiated by spontaneous telomere loss in a human cancer cell line. Neoplasia 2002, 4, 531–538. [Google Scholar] [CrossRef]
- McClintock, B. The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 1950, 36, 344–355. [Google Scholar] [CrossRef]
- Toledo, F. Mécanismes Précoces D’amplification Génique dans les Cellules de Mammifères [Early Mechanims of Gene Amplification in Mammalian Cells]. Ph.D. Thesis, Université Pierre et Marie Curie, Paris, France, 1994. [Google Scholar]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Kuerbitz, S.J.; Plunkett, B.S.; Walsh, W.V.; Kastan, M.B. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc. Natl. Acad. Sci. USA 1992, 89, 7491–7495. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Tainsky, M.A.; Bischoff, F.Z.; Strong, L.C.; Wahl, G.M. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell 1992, 70, 937–948. [Google Scholar] [CrossRef]
- Livingstone, L.R.; White, A.; Sprouse, J.; Livanos, E.; Jacks, T.; Tlsty, T.D. Altered cell cycle arrest and gene amplification potential accompany loss of wild-type p53. Cell 1992, 70, 923–935. [Google Scholar] [CrossRef]
- Hartwell, L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell 1992, 71, 543–546. [Google Scholar] [CrossRef]
- Gisselsson, D.; Höglund, M.; Mertens, F.; Mitelman, F.; Mandahl, N. Chromosomal organization of amplified chromosome 12 sequences in mesenchymal tumors detected by fluorescence in situ hybridization. Genes Chrom. Cancer 1998, 23, 203–212. [Google Scholar] [CrossRef]
- Shuster, M.I.; Han, L.; Le Beau, M.M.; Davis, E.; Sawicki, M.; Lese, C.M.; Park, N.H.; Colicelli, J.; Gollin, S.M. A consistent pattern of RIN1 rearrangements in oral squamous cell carcinoma cell lines supports a breakage-fusion-bridge cycle model for 11q13 amplification. Genes Chrom. Cancer 2000, 28, 153–163. [Google Scholar] [CrossRef]
- Hellman, A.; Zlotorynski, E.; Scherer, S.W.; Cheung, J.; Vincent, J.B.; Smith, D.I.; Trakhtenbrot, L.; Kerem, B. A role for common fragile site induction in amplification of human oncogenes. Cancer Cell 2002, 1, 89–97. [Google Scholar] [CrossRef]
- Ciullo, M.; Debily, M.-A.; Rozier, L.; Autiero, M.; Billault, A.; Mayau, V.; El Marhomy, S.; Guardiola, J.; Bernheim, A.; Coullin, P.; et al. Initiation of the breakage-fusion-bridge mechanism through common fragile site activation in human breast cancer cells: The model of PIP gene duplication from a break at FRA7I. Hum. Mol. Genet. 2002, 11, 2887–2894. [Google Scholar] [CrossRef]
- Reshmi, S.C.; Huang, X.; Schoppy, D.W.; Black, R.C.; Saunders, W.S.; Smith, D.I.; Gollin, S.M. Relationship between FRA11F and 11q13 gene amplification in oral cancer. Genes Chrom. Cancer 2007, 46, 143–154. [Google Scholar] [CrossRef]
- Marotta, M.; Onodera, T.; Johnson, J.; Budd, G.T.; Watanabe, T.; Cui, X.; Giuliano, A.E.; Niida, A.; Tanaka, H. Palindromic amplification of the ERBB2 oncogene in primary HER2-positive breast tumors. Sci. Rep. 2017, 7, 41921. [Google Scholar] [CrossRef] [PubMed]
- Gisselsson, D.; Höglund, M.; Mertens, F.; Johansson, B.; Dal Cin, P.; Van den Berghe, H.; Earnshaw, W.C.; Mitelman, F.; Mandahl, N. The structure and dynamics of ring chromosomes in human neoplastic and non-neoplastic cells. Hum. Genet. 1999, 104, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Reshmi, S.C.; Roychoudhury, S.; Yu, Z.; Feingold, E.; Potter, D.; Saunders, W.S.; Gollin, S.M. Inverted duplication pattern in anaphase bridges confirms the breakage-fusion-bridge (BFB) cycle model for 11q13 amplification. Cytogenet. Genome Res. 2007, 116, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Mills, K.D.; Ferguson, D.O.; Lee, C.; Manis, J.; Fleming, J.; Gao, Y.; Morton, C.C.; Alt, F.W. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 2002, 109, 811–821. [Google Scholar] [CrossRef]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef]
- Difilippantonio, M.J.; Petersen, S.; Chen, H.T.; Johnson, R.; Jasin, M.; Kanaar, R.; Ried, T.; Nussenzweig, A. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J. Exp. Med. 2002, 196, 469–480. [Google Scholar] [CrossRef]
- Lim, G.; Karaskova, J.; Beheshti, B.; Vukovic, B.; Bayani, J.; Selvarajah, S.; Watson, S.K.; Lam, W.L.; Zielenska, M.; Squire, J.A. An integrated mBAND and submegabase resolution tiling set (SMRT) CGH array analysis of focal amplification, microdeletions, and ladder structures consistent with breakage-fusion-bridge cycle events in osteosarcoma. Genes Chrom. Cancer 2005, 42, 392–403. [Google Scholar] [CrossRef]
- Miller, C.T.; Lin, L.; Casper, A.M.; Lim, J.; Thomas, D.G.; Orringer, M.B.; Chang, A.C.; Chambers, A.F.; Giordano, T.J.; Glover, T.W.; et al. Genomic amplification of MET with boundaries within fragile site FRA7G and upregulation of MET pathways in esophageal adenocarcinoma. Oncogene 2006, 25, 409–418. [Google Scholar] [CrossRef]
- Pelliccia, F.; Bosco, N.; Rocchi, A. Breakages at common fragile sites set boundaries of amplified regions in two leukemia cell lines K562—Molecular characterization of FRA2H and localization of a new CFS FRA2S. Cancer Lett. 2010, 299, 37–44. [Google Scholar] [CrossRef]
- Blumrich, A.; Zapatka, M.; Brueckner, L.M.; Zheglo, D.; Schwab, M.; Savelyeva, L. The FRA2C common fragile site maps to the borders of MYCN amplicons in neuroblastoma and is associated with gross chromosomal rearrangements in different cancers. Hum. Mol. Genet. 2011, 20, 1488–1501. [Google Scholar] [CrossRef]
- Kinsella, M.; Bafna, V. Combinatorics of the breakage-fusion-bridge mechanism. J. Comput. Biol. 2012, 19, 662–678. [Google Scholar] [CrossRef] [PubMed]
- Zakov, S.; Kinsella, M.; Bafna, V. An algorithmic approach for breakage-fusion-bridge detection in tumor genomes. Proc. Natl. Acad. Sci. USA 2013, 110, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Zakov, S.; Bafna, V. Reconstructing breakage fusion bridge architectures using noisy copy numbers. J. Comput. Biol. 2015, 22, 577–594. [Google Scholar] [CrossRef] [PubMed]
- Hadi, K.; Yao, X.; Behr, J.M.; Deshpande, A.; Xanthopoulakis, C.; Tian, H.; Kudman, S.; Rosiene, J.; Darmofal, M.; DeRose, J.; et al. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 2020, 183, 197–210.e32. [Google Scholar] [CrossRef] [PubMed]
- Marotta, M.; Chen, X.; Inoshita, A.; Stephens, R.; Budd, G.T.; Crowe, J.P.; Lyons, J.; Kondratova, A.; Tubbs, R.; Tanaka, H. A common copy-number breakpoint of ERBB2 amplification in breast cancer colocalizes with a complex block of segmental duplications. Breast Cancer Res. 2012, 14, R150. [Google Scholar] [CrossRef] [PubMed]
- Pedeutour, F.; Suijkerbuijk, R.F.; Forus, A.; Van Gaal, J.; Van de Klundert, W.; Coindre, J.M.; Nicolo, G.; Collin, F.; Van Haelst, U.; Huffermann, K. Complex composition and co-amplification of SAS and MDM2 in ring and giant rod marker chromosomes in well-differentiated liposarcoma. Genes Chrom. Cancer 1994, 10, 85–94. [Google Scholar] [CrossRef]
- Iwasaki, H.; Ohjimi, Y.; Ishiguro, M.; Isayama, T.; Fujita, C.; Kaneko, Y.; Kikuchi, M.; Shinohara, N. Supernumerary ring chromosomes and nuclear blebs in some low-grade malignant soft tissue tumours: Atypical lipomatous tumours and dermatofibrosarcoma protuberans. Virchows Archiv. 1998, 432, 521–528. [Google Scholar] [CrossRef]
- Gisselsson, D.; Björk, J.; Höglund, M.; Mertens, F.; Dal Cin, P.; Akerman, M.; Mandahl, N. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol. 2001, 158, 199–206. [Google Scholar] [CrossRef]
- Ambros, I.M.; Rumpler, S.; Luegmayr, A.; Hattinger, C.M.; Strehl, S.; Kovar, H.; Gadner, H.; Ambros, P.F. Neuroblastoma cells can actively eliminate supernumerary MYCN gene copies by micronucleus formation—Sign of tumour cell revertance? Eur. J. Cancer 1997, 33, 2043–2049. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Zhang, C.-Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Nones, K.; Waddell, N.; Wayte, N.; Patch, A.-M.; Bailey, P.; Newell, F.; Holmes, O.; Fink, J.L.; Quinn, M.C.J.; Tang, Y.H.; et al. Genomic catastrophes frequently arise in esophageal adenocarcinoma and drive tumorigenesis. Nat. Commun. 2014, 5, 5224. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schwab, C.; Ryan, S.; Papaemmanuil, E.; Robinson, H.M.; Jacobs, P.; Moorman, A.V.; Dyer, S.; Borrow, J.; Griffiths, M.; et al. Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia. Nature 2014, 508, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Cortés-Ciriano, I.; Lee, J.J.-K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.-Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2,658 human cancers using whole-genome sequencing. Nat. Genet. 2020. [Google Scholar] [CrossRef]
- Maciejowski, J.; Li, Y.; Bosco, N.; Campbell, P.J.; de Lange, T. Chromothripsis and Kataegis Induced by Telomere Crisis. Cell 2015, 163, 1641–1654. [Google Scholar] [CrossRef]
- Shimizu, N.; Shingaki, K.; Kaneko-Sasaguri, Y.; Hashizume, T.; Kanda, T. When, where and how the bridge breaks: Anaphase bridge breakage plays a crucial role in gene amplification and HSR generation. Exp. Cell Res. 2005, 302, 233–243. [Google Scholar] [CrossRef]
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet. 2020, 52, 884–890. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Rao, S.S.P.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef]
- Akdemir, K.C.; Le, V.T.; Chandran, S.; Li, Y.; Verhaak, R.G.; Beroukhim, R.; Campbell, P.J.; Chin, L.; Dixon, J.R.; Futreal, P.A.; et al. Disruption of chromatin folding domains by somatic genomic rearrangements in human cancer. Nat. Genet. 2020, 52, 294–305. [Google Scholar] [CrossRef]
- Korbel, J.O.; Campbell, P.J. Criteria for inference of chromothripsis in cancer genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Spies, N.; Weng, Z.; Bishara, A.; McDaniel, J.; Catoe, D.; Zook, J.M.; Salit, M.; West, R.B.; Batzoglou, S.; Sidow, A. Genome-wide reconstruction of complex structural variants using read clouds. Nat. Methods 2017, 14, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [PubMed]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [CrossRef] [PubMed]
- Thway, K. Well-differentiated liposarcoma and dedifferentiated liposarcoma: An updated review. Semin. Diagn. Pathol. 2019, 112–121. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toledo, F. Mechanisms Generating Cancer Genome Complexity: Back to the Future. Cancers 2020, 12, 3783. https://doi.org/10.3390/cancers12123783
Toledo F. Mechanisms Generating Cancer Genome Complexity: Back to the Future. Cancers. 2020; 12(12):3783. https://doi.org/10.3390/cancers12123783
Chicago/Turabian StyleToledo, Franck. 2020. "Mechanisms Generating Cancer Genome Complexity: Back to the Future" Cancers 12, no. 12: 3783. https://doi.org/10.3390/cancers12123783
APA StyleToledo, F. (2020). Mechanisms Generating Cancer Genome Complexity: Back to the Future. Cancers, 12(12), 3783. https://doi.org/10.3390/cancers12123783