Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies

 ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Expression of PD-L1 and CD38 on Tumor Cells and Frequency of PD-1+ T-Cells in MM and Primary Plasma Cell Leukemia Patients
2.1.1. PD-1/PD-L1 Axis and CD38 Expression in MM Patients
2.1.2. PD1/PD-L1 Axis and CD38 Expression in Primary Plasma Cell Leukemia
2.2. Daratumumab-Mediated MM Cell Lysis in Short-Term Ex Vivo Assays
2.2.1. PD-L1 Expression Levels Are Not Associated with the Extent of Daratumumab-Mediated MM Cell Lysis
2.2.2. Effector (NK-Cell, T-Cell, and Monocyte)-to-Target Ratio is Associated with the Extent of Daratumumab-Mediated MM Cell Lysis
2.3. Combining Nivolumab and Daratumumab in Short-Term In Vitro Experiments
2.4. Combined Targeting of PD-1 and CD38 in Mouse Tumor Models
2.4.1. Mechanisms of Action of the Anti-Mouse CD38 Antibody
2.4.2. Enhanced Anti-Tumor Activity with the Combination of Anti-mCD38 and Anti-mPD-1 in a Murine Myeloma Tumor Model
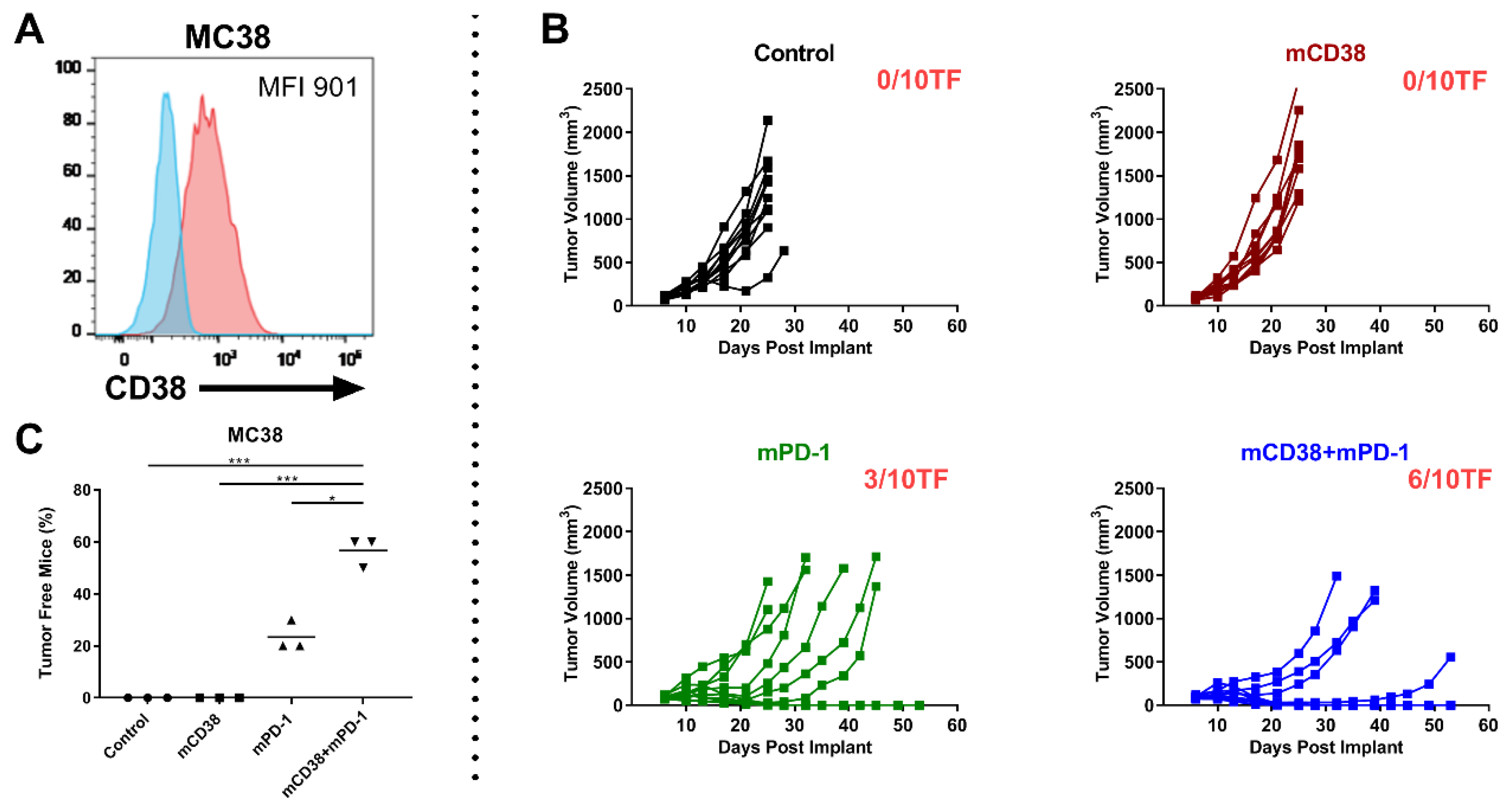
2.4.3. Enhanced Antitumor Activity with the Combination of Anti-mCD38 and Anti-mPD-1 in the CD38-Expressing MC38 Tumor Model
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Antibodies and Reagents
4.3. Bone Marrow Mononuclear Cells
4.4. Flow Cytometric Analysis of BM Samples from MM Patients and Healthy Controls
4.5. Flow Cytometry-Based Ex Vivo Cytotoxicity Assays in BM-MNCs
4.6. Bioluminescence Imaging-Based Cytotoxicity Assays Using Human MM Cell Lines
4.7. In Vitro ADCC and ADCP Assays
4.8. Mice
4.9. Mouse Cell Lines and Culturing Conditions
4.10. Tumor Cell Implantation and Treatment
4.11. Immunophenotypic Analysis
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lokhorst, H.M.; Plesner, T.; Laubach, J.P.; Nahi, H.; Gimsing, P.; Hansson, M.; Minnema, M.C.; Lassen, U.; Krejcik, J.; Palumbo, A.; et al. Targeting CD38 with Daratumumab Monotherapy in Multiple Myeloma. N. Engl. J. Med. 2015, 373, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Lonial, S.; Weiss, B.M.; Usmani, S.Z.; Singhal, S.; Chari, A.; Bahlis, N.J.; Belch, A.; Krishnan, A.; Vescio, R.A.; Mateos, M.V.; et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): An open-label, randomised, phase 2 trial. Lancet 2016, 387, 1551–1560. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): A randomised, open-label, phase 3 study. Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Dimopoulos, M.A.; Cavo, M.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; Kaplan, P.; et al. Daratumumab plus Bortezomib, Melphalan, and Prednisone for Untreated Myeloma. N. Engl. J. Med. 2018, 378, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Dimopoulos, M.A.; Oriol, A.; Nahi, H.; San-Miguel, J.; Bahlis, N.J.; Usmani, S.Z.; Rabin, N.; Orlowski, R.Z.; Komarnicki, M.; Suzuki, K.; et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 1319–1331. [Google Scholar] [CrossRef]
- Palumbo, A.; Chanan-Khan, A.; Weisel, K.; Nooka, A.K.; Masszi, T.; Beksac, M.; Spicka, I.; Hungria, V.; Munder, M.; Mateos, M.V.; et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. N. Engl. J. Med. 2016, 375, 754–766. [Google Scholar] [CrossRef]
- Chari, A.; Suvannasankha, A.; Fay, J.W.; Arnulf, B.; Kaufman, J.L.; Ifthikharuddin, J.J.; Weiss, B.M.; Krishnan, A.; Lentzsch, S.; Comenzo, R.; et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 2017, 130, 974–981. [Google Scholar] [CrossRef]
- De Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Casneuf, T.; van Velzen, J.; van Kessel, B.; Axel, A.E.; Syed, K.; Groen, R.W.; van Duin, M.; Sonneveld, P.; Minnema, M.C.; et al. CD38 expression and complement inhibitors affect response and resistance to daratumumab therapy in myeloma. Blood 2016, 128, 959–970. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Groen, R.W.; Lokhorst, H.M.; van Kessel, B.; Bloem, A.C.; van Velzen, J.; de Jong-Korlaar, R.; Yuan, H.; Noort, W.A.; Klein, S.K.; et al. Upregulation of CD38 expression on multiple myeloma cells by all-trans retinoic acid improves the efficacy of daratumumab. Leukemia 2015, 29, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.; Weiss, B.M.; et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J. Immunomodulatory effects of CD38-targeting antibodies. Immunol. Lett. 2018, 199, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Adams, H.C.; Stevenaert, F.; Krejcik, J.; Van der Borght, K.; Smets, T.; Bald, J.; Abraham, Y.; Ceulemans, H.; Chiu, C.; Vanhoof, G.; et al. High-Parameter Mass Cytometry Evaluation of Relapsed/Refractory Multiple Myeloma Patients Treated with Daratumumab Demonstrates Immune Modulation as a Novel Mechanism of Action. Cytometry A 2019, 95, 279–289. [Google Scholar] [CrossRef]
- Van De Donk, N.W.; Adams, H., III; Vanhoof, G.; Krejcik, J.; Van der Borght, K.; Casneuf, T.; Smets, T.; Axel, A.; Abraham, Y.; Ceulmans, H.; et al. Daratumumab in Combination with Lenalidomide Plus Dexamethasone Results in Persistent Natural Killer (NK) Cells with a Distinct Phenotype and Expansion of Effector Memory T-Cells in Pollux, a Phase 3 Randomized Study. Blood 2017, 130, 3124. [Google Scholar] [CrossRef]
- Casneuf, T.; Adams, H.C.; van de Donk, N.W.C.J.; Abraham, Y.; Bald, J.; Vanhoof, G.; Van der Borght, K.; Smets, T.; Foulk, B.; Nielsen, K.C.; et al. Deep immune profiling of patients treated with lenalidomide and dexamethasone with or without daratumumab. Leukemia 2020. [Google Scholar] [CrossRef]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma cells from multiple myeloma patients express B7-H1 (PD-L1) and increase expression after stimulation with IFN-{gamma} and TLR ligands via a MyD88-, TRAF6-, and MEK-dependent pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Tamura, H.; Ishibashi, M.; Yamashita, T.; Tanosaki, S.; Okuyama, N.; Kondo, A.; Hyodo, H.; Shinya, E.; Takahashi, H.; Dong, H.; et al. Marrow stromal cells induce B7-H1 expression on myeloma cells, generating aggressive characteristics in multiple myeloma. Leukemia 2013, 27, 464–472. [Google Scholar] [CrossRef]
- Görgün, G.; Samur, M.K.; Cowens, K.B.; Paula, S.; Bianchi, G.; Anderson, J.E.; White, R.E.; Singh, A.; Ohguchi, H.; Suzuki, R.; et al. Lenalidomide Enhances Immune Checkpoint Blockade-Induced Immune Response in Multiple Myeloma. Clin. Cancer Res. 2015, 21, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Rosenblatt, J.; Glotzbecker, B.; Mills, H.; Vasir, B.; Tzachanis, D.; Levine, J.D.; Joyce, R.M.; Wellenstein, K.; Keefe, W.; Schickler, M.; et al. PD-1 blockade by CT-011, anti-PD-1 antibody, enhances ex vivo T-cell responses to autologous dendritic cell/myeloma fusion vaccine. J. Immunother. 2011, 34, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Paiva, B.; Azpilikueta, A.; Puig, N.; Ocio, E.M.; Sharma, R.; Oyajobi, B.O.; Labiano, S.; San-Segundo, L.; Rodriguez, A.; Aires-Mejia, I.; et al. PD-L1/PD-1 presence in the tumor microenvironment and activity of PD-1 blockade in multiple myeloma. Leukemia 2015, 29, 2110–2113. [Google Scholar] [CrossRef] [PubMed]
- Kearl, T.J.; Jing, W.; Gershan, J.A.; Johnson, B.D. Programmed death receptor-1/programmed death receptor ligand-1 blockade after transient lymphodepletion to treat myeloma. J. Immunol. 2013, 190, 5620–5628. [Google Scholar] [CrossRef]
- Hallett, W.H.; Jing, W.; Drobyski, W.R.; Johnson, B.D. Immunosuppressive effects of multiple myeloma are overcome by PD-L1 blockade. Biol. Blood Marrow Transplant. 2011, 17, 1133–1145. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Lesokhin, A.M.; Ansell, S.M.; Armand, P.; Scott, E.C.; Halwani, A.; Gutierrez, M.; Millenson, M.M.; Cohen, A.D.; Schuster, S.J.; Lebovic, D.; et al. Nivolumab in Patients With Relapsed or Refractory Hematologic Malignancy: Preliminary Results of a Phase Ib Study. J. Clin. Oncol. 2016, 34, 2698–2704. [Google Scholar] [CrossRef]
- Krejcik, J.; Frerichs, K.A.; Nijhof, I.S.; van Kessel, B.; van Velzen, J.F.; Bloem, A.C.; Broekmans, M.E.C.; Zweegman, S.; van Meerloo, J.; Musters, R.J.P.; et al. Monocytes and Granulocytes Reduce CD38 Expression Levels on Myeloma Cells in Patients Treated with Daratumumab. Clin. Cancer Res. 2017, 23, 7498–7511. [Google Scholar] [CrossRef]
- Nijhof, I.S.; Groen, R.W.; Noort, W.A.; van Kessel, B.; de Jong-Korlaar, R.; Bakker, J.; van Bueren, J.J.; Parren, P.W.; Lokhorst, H.M.; van de Donk, N.W.; et al. Preclinical Evidence for the Therapeutic Potential of CD38-Targeted Immuno-Chemotherapy in Multiple Myeloma Patients Refractory to Lenalidomide and Bortezomib. Clin. Cancer Res. 2015, 21, 2802–2810. [Google Scholar] [CrossRef] [PubMed]
- Alrasheed, N.; Lee, L.; Ghorani, E.; Henry, J.Y.; Conde, L.; Chin, M.; Galas-Filipowicz, D.; Furness, A.J.S.; Chavda, S.J.; Richards, H.; et al. Marrow-Infiltrating Regulatory T Cells Correlate with the Presence of Dysfunctional CD4. Clin. Cancer Res. 2020, 26, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Saudemont, A.; Quesnel, B. In a model of tumor dormancy, long-term persistent leukemic cells have increased B7-H1 and B7.1 expression and resist CTL-mediated lysis. Blood 2004, 104, 2124–2133. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.H.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 impair tumor antigen-specific CD8+; T cells in melanoma patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Schjesvold, F.; Oriol, A.; Karlin, L.; Cavo, M.; Rifkin, R.M.; Yimer, H.A.; LeBlanc, R.; Takezako, N.; McCroskey, R.D.; et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naive multiple myeloma (KEYNOTE-185): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e448–e458. [Google Scholar] [CrossRef]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): A randomised, open-label, phase 3 trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Ong, G.L.; Mattes, M.J. Mouse strains with typical mammalian levels of complement activity. J. Immunol. Methods 1989, 125, 147–158. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef]
- Chillemi, A.; Zaccarello, G.; Quarona, V.; Ferracin, M.; Ghimenti, C.; Massaia, M.; Horenstein, A.L.; Malavasi, F. Anti-CD38 antibody therapy: Windows of opportunity yielded by the functional characteristics of the target molecule. Mol. Med. 2013, 19, 99–108. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Chillemi, A.; Quarona, V.; Chiesa, S.; Imperatori, A.; Zanellato, S.; Mortara, L.; Gattorno, M.; Pistoia, V.; et al. CD56brightCD16- NK Cells Produce Adenosine through a CD38-Mediated Pathway and Act as Regulatory Cells Inhibiting Autologous CD4+ T Cell Proliferation. J. Immunol. 2015, 195, 965–972. [Google Scholar] [CrossRef]
- Lammerts van Bueren, J.; Jakobs, D.; Kaldenhoven, N.; Roza, M.; Hiddingh, S.; Meesters, J.; Voorhorst, M.; Gresnigt, E.; Wiegman, L.; Ortiz Buijsse, A.; et al. Direct in Vitro Comparison of Daratumumab with Surrogate Analogs of CD38 Antibodies MOR03087, SAR650984 and Ab79. Blood 2014, 124, 3474. [Google Scholar] [CrossRef]
- Li, B.; VanRoey, M.; Wang, C.; Chen, T.H.; Korman, A.; Jooss, K. Anti-programmed death-1 synergizes with granulocyte macrophage colony-stimulating factor—Secreting tumor cell immunotherapy providing therapeutic benefit to mice with established tumors. Clin. Cancer Res. 2009, 15, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Bezman, N.A.; Jhatakia, A.; Kearney, A.Y.; Brender, T.; Maurer, M.; Henning, K.; Jenkins, M.R.; Rogers, A.J.; Neeson, P.J.; Korman, A.J.; et al. PD-1 blockade enhances elotuzumab efficacy in mouse tumor models. Blood Adv. 2017, 1, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Van der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. Towards effective immunotherapy of myeloma: Enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 2011, 96, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Van der Veer, M.S.; de Weers, M.; van Kessel, B.; Bakker, J.M.; Wittebol, S.; Parren, P.W.; Lokhorst, H.M.; Mutis, T. The therapeutic human CD38 antibody daratumumab improves the anti-myeloma effect of newly emerging multi-drug therapies. Blood Cancer J. 2011, 1, e41. [Google Scholar] [CrossRef]
- Greco, W.R.; Bravo, G.; Parsons, J.C. The search for synergy: A critical review from a response surface perspective. Pharmacol. Rev. 1995, 47, 331–385. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | NDMM (n = 13) | pPCL (n = 5) | RRMM (n = 31) | |
|---|---|---|---|---|
| Age, median (range) | 75 (31–86) | 65 (57–97) | 66 (46–77) | |
| Male sex, n (%) | 7 (54) | 2 (40) | 16 (52) | |
| M protein, n (%) | ||||
| IgG | 6 (50) | 2 (40) | 22 (71) | |
| IgA | 0 | 0 | 4 (13) | |
| FLC only | 6 (50) | 3 (60) | 5 (16) | |
| Unknown | 1 (8) | 0 | 0 | |
| ISS score, n (%) | ||||
| 1 | 2 (29) | 0 | n.a. | |
| 2 | 3 (43) | 1 (25) | ||
| 3 | 2 (29) | 3 (75) | ||
| Unknown | 6 (46) | 1 (20) | ||
| High risk cytogenetics a, n (%) | 1 (8) | 3 (75) | 6 (23) | |
| Unknown | 1 (8) | 1 (20) | 5 (16) | |
| Del (17p) | 1 (8) | 3 (75) | 4 (15) | |
| t (4;14) | 1 (8) | 1 (25) | 2 (8) | |
| t (14;16) | 0 | 0 | 0 | |
| Prior lines of therapy, median (range) | 0 | 0 | 3 (1–9) | |
| Prior autologous SCT, n (%) | ||||
| Single | n.a. | n.a. | 17 (61) | |
| Double | 2 (7) | |||
| Prior allogeneic SCT, n (%) | n.a. | n.a. | 2 (7) | |
| Lenalidomide, n (%) | ||||
| Exposed | n.a. | n.a. | 27 (87) | |
| Refractory b | 26 (84) | |||
| Pomalidomide, n (%) | ||||
| Exposed | n.a. | n.a. | 16 (52) | |
| Refractory b | 16 (52) | |||
| Bortezomib, n (%) | ||||
| Exposed | n.a. | n.a. | 27 (87) | |
| Refractory | 13 (42) | |||
| Carfilzomib, n (%) | ||||
| Exposed | n.a. | n.a. | 9 (29) | |
| Refractory | 7 (23) | |||
| Daratumumab, n (%) | ||||
| Exposed | n.a. | n.a. | 0 | |
| Refractory | 0 | |||
| IMiD and PI c | ||||
| Exposed | n.a. | n.a. | 27 (87) | |
| Refractory | 18 (58) | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verkleij, C.P.M.; Jhatakia, A.; Broekmans, M.E.C.; Frerichs, K.A.; Zweegman, S.; Mutis, T.; Bezman, N.A.; van de Donk, N.W.C.J. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers 2020, 12, 3713. https://doi.org/10.3390/cancers12123713
Verkleij CPM, Jhatakia A, Broekmans MEC, Frerichs KA, Zweegman S, Mutis T, Bezman NA, van de Donk NWCJ. Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers. 2020; 12(12):3713. https://doi.org/10.3390/cancers12123713
Chicago/Turabian StyleVerkleij, Christie P. M., Amy Jhatakia, Marloes E. C. Broekmans, Kristine A. Frerichs, Sonja Zweegman, Tuna Mutis, Natalie A. Bezman, and Niels W. C. J. van de Donk. 2020. "Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies" Cancers 12, no. 12: 3713. https://doi.org/10.3390/cancers12123713
APA StyleVerkleij, C. P. M., Jhatakia, A., Broekmans, M. E. C., Frerichs, K. A., Zweegman, S., Mutis, T., Bezman, N. A., & van de Donk, N. W. C. J. (2020). Preclinical Rationale for Targeting the PD-1/PD-L1 Axis in Combination with a CD38 Antibody in Multiple Myeloma and Other CD38-Positive Malignancies. Cancers, 12(12), 3713. https://doi.org/10.3390/cancers12123713