Comprehensive Analysis of SWI/SNF Inactivation in Lung Adenocarcinoma Cell Models

 , , , , , , , ,
, , , , , , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
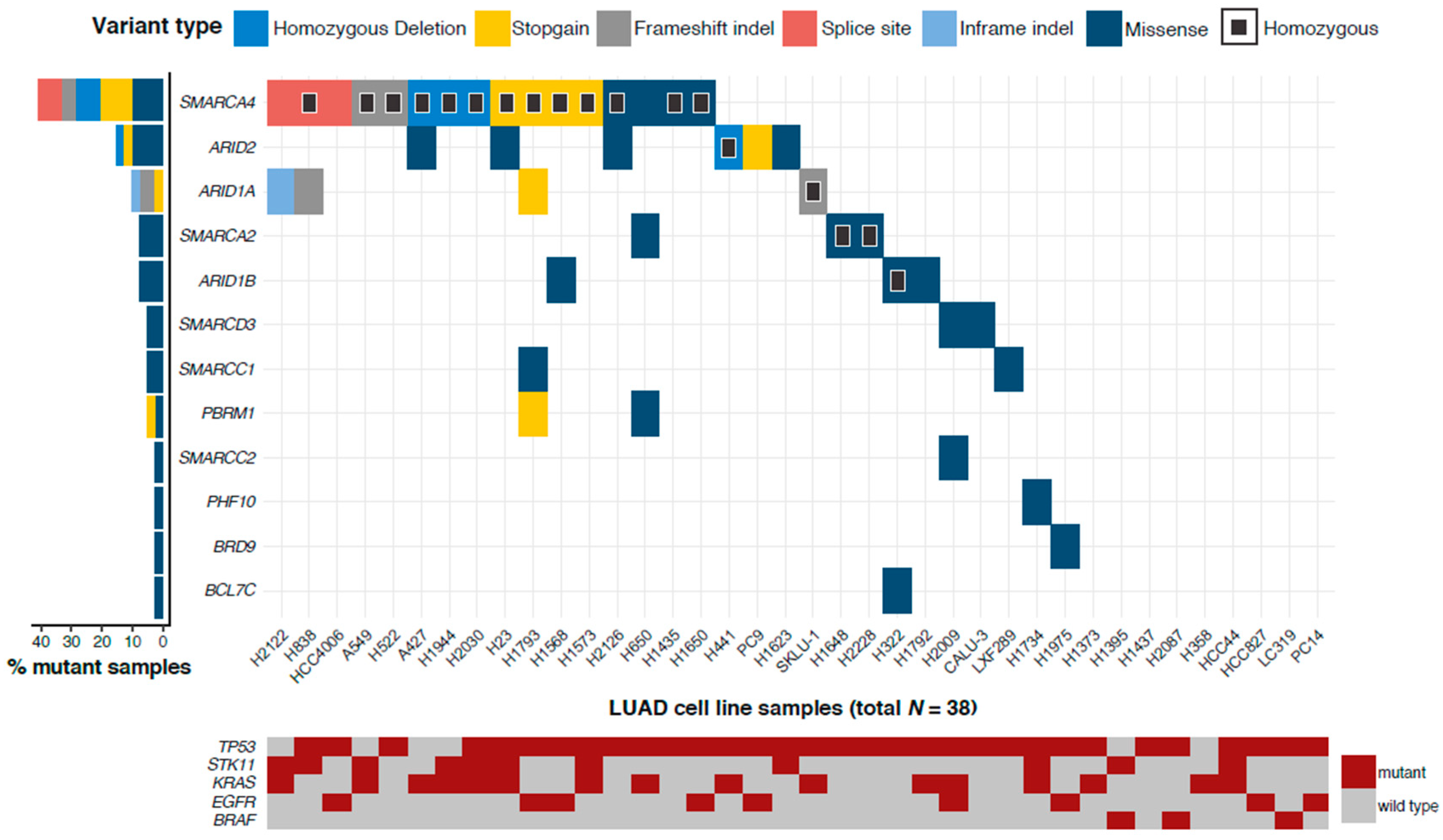
2.1. More than 75% of LUAD Cell Lines Have a Mutated SWI/SNF Subunit
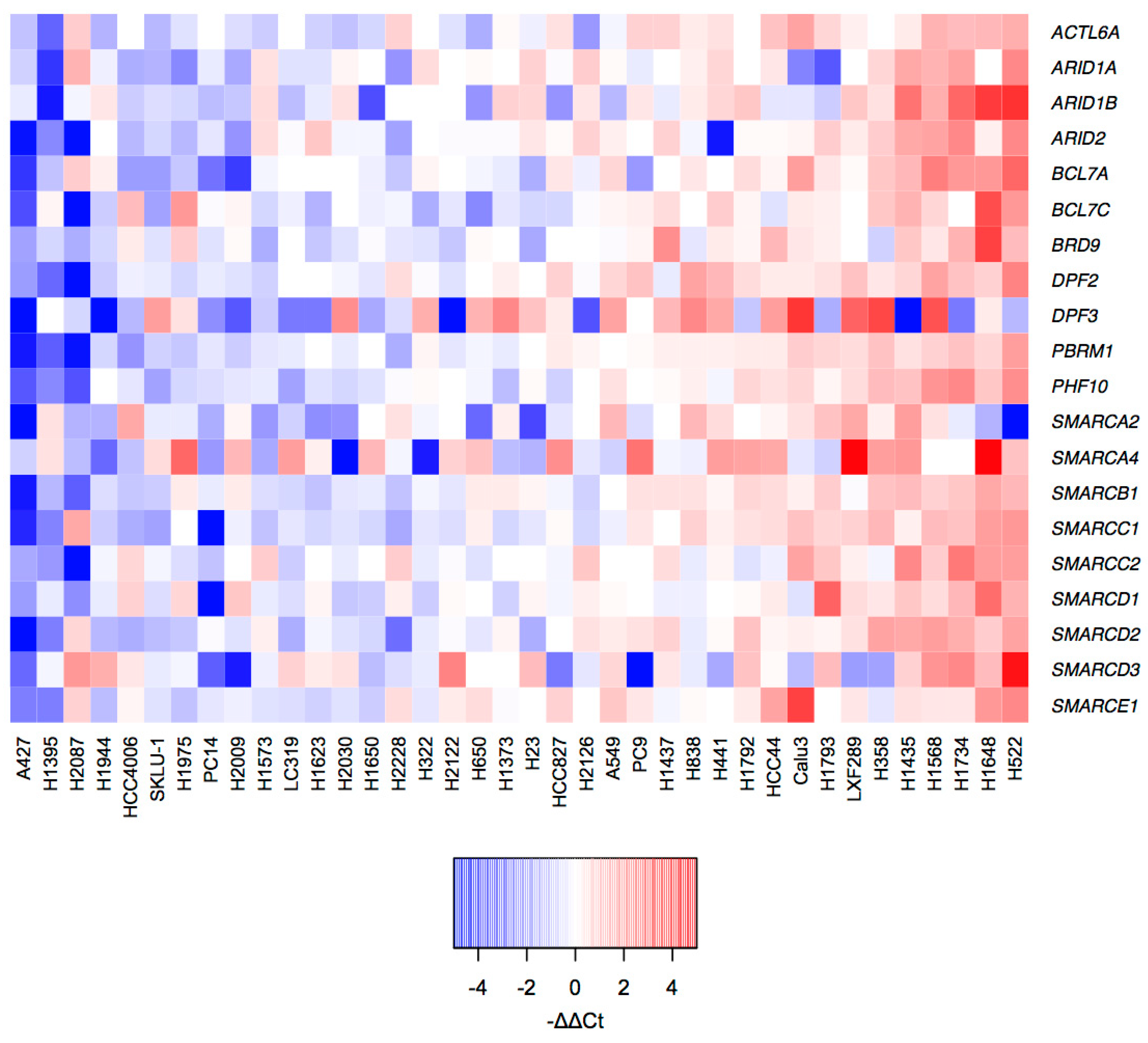
2.2. mRNA Profiles of LUAD Cell Lines Show a Transcriptional De-Regulation of the SWI/SNF Complex
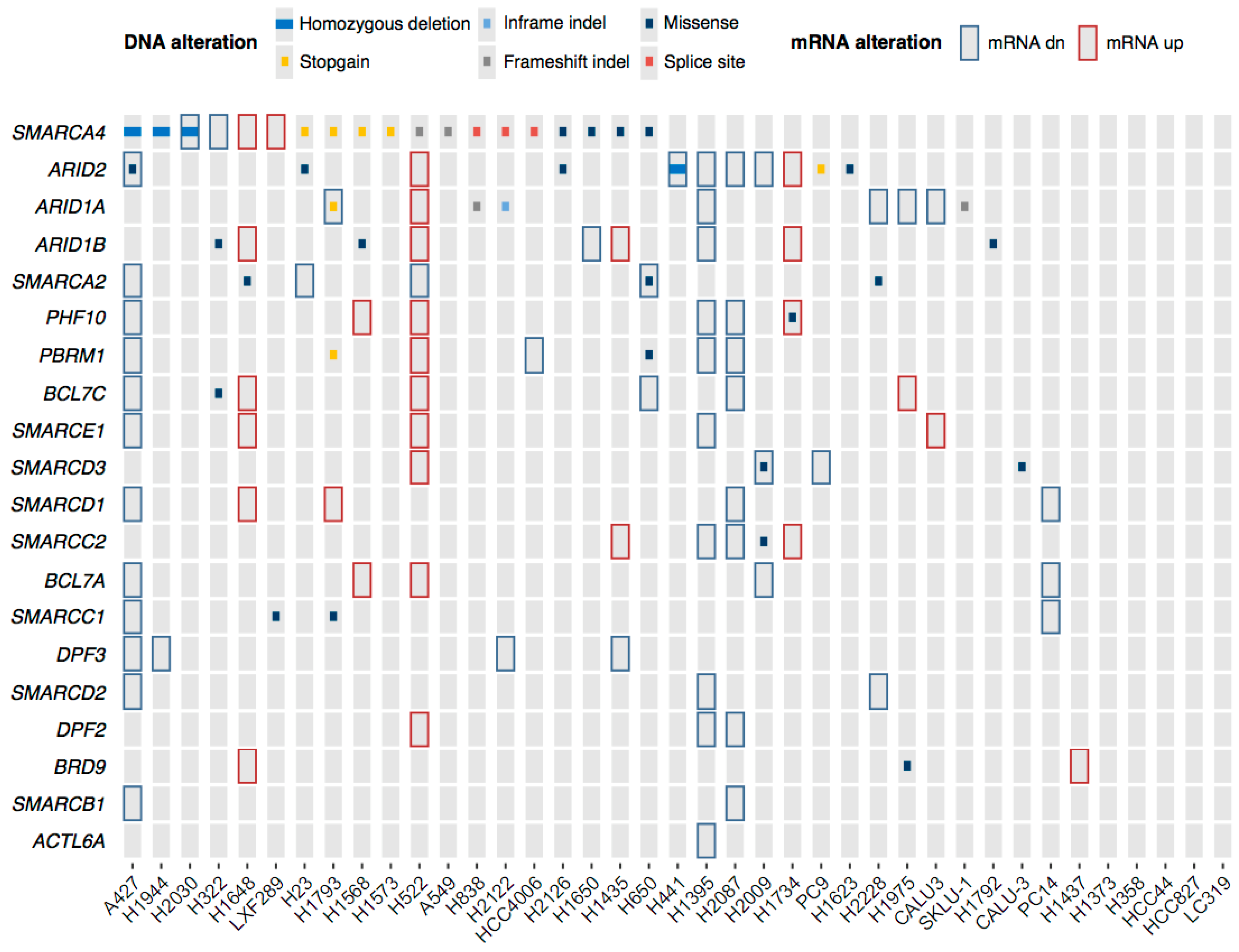
2.3. Genetic and Epigenetic Factors Contribute to the Protein Loss of the ATPases and ARID Subunits
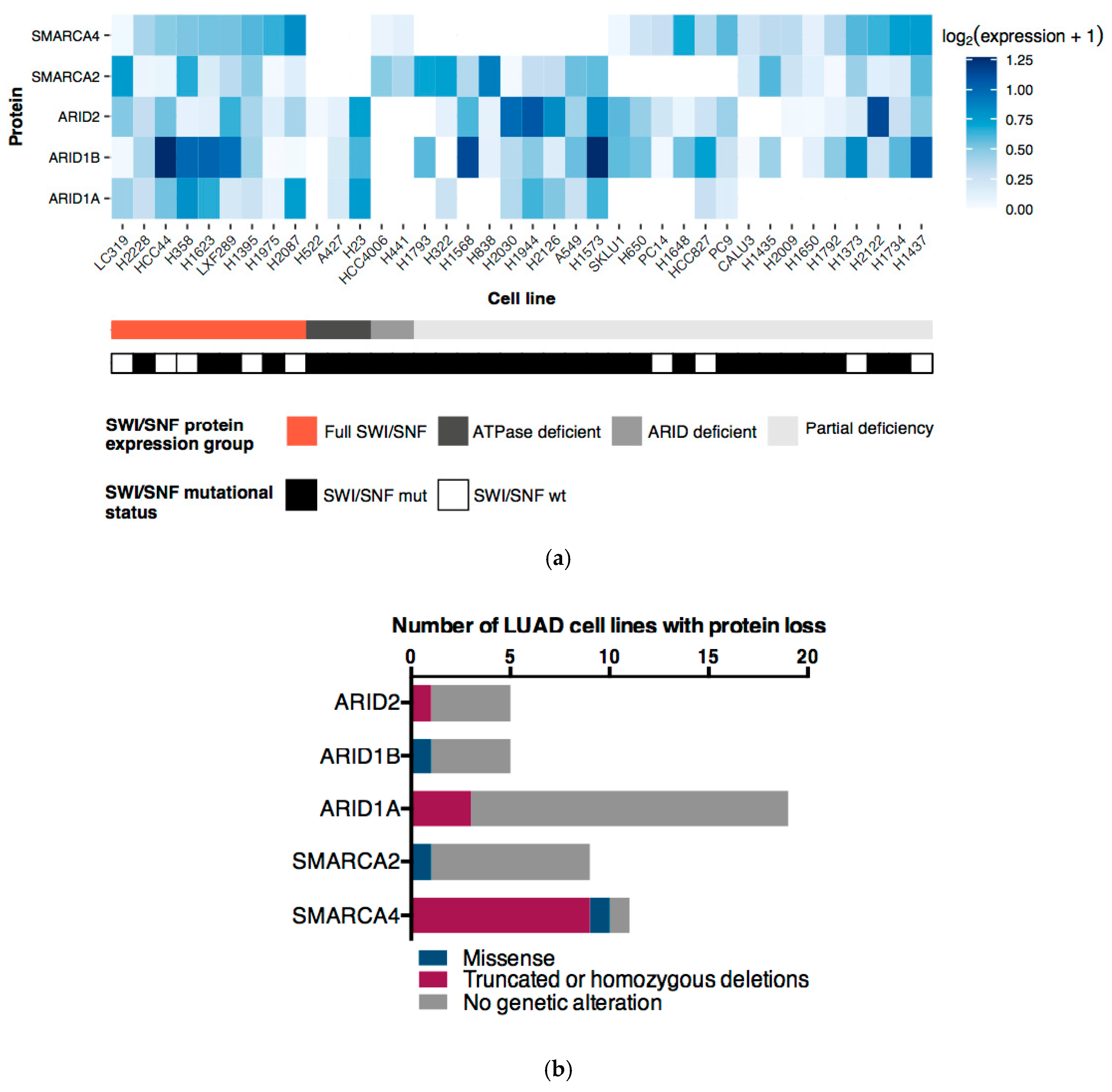
2.4. ATPases and ARID Protein Expression Profiles Define Four LUAD Cell Line Subgroups
3. Discussion
4. Materials and Methods
4.1. Lung Adenocarcinoma Cell Lines
4.2. DNA and RNA Extraction
4.3. Gene Capture and Targeted Sequencing
4.4. Deep Sequencing Data Analysis
4.5. In Silico Analysis of the SWI/SNF Complex in Lung Adenocarcinoma Cell Lines
4.6. Evaluation of the Functional Impact of Missense Mutations
4.7. Real-Time Quantitative Polymerase Chain Reaction
4.8. Western Blot
4.9. TCGA Data Download
4.10. Statistical Analysis
4.11. Data availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- Li, J.; Zhao, W.; Akbani, R.; Liu, W.; Ju, Z.; Ling, S.; Vellano, C.P.; Roebuck, P.; Yu, Q.; Eterovic, A.K.; et al. Characterization of Human Cancer Cell Lines by Reverse-Phase Protein Arrays. Cancer Cell 2017, 31, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Pollack, J.R. The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 2013, 8, e55119. [Google Scholar] [CrossRef] [PubMed]
- Pulice, J.L.; Kadoch, C. Composition and Function of Mammalian SWI/SNF Chromatin Remodeling Complexes in Human Disease. Cold Spring Harb. Symp. Quant. Biol. 2017, 81, 53–60. [Google Scholar] [CrossRef]
- Kaeser, M.D.; Aslanian, A.; Dong, M.-Q.; Yates, J.R., 3rd; Emerson, B.M. BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 2008, 283, 32254–32263. [Google Scholar] [CrossRef]
- Alpsoy, A.; Dykhuizen, E.C. Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 2018, 293, 3892–3903. [Google Scholar] [CrossRef]
- Mashtalir, N.; D’Avino, A.R.; Michel, B.C.; Luo, J.; Pan, J.; Otto, J.E.; Zullow, H.J.; McKenzie, Z.M.; Kubiak, R.L.; Pierre, R.S.; et al. Modular Organization and Assembly of SWI/SNF Family Chromatin Remodeling Complexes. Cell 2018, 175, 1272–1288. [Google Scholar] [CrossRef]
- Centore, R.C.; Sandoval, G.J.; Soares, L.M.M.; Kadoch, C.; Chan, H.M. Mammalian SWI/SNF Chromatin Remodeling Complexes: Emerging Mechanisms and Therapeutic Strategies. Trends Genet. 2020, 36, 936–950. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Vangamudi, B.; Paul, T.A.; Shah, P.K.; Kost-Alimova, M.; Nottebaum, L.; Shi, X.; Zhan, Y.; Leo, E.; Mahadeshwar, H.S.; Protopopov, A.; et al. The SMARCA2/4 ATPase Domain Surpasses the Bromodomain as a Drug Target in SWI/SNF-Mutant Cancers: Insights from cDNA Rescue and PFI-3 Inhibitor Studies. Cancer Res. 2015, 75, 3865–3878. [Google Scholar] [CrossRef] [PubMed]
- Rago, F.; DiMare, M.T.; Elliott, G.; Ruddy, D.A.; Sovath, S.; Kerr, G.; Bhang, H.-E.C.; Jagani, Z. Degron mediated BRM/SMARCA2 depletion uncovers novel combination partners for treatment of BRG1/SMARCA4-mutant cancers. Biochem. Biophys. Res. Commun. 2018, 508, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Farnaby, W.; Koegl, M.; Roy, M.J.; Whitworth, C.; Diers, E.; Trainor, N.; Zollman, D.; Steurer, S.; Karolyi-Oezguer, J.; Riedmueller, C.; et al. BAF complex vulnerabilities in cancer demonstrated via structure-based PROTAC design. Nat. Chem. Biol. 2019, 15, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Papillon, J.P.N.; Nakajima, K.; Adair, C.D.; Hempel, J.; Jouk, A.O.; Karki, R.G.; Mathieu, S.; Möbitz, H.; Ntaganda, R.; Smith, T.; et al. Discovery of Orally Active Inhibitors of Brahma Homolog (BRM)/SMARCA2 ATPase Activity for the Treatment of Brahma Related Gene 1 (BRG1)/SMARCA4-Mutant Cancers. J. Med. Chem. 2018, 61, 10155–10172. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Sun, Y.; Terranova, C.; Khan, F.; Martinez-Ledesma, J.; Gay, J.; Gao, G.; Mullinax, R.A.; Khor, T.; Feng, N.; et al. Mutations in the SWI/SNF complex induce a targetable dependence on oxidative phosphorylation in lung cancer. Nat. Med. 2018, 24, 1047–1057. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef]
- Xue, Y.; Meehan, B.; Fu, Z.; Wang, X.Q.D.; Fiset, P.O.; Rieker, R.; Levins, C.; Kong, T.; Zhu, X.; Morin, G.; et al. SMARCA4 loss is synthetic lethal with CDK4/6 inhibition in non-small cell lung cancer. Nat. Commun. 2019, 10, 557. [Google Scholar] [CrossRef]
- Medina, P.P.; Carretero, J.; Fraga, M.F.; Esteller, M.; Sidransky, D.; Sanchez-Cespedes, M. Genetic and epigenetic screening for gene alterations of the chromatin-remodeling factor, SMARCA4/BRG1, in lung tumors. Genes Chromosomes Cancer 2004, 41, 170–177. [Google Scholar] [CrossRef]
- Medina, P.P.; Carretero, J.; Ballestar, E.; Angulo, B.; Lopez-Rios, F.; Esteller, M.; Sanchez-Cespedes, M. Transcriptional targets of the chromatin-remodelling factor SMARCA4/BRG1 in lung cancer cells. Hum. Mol. 2005, 14, 973–982. [Google Scholar] [CrossRef]
- Medina, P.P.; Romero, O.A.; Kohno, T.; Montuenga, L.M.; Pio, R.; Yokota, J.; Sanchez-Cespedes, M. Frequent BRG1/SMARCA4-inactivating mutations in human lung cancer cell lines. Hum. Mutat. 2008, 29, 617–622. [Google Scholar] [CrossRef]
- Medina, P.P.; Sanchez-Cespedes, M. Involvement of the chromatin-remodeling factor BRG1/SMARCA4 in human cancer. Epigenetics 2008, 3, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–376.e18. [Google Scholar] [CrossRef] [PubMed]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Marquez, S.B.; Thompson, K.W.; Lu, L.; Reisman, D. Beyond Mutations: Additional Mechanisms and Implications of SWI/SNF Complex Inactivation. Front. Oncol. 2015, 4, 372. [Google Scholar] [CrossRef]
- Coira, I.F.; Rufino-Palomares, E.E.; Romero, O.A.; Peinado, P.; Metheetrairut, C.; Boyero-Corral, L.; Carretero, J.; Farez-Vidal, E.; Cuadros, M.; Reyes-Zurita, F.J.; et al. Expression inactivation of SMARCA4 by microRNAs in lung tumors. Hum. Mol. Genet. 2015, 24, 1400–1409. [Google Scholar] [CrossRef]
- Peinado, P.; Andrades, A.; Cuadros, M.; Rodriguez, M.I.; Coira, I.F.; Garcia, D.J.; Benitez-Cantos, M.S.; Cano, C.; Rufino-Palomares, E. Integrative multi-omic analysis of the SWI/SNF complex defines a new clinical subgroup of lung adenocarcinoma patients. Manuscript in preparation.
- Cancer Cell Line Encyclopedia (CCLE). Available online: https://portals.broadinstitute.org/ccle (accessed on 18 July 2018).
- van de Haar, J.; Canisius, S.; Yu, M.K.; Voest, E.E.; Wessels, L.F.A.; Ideker, T. Identifying Epistasis in Cancer Genomes: A Delicate Affair. Cell 2019, 177, 1375–1383. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef]
- Reisman, D.N.; Sciarrotta, J.; Wang, W. Loss of BRG1/BRM in Human Lung Cancer Cell Lines and Primary Lung Cancers: Correlation with Poor Prognosis. Cancer Res. 2003, 63, 560–566. [Google Scholar] [PubMed]
- Fukuoka, J.; Fujii, T.; Shih, J.H. Chromatin Remodeling Factors and BRM/BRG1 Expression as Prognostic Indicators in Non-Small Cell Lung Cancer. Clin. Cancer Res. 2004, 10, 4314–4324. [Google Scholar] [CrossRef] [PubMed]
- Marquez-Vilendrer, S.B.; Rai, S.K.; Gramling, S.J.; Lu, L.; Reisman, D.N. Loss of the SWI/SNF ATPase subunits BRM and BRG1 drives lung cancer development. Oncoscience 2016, 3, 322–336. [Google Scholar] [CrossRef] [PubMed]
- Oike, T.; Ogiwara, H.; Tominaga, Y.; Ito, K.; Ando, O.; Tsuta, K.; Mizukami, T.; Shimada, Y.; Isomura, H.; Komachi, M.; et al. A Synthetic Lethality-Based Strategy to Treat Cancers Harboring a Genetic Deficiency in the Chromatin Remodeling Factor BRG1. Cancer Res. 2013, 73, 5508–5518. [Google Scholar] [CrossRef] [PubMed]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef]
- Savas, S.; Skardasi, G. The SWI/SNF complex subunit genes—Their functions, variations, and links to risk and survival outcomes in human cancers. Crit. Rev. Oncol. Hematol. 2018, 123, 114–131. [Google Scholar] [CrossRef]
- Michel, B.C.; D’Avino, A.R.; Cassel, S.H.; Mashtalir, N.; McKenzie, Z.M.; McBride, M.J.; Valencia, A.M.; Zhou, Q.; Bocker, M.; Soares, L.M.M.; et al. A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 2018, 20, 1410–1420. [Google Scholar] [CrossRef]
- Wang, X.; Sansam, C.G.; Thom, C.S.; Metzger, D.; Evans, J.A.; Nguyen, P.T.L.; Roberts, C.W.M. Oncogenesis caused by loss of the SNF5 tumor suppressor is dependent on activity of BRG1, the ATPase of the SWI/SNF chromatin remodeling complex. Cancer Res. 2009, 69, 8094–8101. [Google Scholar] [CrossRef]
- Hoffman, G.R.; Rahal, R.; Buxton, F.; Xiang, K.; McAllister, G.; Frias, E.; Bagdasarian, L.; Huber, J.; Lindeman, A.; Chen, D.; et al. Functional epigenetics approach identifies BRM/SMARCA2 as a critical synthetic lethal target in BRG1-deficient cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 3128–3133. [Google Scholar] [CrossRef]
- Schiaffino-Ortega, S.; Balinas, C.; Cuadros, M.; Medina, P.P. SWI/SNF proteins as targets in cancer therapy. J. Hematol. Oncol. 2014, 7, 81. [Google Scholar] [CrossRef][Green Version]
- Mittal, P.; Roberts, C.W.M. The SWI/SNF complex in cancer—Biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 2020, 17, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Mathur, R. ARID1A loss in Cancer: Towards a mechanistic understanding. Pharm. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Nieto, S.; Sanchez-Cespedes, M. BRG1 and LKB1: Tales of two tumor suppressor genes on chromosome 19p and lung cancer. Carcinogenesis 2009, 30, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Herpel, E.; Rieker, R.J.; Dienemann, H.; Muley, T.; Meister, M.; Hartmann, A.; Warth, A.; Agaimy, A. SMARCA4 and SMARCA2 Deficiency in Non-small Cell Lung Cancer: Immunohistochemical Survey of 316 Consecutive Specimens. Ann. Diagn. Pathol. 2016, 26, 47–51. [Google Scholar] [CrossRef]
- Huber, W.; Carey, V.; Gentleman, R.; Anders, S.; Carlson, M.; Carvalho, B.S.; Bravo, H.C.; Davis, S.; Gatto, L.; Girke, T.; et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods 2015, 12, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Genomic Data Commons (GDC). Available online: https://portal.gdc.cancer.gov/ (accessed on 18 July 2018).
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2018. Available online: https://www.R-project.org/ (accessed on 18 July 2018).





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peinado, P.; Andrades, A.; Cuadros, M.; Rodriguez, M.I.; Coira, I.F.; Garcia, D.J.; Álvarez-Perez, J.C.; Baliñas-Gavira, C.; Arenas, A.M.; Patiño-Mercau, J.R.; et al. Comprehensive Analysis of SWI/SNF Inactivation in Lung Adenocarcinoma Cell Models. Cancers 2020, 12, 3712. https://doi.org/10.3390/cancers12123712
Peinado P, Andrades A, Cuadros M, Rodriguez MI, Coira IF, Garcia DJ, Álvarez-Perez JC, Baliñas-Gavira C, Arenas AM, Patiño-Mercau JR, et al. Comprehensive Analysis of SWI/SNF Inactivation in Lung Adenocarcinoma Cell Models. Cancers. 2020; 12(12):3712. https://doi.org/10.3390/cancers12123712
Chicago/Turabian StylePeinado, Paola, Alvaro Andrades, Marta Cuadros, Maria Isabel Rodriguez, Isabel F. Coira, Daniel J. Garcia, Juan Carlos Álvarez-Perez, Carlos Baliñas-Gavira, Alberto M. Arenas, Juan Rodrigo Patiño-Mercau, and et al. 2020. "Comprehensive Analysis of SWI/SNF Inactivation in Lung Adenocarcinoma Cell Models" Cancers 12, no. 12: 3712. https://doi.org/10.3390/cancers12123712
APA StylePeinado, P., Andrades, A., Cuadros, M., Rodriguez, M. I., Coira, I. F., Garcia, D. J., Álvarez-Perez, J. C., Baliñas-Gavira, C., Arenas, A. M., Patiño-Mercau, J. R., Sanjuan-Hidalgo, J., Romero, O. A., Montuenga, L. M., Carretero, J., Sanchez-Cespedes, M., & Medina, P. P. (2020). Comprehensive Analysis of SWI/SNF Inactivation in Lung Adenocarcinoma Cell Models. Cancers, 12(12), 3712. https://doi.org/10.3390/cancers12123712