MSI1 Promotes the Expression of the GBM Stem Cell Marker CD44 by Impairing miRNA-Dependent Degradation

 ,
,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
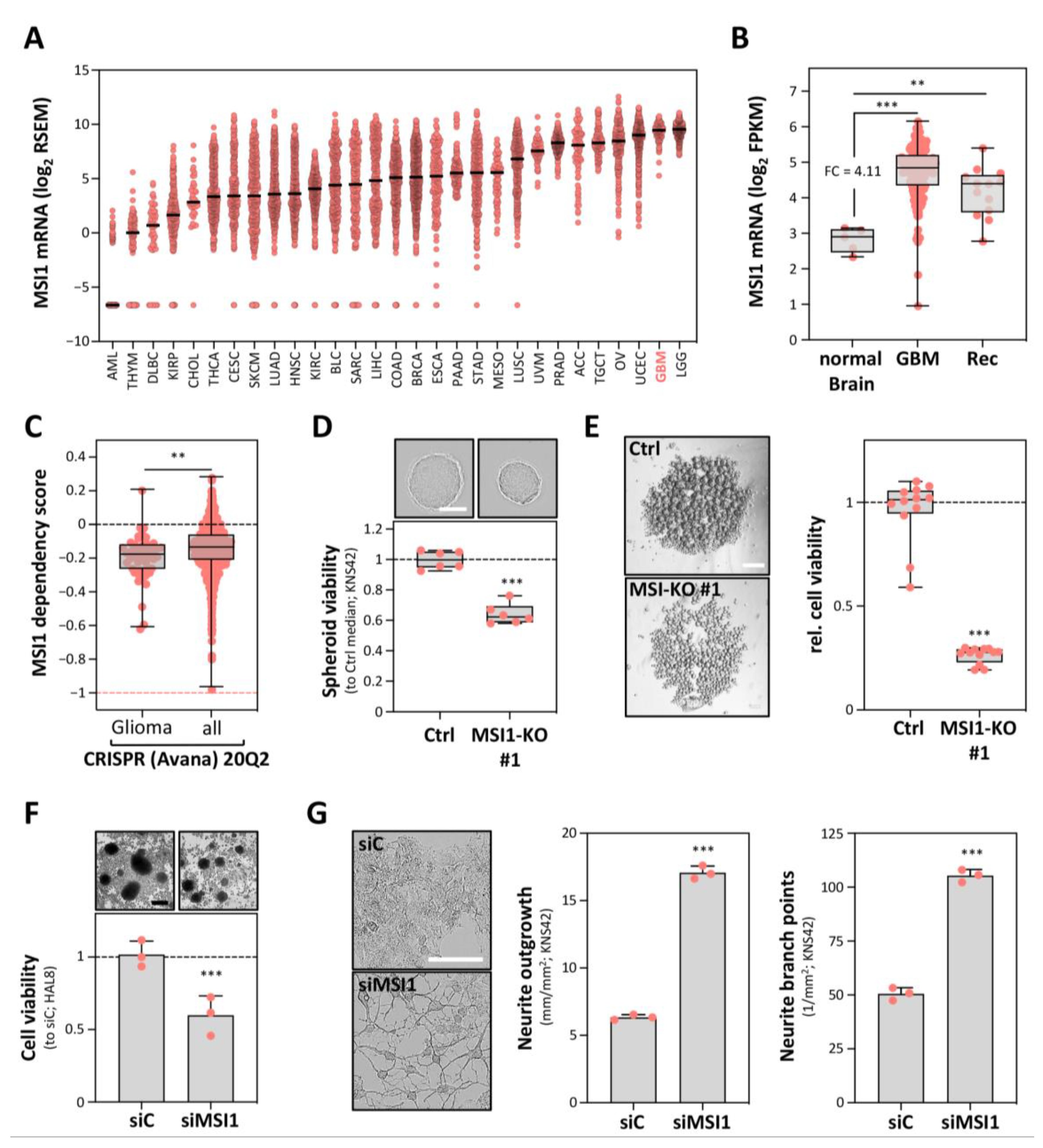
2.1. MSI1 Conveys a Pro-Oncogenic Role in GBM Cells In Vitro
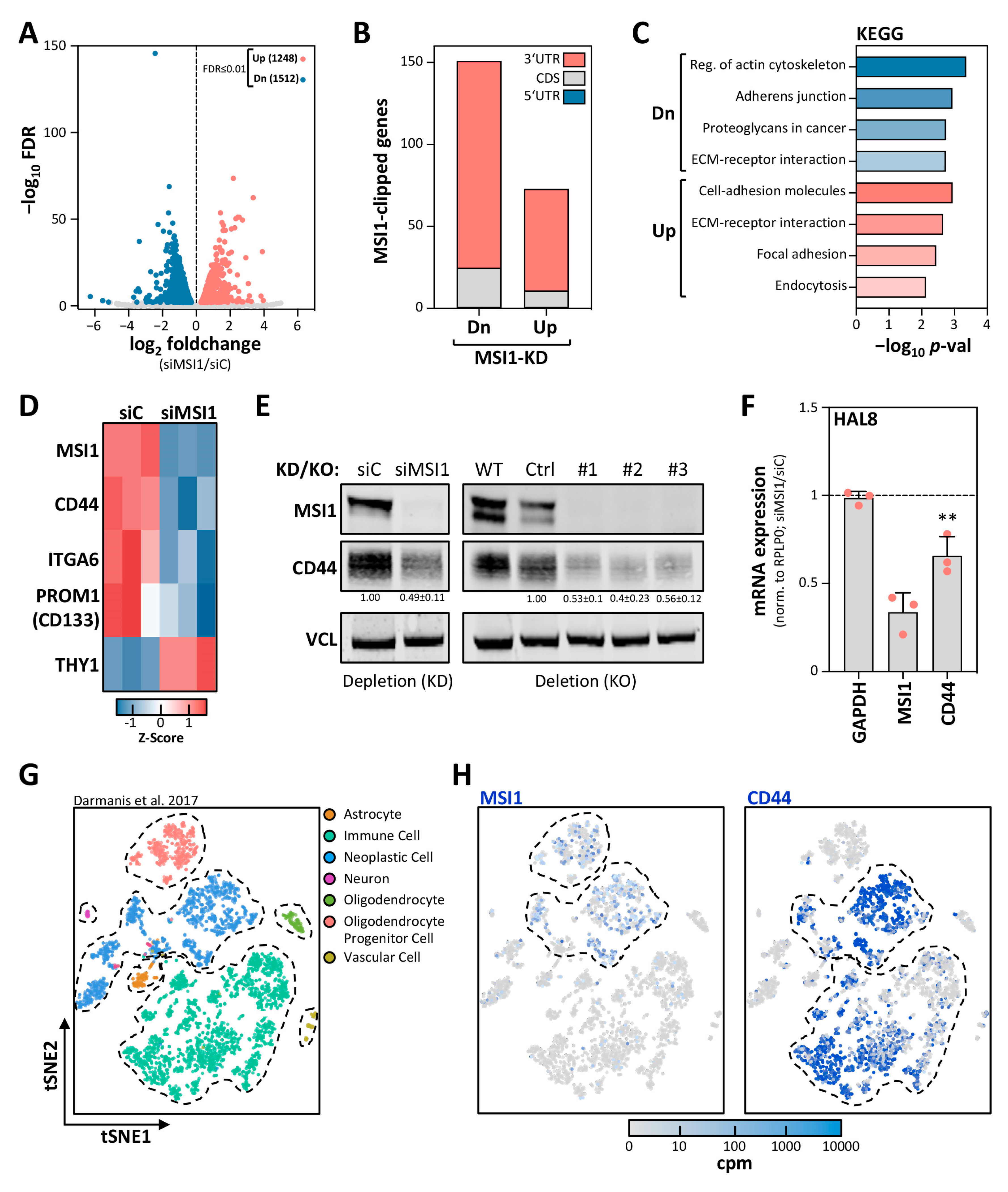
2.2. MSI1 Promotes CD44 Expression in GBM Cells
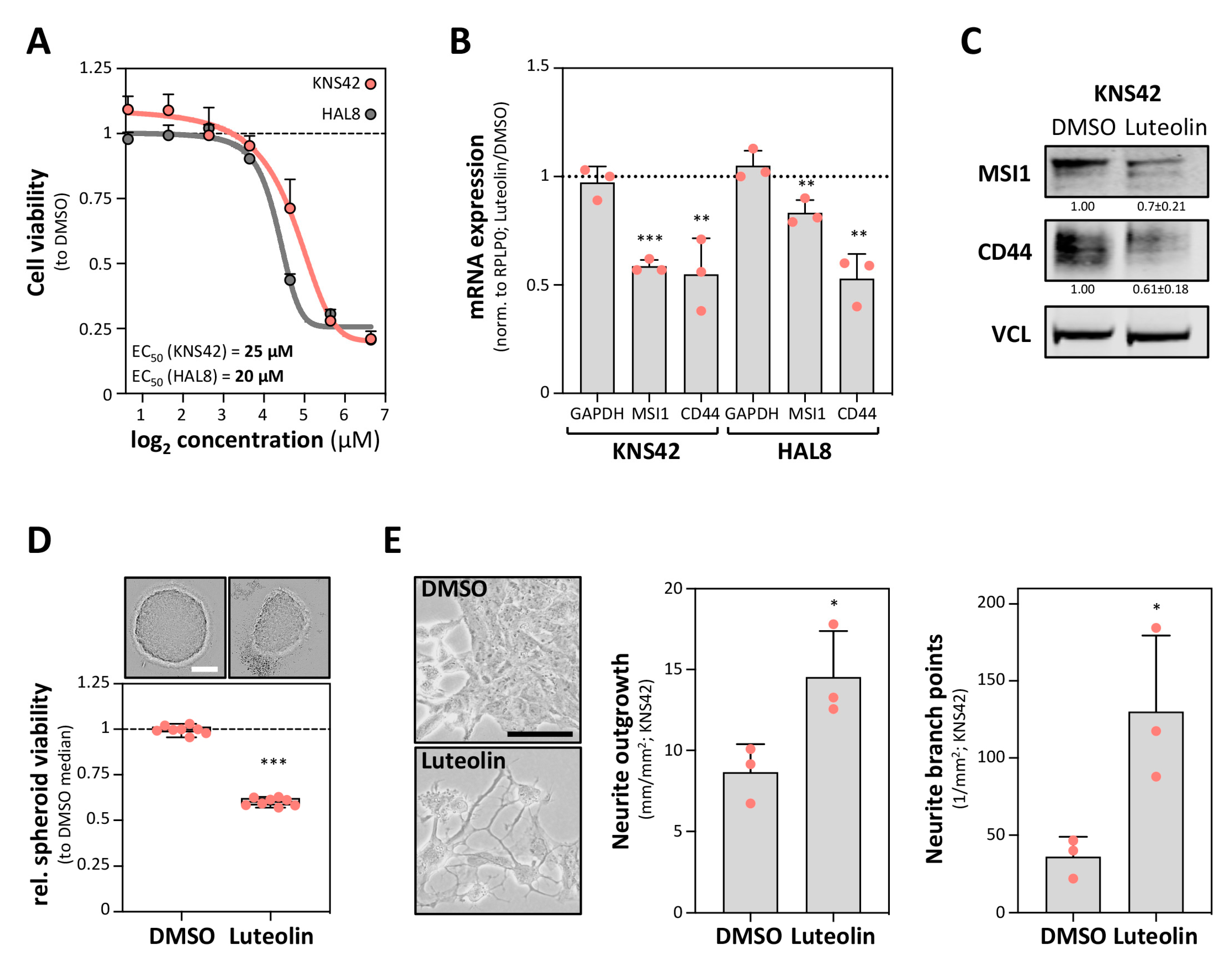
2.3. The MSI1 Inhibitor Luteolin Impairs CD44 Expression in GBM
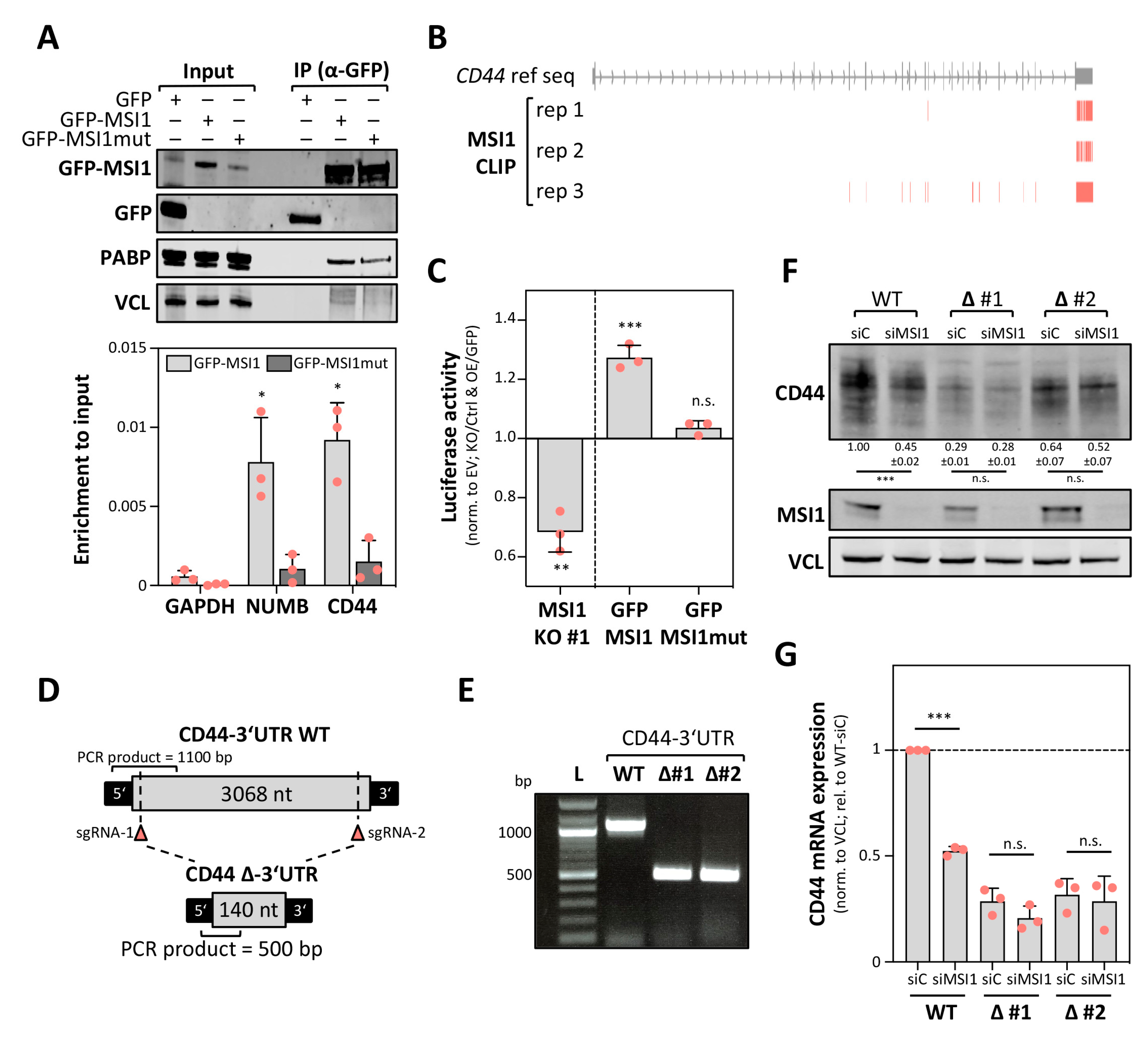
2.4. MSI1 Promotes CD44 Expression in a 3′UTR Dependent Manner
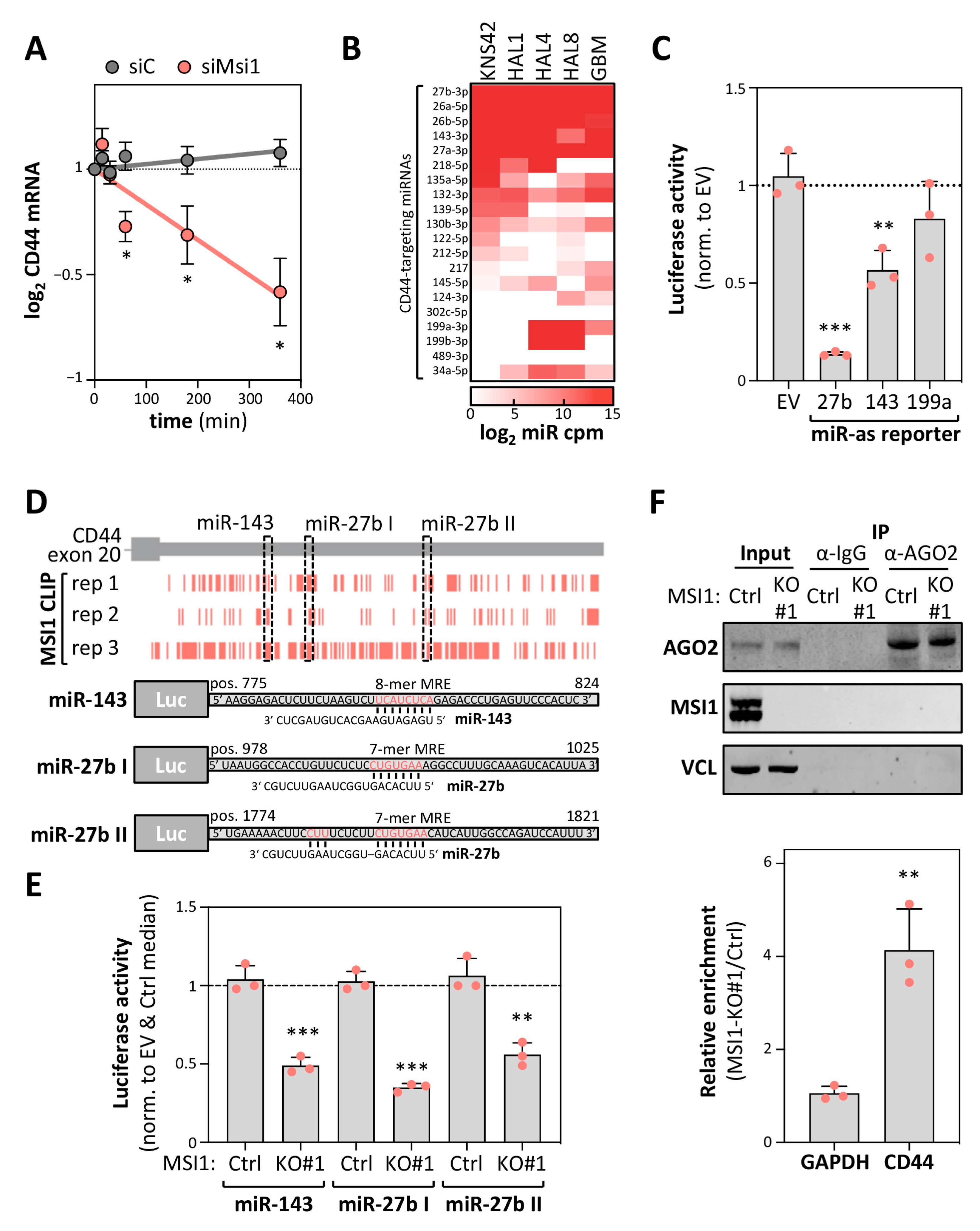
2.5. MSI1 Controls CD44 mRNA Turnover in an miRNA-Dependent Manner
3. Discussion
4. Materials and Methods
4.1. Isolation and Cultivation of Primary Tumorspheres
4.2. Adherent Cell Culture and Transfection
4.3. CRISPR/Cas9 and Lentiviral Transduction
4.4. Cell Viability, Spheroid Growth and Anoikis Resistance
4.5. Neurite Outgrowth Analysis
4.6. Luciferase Assay
4.7. Inhibition of RNA Synthesis
4.8. Extraction of RNA and cDNA Synthesis
4.9. RIP and RT-qPCR
4.10. Western Blotting
4.11. MSI1-CLIP Data Analysis
4.12. Deep-Sequencing and Differential Gene Expression
4.13. Publicaly Available Data
4.14. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar] [PubMed]
- Couturier, C.P.; Ayyadhury, S.; Le, P.U.; Nadaf, J.; Monlong, J.; Riva, G.; Allache, R.; Baig, S.; Yan, X.; Bourgey, M.; et al. Single-cell RNA-seq reveals that glioblastoma recapitulates a normal neurodevelopmental hierarchy. Nat. Commun. 2020, 11, 3406. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Tokunaga, A.; Yoshida, T.; Hashimoto, M.; Mikoshiba, K.; Weinmaster, G.; Nakafuku, M.; Okano, H. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol. Cell Biol. 2001, 21, 3888–3900. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Imai, T.; Hamaguchi, K.; Okabe, M.; Aruga, J.; Nakajima, K.; Yasutomi, D.; Nagata, T.; Kurihara, Y.; Uesugi, S.; et al. Mouse-Musashi-1, a neural RNA-binding protein highly enriched in the mammalian CNS stem cell. Dev. Biol. 1996, 176, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Penalva, L.O.; Yuan, H.; Linnoila, R.I.; Lu, J.; Okano, H.; Glazer, R.I. Musashi1 regulates breast tumor cell proliferation and is a prognostic indicator of poor survival. Mol. Cancer 2010, 9, 221. [Google Scholar] [CrossRef]
- Wang, X.Y.; Yu, H.; Linnoila, R.I.; Li, L.; Li, D.; Mo, B.; Okano, H.; Penalva, L.O.; Glazer, R.I. Musashi1 as a potential therapeutic target and diagnostic marker for lung cancer. Oncotarget 2013, 4, 739–750. [Google Scholar] [CrossRef]
- Kanemura, Y.; Mori, K.; Sakakibara, S.; Fujikawa, H.; Hayashi, H.; Nakano, A.; Matsumoto, T.; Tamura, K.; Imai, T.; Ohnishi, T.; et al. Musashi1, an evolutionarily conserved neural RNA-binding protein, is a versatile marker of human glioma cells in determining their cellular origin, malignancy, and proliferative activity. Differentiation 2001, 68, 141–152. [Google Scholar] [CrossRef]
- Chiou, G.Y.; Yang, T.W.; Huang, C.C.; Tang, C.Y.; Yen, J.Y.; Tsai, M.C.; Chen, H.Y.; Fadhilah, N.; Lin, C.C.; Jong, Y.J. Musashi-1 promotes a cancer stem cell lineage and chemoresistance in colorectal cancer cells. Sci. Rep. 2017, 7, 2172. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Cambuli, F.M.; Correa, B.R.; Rezza, A.; Burns, S.C.; Qiao, M.; Uren, P.J.; Kress, E.; Boussouar, A.; Galante, P.A.; Penalva, L.O.; et al. A Mouse Model of Targeted Musashi1 Expression in Whole Intestinal Epithelium Suggests Regulatory Roles in Cell Cycle and Stemness. Stem. Cells 2015, 33, 3621–3634. [Google Scholar] [CrossRef] [PubMed]
- Uren, P.J.; Vo, D.T.; de Araujo, P.R.; Potschke, R.; Burns, S.C.; Bahrami-Samani, E.; Qiao, M.; de Sousa Abreu, R.; Nakaya, H.I.; Correa, B.R.; et al. RNA-Binding Protein Musashi1 Is a Central Regulator of Adhesion Pathways in Glioblastoma. Mol. Cell Biol. 2015, 35, 2965–2978. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.G.; Park, F.D.; Koechlein, C.S.; Kritzik, M.; Reya, T. Musashi signaling in stem cells and cancer. Annu. Rev. Cell Dev. Biol. 2015, 31, 249–267. [Google Scholar] [CrossRef] [PubMed]
- Hattori, A.; Buac, K.; Ito, T. Regulation of Stem Cell Self-Renewal and Oncogenesis by RNA-Binding Proteins. Adv. Exp. Med. Biol. 2016, 907, 153–188. [Google Scholar] [CrossRef] [PubMed]
- Potschke, R.; Gielen, G.; Pietsch, T.; Kramm, C.; Klusmann, J.H.; Huttelmaier, S.; Kuhnol, C.D. Musashi1 enhances chemotherapy resistance of pediatric glioblastoma cells in vitro. Pediatr. Res. 2020, 87, 669–676. [Google Scholar] [CrossRef]
- Glazer, R.I.; Vo, D.T.; Penalva, L.O. Musashi1: An RBP with versatile functions in normal and cancer stem cells. Front. Biosci. 2012, 17, 54–64. [Google Scholar] [CrossRef]
- Yi, C.; Li, G.; Ivanov, D.N.; Wang, Z.; Velasco, M.X.; Hernandez, G.; Kaundal, S.; Villarreal, J.; Gupta, Y.K.; Qiao, M.; et al. Luteolin inhibits Musashi1 binding to RNA and disrupts cancer phenotypes in glioblastoma cells. RNA Biol. 2018, 15, 1420–1432. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Toda, M.; Iizuka, Y.; Yu, W.; Imai, T.; Ikeda, E.; Yoshida, K.; Kawase, T.; Kawakami, Y.; Okano, H.; Uyemura, K. Expression of the neural RNA-binding protein Musashi1 in human gliomas. Glia 2001, 34, 1–7. [Google Scholar] [CrossRef]
- Kudinov, A.E.; Karanicolas, J.; Golemis, E.A.; Boumber, Y. Musashi RNA-Binding Proteins as Cancer Drivers and Novel Therapeutic Targets. Clin. Cancer Res. 2017, 23, 2143–2153. [Google Scholar] [CrossRef] [PubMed]
- Meyers, R.M.; Bryan, J.G.; McFarland, J.M.; Weir, B.A.; Sizemore, A.E.; Xu, H.; Dharia, N.V.; Montgomery, P.G.; Cowley, G.S.; Pantel, S.; et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet. 2017, 49, 1779–1784. [Google Scholar] [CrossRef] [PubMed]
- Velasco, M.X.; Kosti, A.; Guardia, G.D.A.; Santos, M.C.; Tegge, A.; Qiao, M.; Correa, B.R.S.; Hernandez, G.; Kokovay, E.; Galante, P.A.F.; et al. Antagonism between the RNA-binding protein Musashi1 and miR-137 and its potential impact on neurogenesis and glioblastoma development. RNA 2019, 25, 768–782. [Google Scholar] [CrossRef]
- Chen, H.Y.; Wang, M.L.; Laurent, B.; Hsu, C.H.; Chen, M.T.; Lin, L.T.; Shen, J.; Chang, W.C.; Hsu, J.; Hung, M.C.; et al. Musashi-1 promotes stress-induced tumor progression through recruitment of AGO2. Theranostics 2020, 10, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Lathia, J.D.; Gallagher, J.; Heddleston, J.M.; Wang, J.; Eyler, C.E.; Macswords, J.; Wu, Q.; Vasanji, A.; McLendon, R.E.; Hjelmeland, A.B.; et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem. Cell 2010, 6, 421–432. [Google Scholar] [CrossRef]
- Papatheodorou, I.; Moreno, P.; Manning, J.; Fuentes, A.M.; George, N.; Fexova, S.; Fonseca, N.A.; Fullgrabe, A.; Green, M.; Huang, N.; et al. Expression Atlas update: From tissues to single cells. Nucleic. Acids Res. 2020, 48, D77–D83. [Google Scholar] [CrossRef]
- Babicki, S.; Arndt, D.; Marcu, A.; Liang, Y.; Grant, J.R.; Maciejewski, A.; Wishart, D.S. Heatmapper: Web-enabled heat mapping for all. Nucleic. Acids Res. 2016, 44, W147–W153. [Google Scholar] [CrossRef]
- Kurihara, Y.; Nagata, T.; Imai, T.; Hiwatashi, A.; Horiuchi, M.; Sakakibara, S.; Katahira, M.; Okano, H.; Uesugi, S. Structural properties and RNA-binding activities of two RNA recognition motifs of a mouse neural RNA-binding protein, mouse-Musashi-1. Gene 1997, 186, 21–27. [Google Scholar] [CrossRef]
- Kawahara, H.; Imai, T.; Imataka, H.; Tsujimoto, M.; Matsumoto, K.; Okano, H. Neural RNA-binding protein Musashi1 inhibits translation initiation by competing with eIF4G for PABP. J. Cell Biol. 2008, 181, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Chavali, P.L.; Stojic, L.; Meredith, L.W.; Joseph, N.; Nahorski, M.S.; Sanford, T.J.; Sweeney, T.R.; Krishna, B.A.; Hosmillo, M.; Firth, A.E.; et al. Neurodevelopmental protein Musashi-1 interacts with the Zika genome and promotes viral replication. Science 2017, 357, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Cullen, B.R. Reduced expression of brain-enriched microRNAs in glioblastomas permits targeted regulation of a cell death gene. PLoS ONE 2011, 6, e24248. [Google Scholar] [CrossRef] [PubMed]
- Hua, D.; Mo, F.; Ding, D.; Li, L.; Han, X.; Zhao, N.; Foltz, G.; Lin, B.; Lan, Q.; Huang, Q. A catalogue of glioblastoma and brain MicroRNAs identified by deep sequencing. OMICS 2012, 16, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, D.; Nie, J.; Zhou, S.; Wang, J.; Tang, Q.; Yang, X. MicroRNA143 targets CD44 to inhibit breast cancer progression and stem cell-like properties. Mol. Med. Rep. 2016, 13, 5193–5199. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, B.; Jin, Y.; Wu, Q.; Cao, L. MiR-27b targets PI3K p110alpha to inhibit proliferation and migration in colorectal cancer stem cell. Am. J. Transl. Res. 2019, 11, 5988–5997. [Google Scholar]
- Henry, J.C.; Park, J.K.; Jiang, J.; Kim, J.H.; Nagorney, D.M.; Roberts, L.R.; Banerjee, S.; Schmittgen, T.D. miR-199a-3p targets CD44 and reduces proliferation of CD44 positive hepatocellular carcinoma cell lines. Biochem. Biophys Res. Commun. 2010, 403, 120–125. [Google Scholar] [CrossRef]
- Muller, S.; Glass, M.; Singh, A.K.; Haase, J.; Bley, N.; Fuchs, T.; Lederer, M.; Dahl, A.; Huang, H.; Chen, J.; et al. IGF2BP1 promotes SRF-dependent transcription in cancer in a m6A- and miRNA-dependent manner. Nucleic. Acids Res. 2019, 47, 375–390. [Google Scholar] [CrossRef]
- Yan, Y.; Zuo, X.; Wei, D. Concise Review: Emerging Role of CD44 in Cancer Stem Cells: A Promising Biomarker and Therapeutic Target. Stem. Cells Transl. Med. 2015, 4, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Sakakibara, S.; Okano, H. Expression of neural RNA-binding proteins in the postnatal CNS: Implications of their roles in neuronal and glial cell development. J. Neurosci. 1997, 17, 8300–8312. [Google Scholar] [CrossRef] [PubMed]
- Kuwako, K.; Kakumoto, K.; Imai, T.; Igarashi, M.; Hamakubo, T.; Sakakibara, S.; Tessier-Lavigne, M.; Okano, H.J.; Okano, H. Neural RNA-binding protein Musashi1 controls midline crossing of precerebellar neurons through posttranscriptional regulation of Robo3/Rig-1 expression. Neuron 2010, 67, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Iacopino, F.; Angelucci, C.; Piacentini, R.; Biamonte, F.; Mangiola, A.; Maira, G.; Grassi, C.; Sica, G. Isolation of cancer stem cells from three human glioblastoma cell lines: Characterization of two selected clones. PLoS ONE 2014, 9, e105166. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, S.; Ajay, A.K.; Trivedi, P.; Seematti, J.; Pellegrini, K.; Craciun, F.; Vaidya, V.S. RNA-binding Protein Musashi Homologue 1 Regulates Kidney Fibrosis by Translational Inhibition of p21 and Numb mRNA. J. Biol. Chem. 2016, 291, 14085–14094. [Google Scholar] [CrossRef] [PubMed]
- Haase, J.; Misiak, D.; Bauer, M.; Pazaitis, N.; Braun, J.; Potschke, R.; Mensch, A.; Bell, J.L.; Dralle, H.; Siebolts, U.; et al. IGF2BP1 is the first positive marker for anaplastic thyroid carcinoma diagnosis. Mod. Pathol. 2020, 1–10. [Google Scholar] [CrossRef]
- Degrauwe, N.; Suva, M.L.; Janiszewska, M.; Riggi, N.; Stamenkovic, I. IMPs: An RNA-binding protein family that provides a link between stem cell maintenance in normal development and cancer. Genes Dev. 2016, 30, 2459–2474. [Google Scholar] [CrossRef]
- Busch, B.; Bley, N.; Muller, S.; Glass, M.; Misiak, D.; Lederer, M.; Vetter, M.; Strauss, H.G.; Thomssen, C.; Huttelmaier, S. The oncogenic triangle of HMGA2, LIN28B and IGF2BP1 antagonizes tumor-suppressive actions of the let-7 family. Nucleic. Acids Res. 2016, 44, 3845–3864. [Google Scholar] [CrossRef]
- Takahashi, R.U.; Miyazaki, H.; Takeshita, F.; Yamamoto, Y.; Minoura, K.; Ono, M.; Kodaira, M.; Tamura, K.; Mori, M.; Ochiya, T. Loss of microRNA-27b contributes to breast cancer stem cell generation by activating ENPP1. Nat. Commun. 2015, 6, 7318. [Google Scholar] [CrossRef]
- Huang, S.; Guo, W.; Tang, Y.; Ren, D.; Zou, X.; Peng, X. miR-143 and miR-145 inhibit stem cell characteristics of PC-3 prostate cancer cells. Oncol. Rep. 2012, 28, 1831–1837. [Google Scholar] [CrossRef]
- Liu, C.; Kelnar, K.; Liu, B.; Chen, X.; Calhoun-Davis, T.; Li, H.; Patrawala, L.; Yan, H.; Jeter, C.; Honorio, S.; et al. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat. Med. 2011, 17, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Jeyapalan, Z.; Deng, Z.; Shatseva, T.; Fang, L.; He, C.; Yang, B.B. Expression of CD44 3′-untranslated region regulates endogenous microRNA functions in tumorigenesis and angiogenesis. Nucleic. Acids Res. 2011, 39, 3026–3041. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, L.; Andruska, N.; Mao, C.; Le, J.; Shapiro, D.J. A Novel IMP1 Inhibitor, BTYNB, Targets c-Myc and Inhibits Melanoma and Ovarian Cancer Cell Proliferation. Transl. Oncol. 2017, 10, 818–827. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, H.; Jia, Y.; Pan, H.; Ding, H. Luteolin induces apoptosis by ROS/ER stress and mitochondrial dysfunction in gliomablastoma. Cancer Chemother. Pharmacol. 2017, 79, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, H.; Jia, Y.; Ding, H.; Zhang, L.; Pan, H. Luteolin reduces migration of human glioblastoma cell lines via inhibition of the p-IGF-1R/PI3K/AKT/mTOR signaling pathway. Oncol. Lett. 2017, 14, 3545–3551. [Google Scholar] [CrossRef]
- Liu, Y.; Lang, T.; Jin, B.; Chen, F.; Zhang, Y.; Beuerman, R.W.; Zhou, L.; Zhang, Z. Luteolin inhibits colorectal cancer cell epithelial-to-mesenchymal transition by suppressing CREB1 expression revealed by comparative proteomics study. J. Proteom. 2017, 161, 1–10. [Google Scholar] [CrossRef]
- Cook, M.T.; Liang, Y.; Besch-Williford, C.; Hyder, S.M. Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer 2017, 9, 9–19. [Google Scholar] [CrossRef]
- Zang, M.D.; Hu, L.; Fan, Z.Y.; Wang, H.X.; Zhu, Z.L.; Cao, S.; Wu, X.Y.; Li, J.F.; Su, L.P.; Li, C.; et al. Luteolin suppresses gastric cancer progression by reversing epithelial-mesenchymal transition via suppression of the Notch signaling pathway. J. Transl. Med. 2017, 15, 52. [Google Scholar] [CrossRef]
- Chakrabarti, M.; Ray, S.K. Synergistic anti-tumor actions of luteolin and silibinin prevented cell migration and invasion and induced apoptosis in glioblastoma SNB19 cells and glioblastoma stem cells. Brain Res. 2015, 1629, 85–93. [Google Scholar] [CrossRef]
- Sonoki, H.; Tanimae, A.; Endo, S.; Matsunaga, T.; Furuta, T.; Ichihara, K.; Ikari, A. Kaempherol and Luteolin Decrease Claudin-2 Expression Mediated by Inhibition of STAT3 in Lung Adenocarcinoma A549 Cells. Nutrients 2017, 9, 597. [Google Scholar] [CrossRef]
- Azari, H.; Millette, S.; Ansari, S.; Rahman, M.; Deleyrolle, L.P.; Reynolds, B.A. Isolation and expansion of human glioblastoma multiforme tumor cells using the neurosphere assay. J. Vis. Exp. 2011, 10, e3633. [Google Scholar] [CrossRef] [PubMed]
- Klages, N.; Zufferey, R.; Trono, D. A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol. Ther. 2000, 2, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.; Bley, N.; Glass, M.; Busch, B.; Rousseau, V.; Misiak, D.; Fuchs, T.; Lederer, M.; Huttelmaier, S. IGF2BP1 enhances an aggressive tumor cell phenotype by impairing miRNA-directed downregulation of oncogenic factors. Nucleic. Acids Res. 2018, 46, 6285–6303. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Yates, A.; Akanni, W.; Amode, M.R.; Barrell, D.; Billis, K.; Carvalho-Silva, D.; Cummins, C.; Clapham, P.; Fitzgerald, S.; Gil, L.; et al. Ensembl 2016. Nucleic. Acids Res. 2016, 44, D710–D716. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic. Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, l1. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pötschke, R.; Haase, J.; Glaß, M.; Simmermacher, S.; Misiak, C.; Penalva, L.O.F.; Kühnöl, C.D.; Hüttelmaier, S. MSI1 Promotes the Expression of the GBM Stem Cell Marker CD44 by Impairing miRNA-Dependent Degradation. Cancers 2020, 12, 3654. https://doi.org/10.3390/cancers12123654
Pötschke R, Haase J, Glaß M, Simmermacher S, Misiak C, Penalva LOF, Kühnöl CD, Hüttelmaier S. MSI1 Promotes the Expression of the GBM Stem Cell Marker CD44 by Impairing miRNA-Dependent Degradation. Cancers. 2020; 12(12):3654. https://doi.org/10.3390/cancers12123654
Chicago/Turabian StylePötschke, Rebecca, Jacob Haase, Markus Glaß, Sebastian Simmermacher, Claudia Misiak, Luiz O. F. Penalva, Caspar D. Kühnöl, and Stefan Hüttelmaier. 2020. "MSI1 Promotes the Expression of the GBM Stem Cell Marker CD44 by Impairing miRNA-Dependent Degradation" Cancers 12, no. 12: 3654. https://doi.org/10.3390/cancers12123654
APA StylePötschke, R., Haase, J., Glaß, M., Simmermacher, S., Misiak, C., Penalva, L. O. F., Kühnöl, C. D., & Hüttelmaier, S. (2020). MSI1 Promotes the Expression of the GBM Stem Cell Marker CD44 by Impairing miRNA-Dependent Degradation. Cancers, 12(12), 3654. https://doi.org/10.3390/cancers12123654