Platelet-Derived GARP Induces Peripheral Regulatory T Cells—Potential Impact on T Cell Suppression in Patients with Melanoma-Associated Thrombocytosis

 ,
,  ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Expression of GARP on Platelets
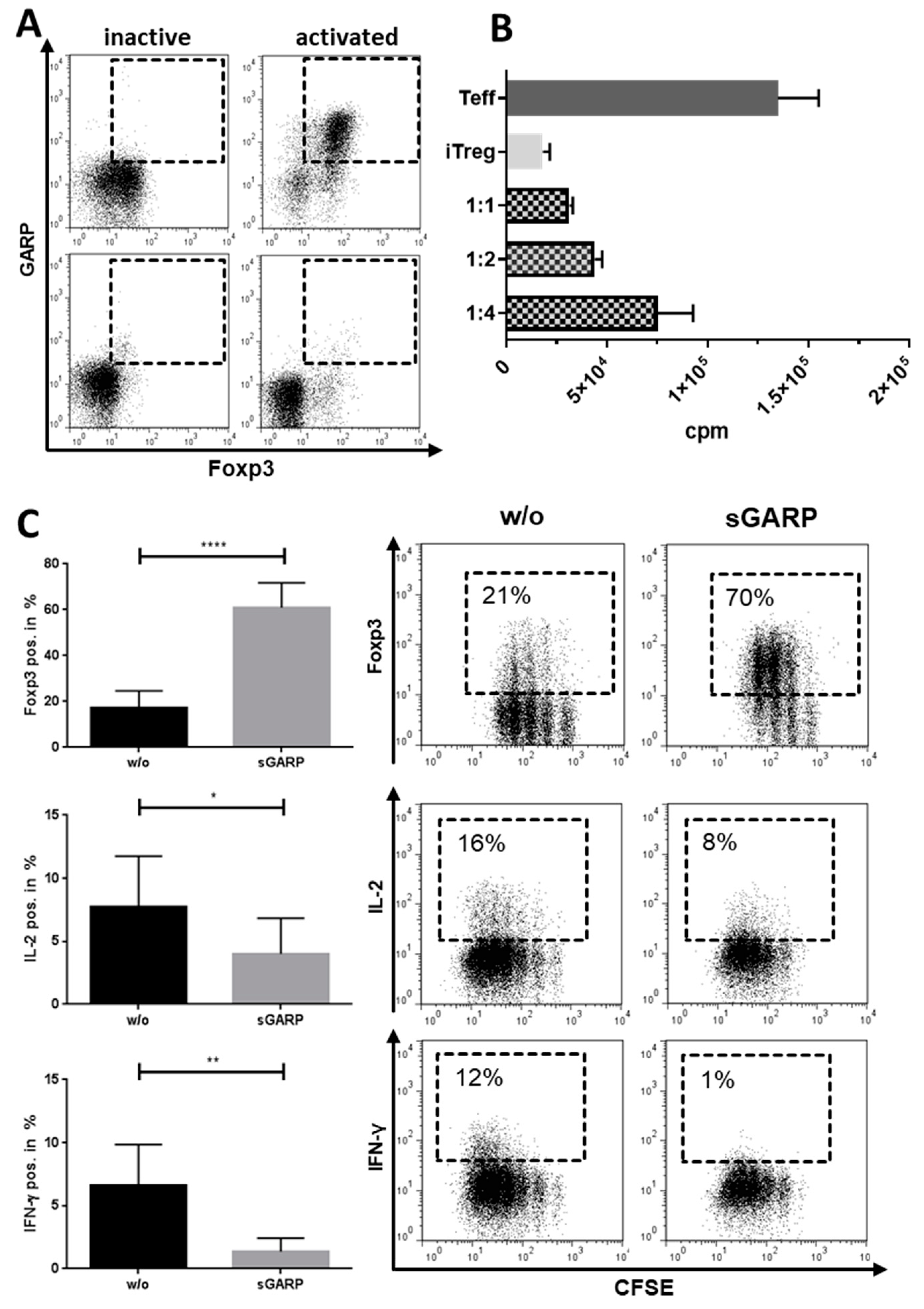
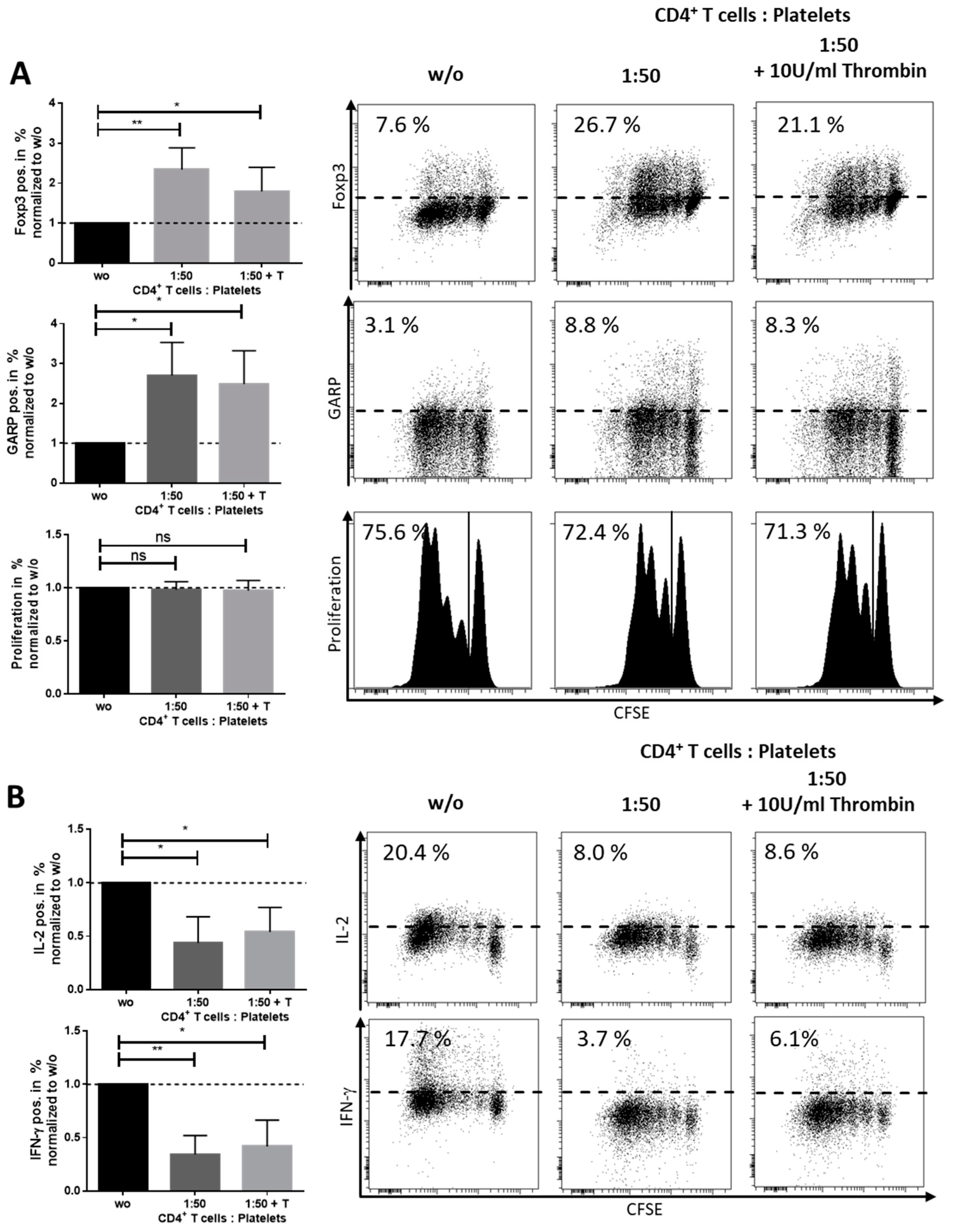
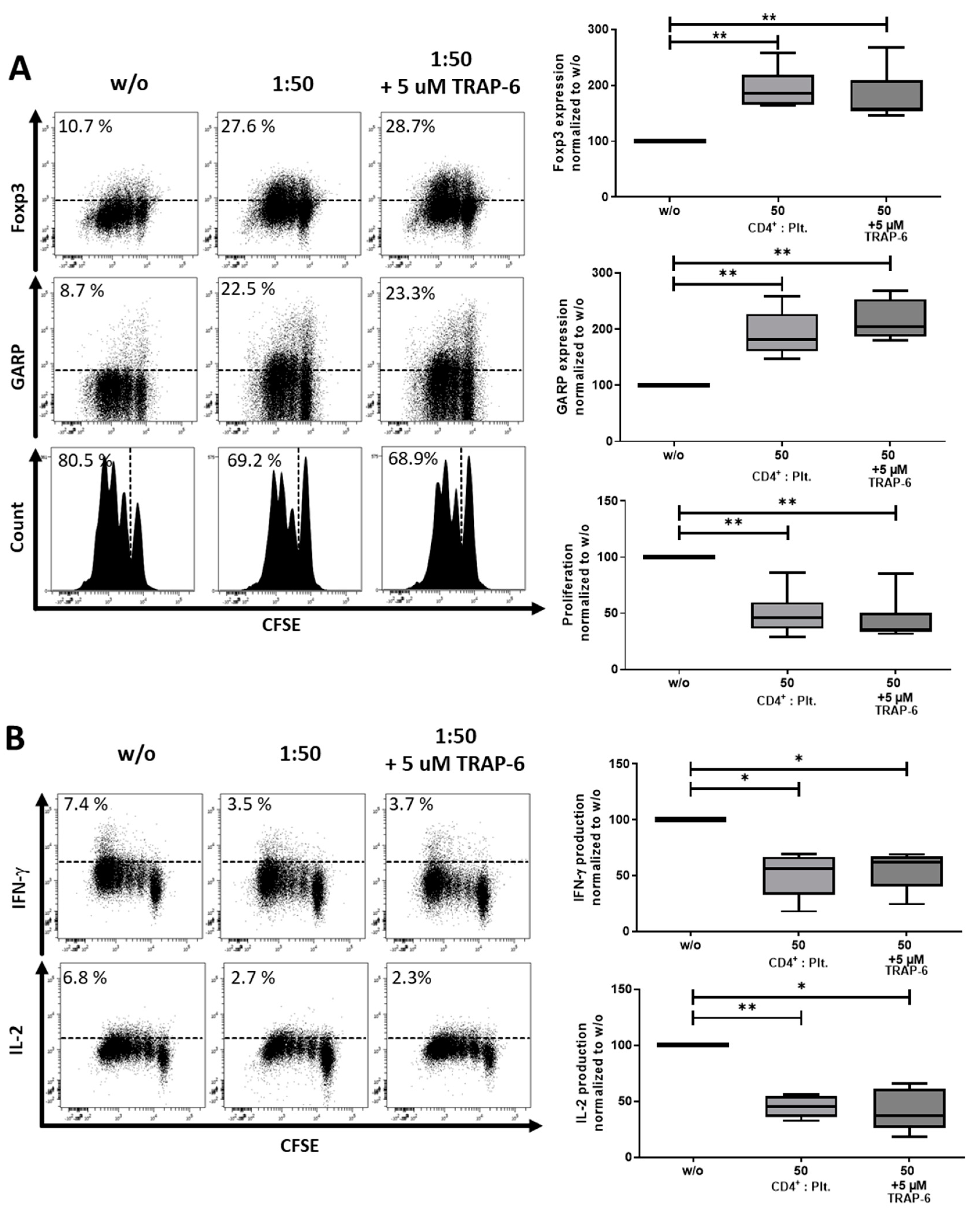
2.2. Platelet-Derived GARP Induced Peripheral Regulatory T cells
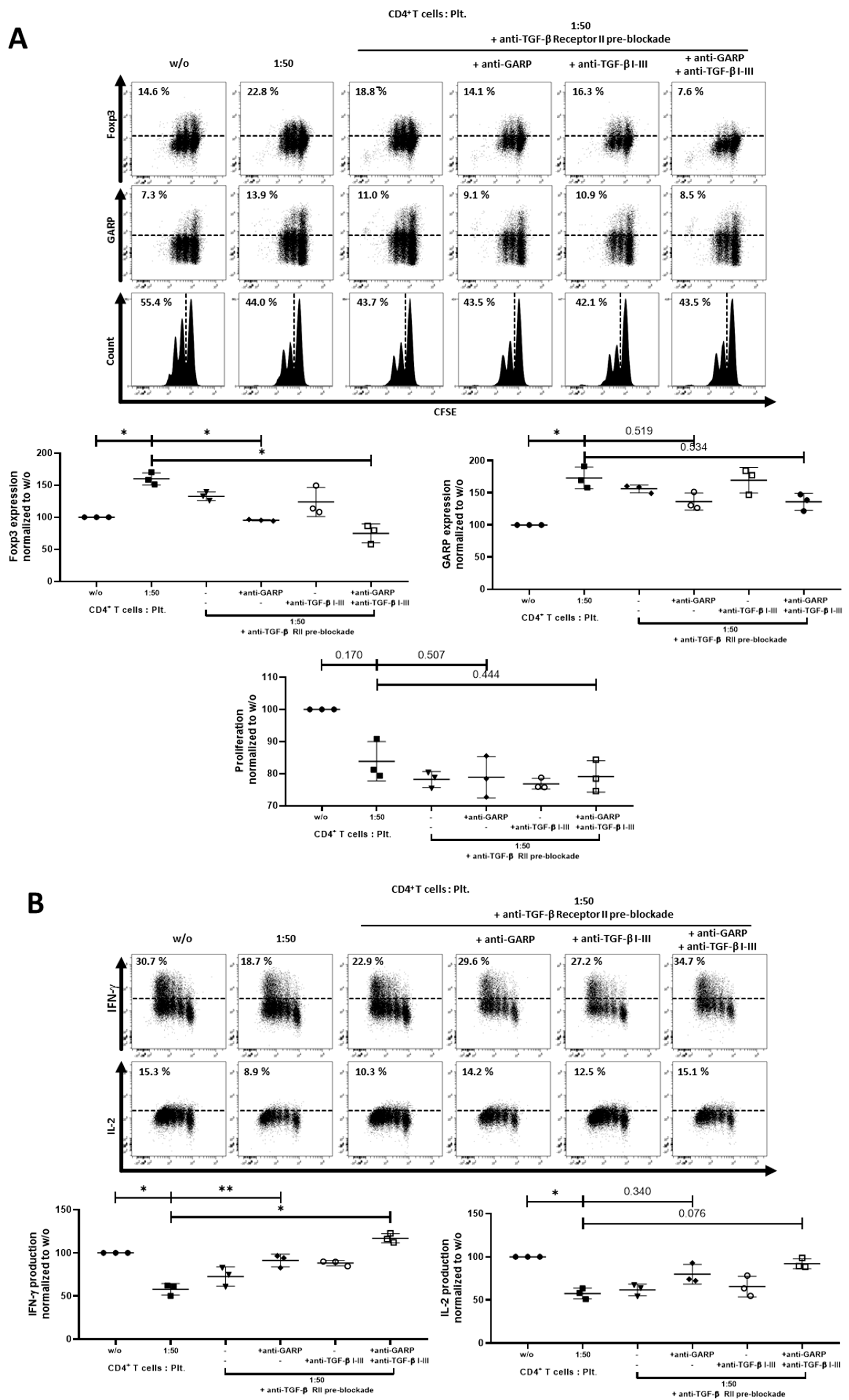
2.3. Role of TGF-βand Platelet-Conditioned Medium in Platelet-Mediated iTreg Induction
2.4. Correlation of Thrombocytosis and Prognosis—Clinical Impact of Platelets Expressing Increased Levels of GARP
3. Discussion
4. Materials and Methods
4.1. Isolation of Pre-Activated and Resting Platelets and Preparation of Platelet-Conditioned Medium
4.2. Isolation and Stimulation of Human T Cell Populations
4.3. Enzyme-Linked Immunosorbent Assay
4.4. Flow Cytometry
4.5. Patients
4.6. Statistics
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B

Appendix C

Appendix D

Appendix E

Appendix F

Appendix G

References
- Schupp, J.; Krebs, F.K.; Zimmer, N.; Trzeciak, E.; Schuppan, D.; Tuettenberg, A. Targeting myeloid cells in the tumor sustaining microenvironment. Cell. Immunol. 2019, 343, 103713. [Google Scholar] [CrossRef] [PubMed]
- Yap, M.L.; McFadyen, J.D.; Wang, X.; Ziegler, M.; Chen, Y.-C.; Willcox, A.; Nowell, C.J.; Scott, A.M.; Sloan, E.K.; Hogarth, P.M.; et al. Activated platelets in the tumor microenvironment for targeting of antibody-drug conjugates to tumors and metastases. Theranostics 2019, 9, 1154–1169. [Google Scholar] [CrossRef] [PubMed]
- Huong, P.T.; Nguyen, L.T.; Nguyen, X.-B.; Lee, S.K.; Bach, D.-H. The Role of Platelets in the Tumor-Microenvironment and the Drug Resistance of Cancer Cells. Cancers 2019, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- McAllister, S.S.; Weinberg, R.A. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat. Cell Biol. 2014, 16, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirousková, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Camerer, E.; Qazi, A.A.; Duong, D.N.; Cornelissen, I.; Advincula, R.; Coughlin, S.R. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood 2004, 104, 397–401. [Google Scholar] [CrossRef]
- Orellana, R.; Kato, S.; Erices, R.; Bravo, M.L.; Gonzalez, P.; Oliva, B.; Cubillos, S.; Valdivia, A.; Ibañez, C.; Brañes, J.; et al. Platelets enhance tissue factor protein and metastasis initiating cell markers, and act as chemoattractants increasing the migration of ovarian cancer cells. BMC Cancer 2015, 15, 290. [Google Scholar] [CrossRef]
- Karpatkin, S.; Pearlstein, E.; Ambrogio, C.; Coller, B.S. Role of adhesive proteins in platelet tumor interaction in vitro and metastasis formation in vivo. J. Clin. Invest. 1988, 81, 1012–1019. [Google Scholar] [CrossRef]
- Stone, R.L.; Nick, A.M.; McNeish, I.A.; Balkwill, F.; Han, H.D.; Bottsford-Miller, J.; Rupairmoole, R.; Armaiz-Pena, G.N.; Pecot, C.V.; Coward, J.; et al. Paraneoplastic thrombocytosis in ovarian cancer. N. Engl. J. Med. 2012, 366, 610–618. [Google Scholar] [CrossRef]
- Sierko, E.; Wojtukiewicz, M.Z. Platelets and angiogenesis in malignancy. Semin. Thromb. Hemost. 2004, 30, 95–108. [Google Scholar] [CrossRef]
- Schumacher, D.; Strilic, B.; Sivaraj, K.K.; Wettschureck, N.; Offermanns, S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Cancer Cell 2013, 24, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev 2017, 36, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.A. Cytokines in Cancer Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Z.; Xu, R. Human Cancer and Platelet Interaction, a Potential Therapeutic Target. Int. J. Mol. Sci. 2018, 19, 1246. [Google Scholar] [CrossRef] [PubMed]
- Li, N. Platelets in cancer metastasis: To help the "villain" to do evil. Int. J. Cancer 2016, 138, 2078–2087. [Google Scholar] [CrossRef] [PubMed]
- Buergy, D.; Wenz, F.; Groden, C.; Brockmann, M.A. Tumor-platelet interaction in solid tumors. Int. J. Cancer 2012, 130, 2747–2760. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053-61. [Google Scholar] [CrossRef]
- Mezouar, S.; Frère, C.; Darbousset, R.; Mege, D.; Crescence, L.; Dignat-George, F.; Panicot-Dubois, L.; Dubois, C. Role of platelets in cancer and cancer-associated thrombosis: Experimental and clinical evidences. Thrombosis Research 2016, 139, 65–76. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, W.-J.; Cai, H.-Q.; Liu, H.-L.; Peng, L.; Li, C.-H.; Ye, L.-Y.; Xu, S.-Q.; Yang, Z.-H.; Lou, J.-N. Platelet adhesion and fusion to endothelial cell facilitate the metastasis of tumor cell in hypoxia-reoxygenation condition. Clin. Exp. Metastasis 2011, 28, 1–12. [Google Scholar] [CrossRef]
- Marcolino, E.; Siddiqui, Y.H.; van den Bosch, M.; Poole, A.W.; Jayaraman, P.-S.; Gaston, K. Blood platelets stimulate cancer extravasation through TGFβ-mediated downregulation of PRH/HHEX. Oncogenesis 2020, 9, 10. [Google Scholar] [CrossRef]
- Ballerini, P.; Dovizio, M.; Bruno, A.; Tacconelli, S.; Patrignani, P. P2Y12 Receptors in Tumorigenesis and Metastasis. Front. Pharmacol. 2018, 9, 66. [Google Scholar] [CrossRef]
- Li, W.-H.; Qiu, Y.; Zhang, H.-Q.; Liu, Y.; You, J.-F.; Tian, X.-X.; Fang, W.-G. P2Y2 receptor promotes cell invasion and metastasis in prostate cancer cells. Br. J. Cancer 2013, 109, 1666–1675. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Novitskiy, S.V.; Pickup, M.W.; Chytil, A.; Polosukhina, D.; Owens, P.; Moses, H.L. Deletion of TGF-β signaling in myeloid cells enhances their anti-tumorigenic properties. J. Leukoc. Biol. 2012, 92, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Gara, S.K.; Achyut, B.R.; Li, Z.; Yan, H.H.; Day, C.-P.; Weiss, J.M.; Trinchieri, G.; Morris, J.C.; Yang, L. TGF-β signaling in myeloid cells is required for tumor metastasis. Cancer Discov. 2013, 3, 936–951. [Google Scholar] [CrossRef] [PubMed]
- Leblanc, R.; Peyruchaud, O. Metastasis: new functional implications of platelets and megakaryocytes. Blood 2016, 128, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Cravioto-Villanueva, A.; Luna-Perez, P.; La Gutierrez-de Barrera, M.; Martinez-Gómez, H.; Maffuz, A.; Rojas-Garcia, P.; Perez-Alvarez, C.; Rodriguez-Ramirez, S.; Rodriguez-Antezana, E.; Ramirez-Ramirez, L. Thrombocytosis as a predictor of distant recurrence in patients with rectal cancer. Arch. Med. Res. 2012, 43, 305–311. [Google Scholar] [CrossRef]
- Weber, M.R.; Zuka, M.; Lorger, M.; Tschan, M.; Torbett, B.E.; Zijlstra, A.; Quigley, J.P.; Staflin, K.; Eliceiri, B.P.; Krueger, J.S.; et al. Activated tumor cell integrin αvβ3 cooperates with platelets to promote extravasation and metastasis from the blood stream. Thromb. Res. 2016, 140, S27–S36. [Google Scholar] [CrossRef]
- Heinmöller, E.; Weinel, R.J.; Heidtmann, H.H.; Salge, U.; Seitz, R.; Schmitz, I.; Müller, K.M.; Zirngibl, H. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J. Cancer Res. Clin. Oncol. 1996, 122, 735–744. [Google Scholar] [CrossRef]
- Honn, K.V.; Tang, D. Hemostasis and malignancy: an overview. Cancer Metastasis Rev. 1992, 11, 223–226. [Google Scholar] [CrossRef]
- Wilson, E.B.; El-Jawhari, J.J.; Neilson, A.L.; Hall, G.D.; Melcher, A.A.; Meade, J.L.; Cook, G.P. Human tumour immune evasion via TGF-β blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS ONE 2011, 6, e22842. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Männel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar] [PubMed]
- Guo, S.-W.; Du, Y.; Liu, X. Platelet-derived TGF-β1 mediates the down-modulation of NKG2D expression and may be responsible for impaired natural killer (NK) cytotoxicity in women with endometriosis. Hum. Reprod. 2016, 31, 1462–1474. [Google Scholar] [CrossRef] [PubMed]
- Placke, T.; Örgel, M.; Schaller, M.; Jung, G.; Rammensee, H.-G.; Kopp, H.-G.; Salih, H.R. Platelet-derived MHC class I confers a pseudonormal phenotype to cancer cells that subverts the antitumor reactivity of natural killer immune cells. Cancer Res. 2012, 72, 440–448. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, J.; Dong, X.; Shi, M.; Lu, C.; Springer, T.A. GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 2012, 23, 1129–1139. [Google Scholar] [CrossRef]
- Zimmer, N.; Kim, E.; Sprang, B.; Leukel, P.; Khafaji, F.; Ringel, F.; Sommer, C.; Tuettenberg, J.; Tuettenberg, A. GARP as an Immune Regulatory Molecule in the Tumor Microenvironment of Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 3676. [Google Scholar] [CrossRef]
- Hahn, S.A.; Stahl, H.F.; Becker, C.; Correll, A.; Schneider, F.-J.; Tuettenberg, A.; Jonuleit, H. Soluble GARP has potent antiinflammatory and immunomodulatory impact on human CD4⁺ T cells. Blood 2013, 122, 1182–1191. [Google Scholar] [CrossRef]
- Hahn, S.A.; Neuhoff, A.; Landsberg, J.; Schupp, J.; Eberts, D.; Leukel, P.; Bros, M.; Weilbaecher, M.; Schuppan, D.; Grabbe, S.; et al. A key role of GARP in the immune suppressive tumor microenvironment. Oncotarget 2016, 7, 42996–43009. [Google Scholar] [CrossRef]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef]
- Gaertner, F.; Massberg, S. Patrolling the vascular borders: platelets in immunity to infection and cancer. Nat Rev Immunol 2019, 19, 747–760. [Google Scholar] [CrossRef]
- Metelli, A.; Wu, B.X.; Riesenberg, B.; Guglietta, S.; Huck, J.D.; Mills, C.; Li, A.; Rachidi, S.; Krieg, C.; Rubinstein, M.P.; et al. Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-β. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, L.; Schön, M.P. Deadly allies: the fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yao, X.; Cen, D.; Zhi, Y.; Zhu, N.; Xu, L. Prognostic role of pretreatment thrombocytosis on survival in patients with cervical cancer: a systematic review and meta-analysis. World J. Surg. Oncol. 2019, 17, 132. [Google Scholar] [CrossRef] [PubMed]
- Vermeersch, E.; Denorme, F.; Maes, W.; de Meyer, S.F.; Vanhoorelbeke, K.; Edwards, J.; Shevach, E.M.; Unutmaz, D.; Fujii, H.; Deckmyn, H.; et al. The role of platelet and endothelial GARP in thrombosis and hemostasis. PLoS ONE 2017, 12, e0173329. [Google Scholar] [CrossRef] [PubMed]
- Palacios-Acedo, A.L.; Mège, D.; Crescence, L.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Platelets, Thrombo-Inflammation, and Cancer: Collaborating With the Enemy. Front. Immunol. 2019, 10, 1805. [Google Scholar] [CrossRef]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, M.; Li, J.; Yang, R.; Du, J.; Luo, Y. High Platelet Levels Attenuate the Efficacy of Platinum-Based Treatment in Non-Small Cell Lung Cancer. Cell. Physiol. Biochem. 2018, 48, 2456–2469. [Google Scholar] [CrossRef]
- Mercier, J.; Voutsadakis, I.A. The platelets-neutrophils to lymphocytes ratio: a new prognostic marker in metastatic colorectal cancer. J. Gastrointest. Oncol. 2018, 9, 478–486. [Google Scholar] [CrossRef]
- Takenaka, Y.; Oya, R.; Kitamiura, T.; Ashida, N.; Shimizu, K.; Takemura, K.; Yamamoto, Y.; Uno, A. Platelet count and platelet-lymphocyte ratio as prognostic markers for head and neck squamous cell carcinoma: Meta-analysis. Head Neck 2018, 40, 2714–2723. [Google Scholar] [CrossRef]
- Brockmann, M.A.; Giese, A.; Mueller, K.; Kaba, F.J.; Lohr, F.; Weiss, C.; Gottschalk, S.; Nolte, I.; Leppert, J.; Tuettenberg, J.; et al. Preoperative thrombocytosis predicts poor survival in patients with glioblastoma. Neuro-oncology. 2007, 9, 335–342. [Google Scholar] [CrossRef]
- Hefler-Frischmuth, K.; Grimm, C.; Gensthaler, L.; Reiser, E.; Schwameis, R.; Hefler, L.A. Prognostic value of preoperative hyponatremia and thrombocytosis in patients with epithelial ovarian cancer. Wien. Klin. Wochenschr. 2018, 130, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.N.; Afshar-Kharghan, V.; Sood, A.K. Platelet effects on ovarian cancer. Semin. Oncol. 2014, 41, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Ho-Tin-Noé, B.; Carbo, C.; Demers, M.; Cifuni, S.M.; Goerge, T.; Wagner, D.D. Innate immune cells induce hemorrhage in tumors during thrombocytopenia. Am. J. Pathol. 2009, 175, 1699–1708. [Google Scholar] [CrossRef] [PubMed]
- Kopp, H.-G.; Placke, T.; Salih, H.R. Platelet-derived transforming growth factor-beta down-regulates NKG2D thereby inhibiting natural killer cell antitumor reactivity. Cancer Res. 2009, 69, 7775–7783. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Hynes, R.O. The initial hours of metastasis: the importance of cooperative host-tumor cell interactions during hematogenous dissemination. Cancer Discov. 2012, 2, 1091–1099. [Google Scholar] [CrossRef]
- Kumm, E.J.; Pagel, O.; Gambaryan, S.; Walter, U.; Zahedi, R.P.; Smolenski, A.; Jurk, K. The Cell Cycle Checkpoint System MAST(L)-ENSA/ARPP19-PP2A is Targeted by cAMP/PKA and cGMP/PKG in Anucleate Human Platelets. Cells 2020, 9, 472. [Google Scholar] [CrossRef]
- Tuettenberg, A.; Huter, E.; Hubo, M.; Horn, J.; Knop, J.; Grimbacher, B.; Kroczek, R.A.; Stoll, S.; Jonuleit, H. The role of ICOS in directing T cell responses: ICOS-dependent induction of T cell anergy by tolerogenic dendritic cells. J. Immunol. 2009, 182, 3349–3356. [Google Scholar] [CrossRef]
- Kotecha, N.; Krutzik, P.O.; Irish, J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010, Chapter 10, Unit10.17. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
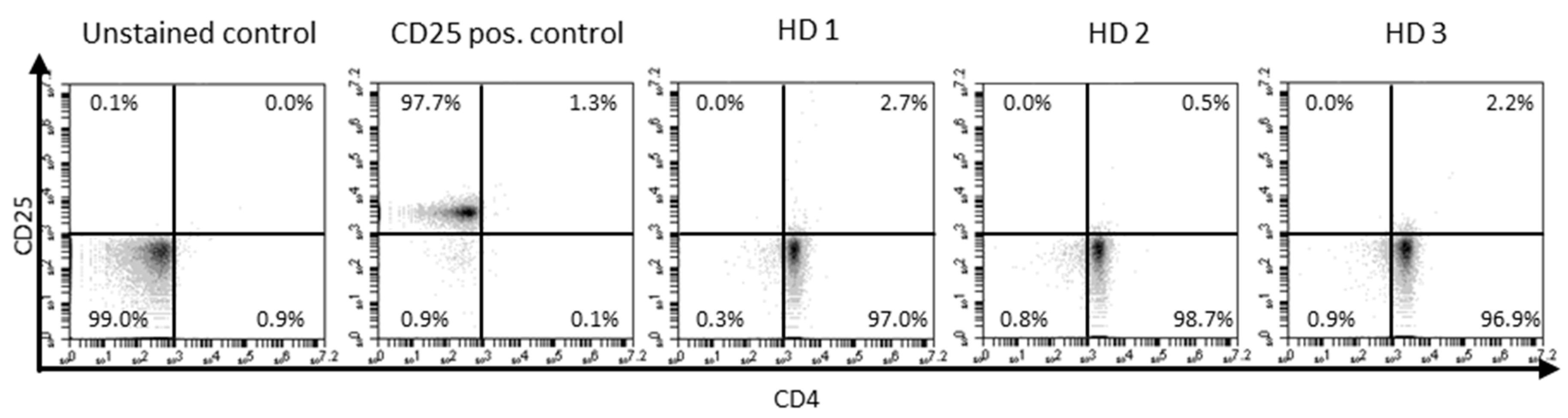{kind=link}
{kind=link}
| Cohort 1. Analysis of Platelet GARP and CD62p Expression Levels | n | % |
| Patients | 35 | 100 |
| Stage I | 18 | 51 |
| Stage IV (responders) | 17 (16) | 49 |
| Cohort 2. Retrospective Analysis of Platelet Count and PLR | n | % |
| Patients | 39 | 100 |
| Responder | 11 | 28 |
| Non-responder | 25 | 64 |
| Unknown outcome, lost to follow-up | 3 | 8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmer, N.; Krebs, F.K.; Zimmer, S.; Mitzel-Rink, H.; Kumm, E.J.; Jurk, K.; Grabbe, S.; Loquai, C.; Tuettenberg, A. Platelet-Derived GARP Induces Peripheral Regulatory T Cells—Potential Impact on T Cell Suppression in Patients with Melanoma-Associated Thrombocytosis. Cancers 2020, 12, 3653. https://doi.org/10.3390/cancers12123653
Zimmer N, Krebs FK, Zimmer S, Mitzel-Rink H, Kumm EJ, Jurk K, Grabbe S, Loquai C, Tuettenberg A. Platelet-Derived GARP Induces Peripheral Regulatory T Cells—Potential Impact on T Cell Suppression in Patients with Melanoma-Associated Thrombocytosis. Cancers. 2020; 12(12):3653. https://doi.org/10.3390/cancers12123653
Chicago/Turabian StyleZimmer, Niklas, Franziska K. Krebs, Sophia Zimmer, Heidrun Mitzel-Rink, Elena J. Kumm, Kerstin Jurk, Stephan Grabbe, Carmen Loquai, and Andrea Tuettenberg. 2020. "Platelet-Derived GARP Induces Peripheral Regulatory T Cells—Potential Impact on T Cell Suppression in Patients with Melanoma-Associated Thrombocytosis" Cancers 12, no. 12: 3653. https://doi.org/10.3390/cancers12123653
APA StyleZimmer, N., Krebs, F. K., Zimmer, S., Mitzel-Rink, H., Kumm, E. J., Jurk, K., Grabbe, S., Loquai, C., & Tuettenberg, A. (2020). Platelet-Derived GARP Induces Peripheral Regulatory T Cells—Potential Impact on T Cell Suppression in Patients with Melanoma-Associated Thrombocytosis. Cancers, 12(12), 3653. https://doi.org/10.3390/cancers12123653