Global Autozygosity Is Associated with Cancer Risk, Mutational Signature and Prognosis



 , ,
, , 
Abstract
Simple Summary
Abstract
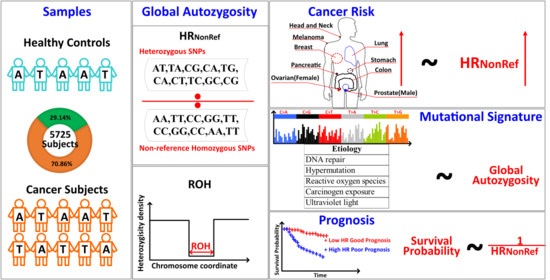
1. Introduction
2. Results
2.1. HRNonRef vs. HRMinor vs. ROH
2.2. Global Autozygosity and Cancer Risk
2.3. Mutational Signatures and Survival Analysis
3. Discussion
4. Materials and Methods
4.1. Genotyping Data Acquisition and Imputation
4.2. Somatic Mutation Data Acquisition and Mutational Signature Computation
4.3. HR and ROH Computation
4.4. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.K.; Esko, T.; Mattsson, H.; Eklund, N.; Gandin, I.; Nutile, T.; Jackson, A.U.; Schurmann, C.; Smith, A.V.; Zhang, W.; et al. Directional dominance on stature and cognition in diverse human populations. Nature 2015, 523, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Ghani, M.; Reitz, C.; Cheng, R.; Vardarajan, B.N.; Jun, G.; Sato, C.; Naj, A.; Rajbhandary, R.; Wang, L.S.; Valladares, O.; et al. Association of long runs of homozygosity with alzheimer disease among african american individuals. JAMA Neurol. 2015, 72, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. Plink: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Narasimhan, V.; Danecek, P.; Scally, A.; Xue, Y.; Tyler-Smith, C.; Durbin, R. Bcftools/roh: A hidden markov model approach for detecting autozygosity from next-generation sequencing data. Bioinformatics 2016, 32, 1749–1751. [Google Scholar] [CrossRef]
- Samuels, D.C.; Wang, J.; Ye, F.; He, J.; Levinson, R.T.; Sheng, Q.H.; Zhao, S.L.; Capra, J.A.; Shyr, Y.; Zheng, W.; et al. Heterozygosity ratio, a robust global genomic measure of autozygosity and its association with height and disease risk. Genetics 2016, 204, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Vine, A.E.; McQuillin, A.; Bass, N.J.; Pereira, A.; Kandaswamy, R.; Robinson, M.; Lawrence, J.; Anjorin, A.; Sklar, P.; Gurling, H.M.D.; et al. No evidence for excess runs of homozygosity in bipolar disorder. Psychiatr. Genet. 2009, 19, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Sims, R.; Dwyer, S.; Harold, D.; Gerrish, A.; Hollingworth, P.; Chapman, J.; Jones, N.; Abraham, R.; Ivanov, D.; Pahwa, J.S.; et al. No evidence that extended tracts of homozygosity are associated with alzheimer’s disease. Am. J. Med. Genet. B 2011, 156B, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Heron, E.A.; Cormican, P.; Donohoe, G.; O’Neill, F.A.; Kendler, K.S.; Riley, B.P.; Gill, M.; Corvin, A.P.; Morris, D.W.; Wellcome Trust Case, C. No evidence that runs of homozygosity are associated with schizophrenia in an irish genome-wide association dataset. Schizophr. Res. 2014, 154, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Ye, F.; Sheng, Q.H.; Clark, T.; Samuels, D.C. Three-stage quality control strategies for DNA re-sequencing data. Brief. Bioinform. 2014, 15, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Raskin, L.; Samuels, D.C.; Shyr, Y.; Guo, Y. Genome measures used for quality control are dependent on gene function and ancestry. Bioinformatics 2015, 31, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, H.; Chen, B.; Figlioli, G.; Elisei, R.; Romei, C.; Cipollini, M.; Cristaudo, A.; Bambi, F.; Hoffmann, P.; Herms, S.; et al. Runs of homozygosity and inbreeding in thyroid cancer. BMC Cancer 2016, 16, 227. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Sud, A.; Litchfield, K.; Levy, M.; Holroyd, A.; Broderick, P.; Kote-Jarai, Z.; Dunning, A.M.; Muir, K.; Peto, J.; et al. Runs of homozygosity and testicular cancer risk. Andrology 2019, 7, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, E.N.; Huang, M.N.; Mahto, U.; Barnes, M.; Stratton, M.R.; Rozen, S.G.; Alexandrov, L.B. Sigprofilermatrixgenerator: A tool for visualizing and exploring patterns of small mutational events. BMC Genom. 2019, 20, 685. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing mutational signatures in human cancer cell lines reveals episodic apobec mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef]
- Letouze, E.; Shinde, J.; Renault, V.; Couchy, G.; Blanc, J.F.; Tubacher, E.; Bayard, Q.; Bacq, D.; Meyer, V.; Semhoun, J.; et al. Mutational signatures reveal the dynamic interplay of risk factors and cellular processes during liver tumorigenesis. Nat. Commun. 2017, 8, 1315. [Google Scholar] [CrossRef]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kubler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476–1486. [Google Scholar] [CrossRef]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013, 9, e1003348. [Google Scholar] [CrossRef]
- Goode, L.L.; Chenevix-Trench, G.; Song, H.; Ramus, S.J.; Notaridou, M.; Lawrenson, K.; Widschwendter, M.; Vierkant, R.A.; Larson, M.C.; Kjaer, S.K.; et al. A genome-wide association study identifies susceptibility loci for ovarian cancer at 2q31 and 8q24. Nat. Genet. 2010, 42, 874–879. [Google Scholar] [CrossRef] [PubMed]
- Eeles, R.A.; Kote-Jarai, Z.; Al Olama, A.A.; Giles, G.G.; Guy, M.; Severi, G.; Muir, K.; Hopper, J.L.; Henderson, B.E.; Haiman, C.A.; et al. Identification of seven new prostate cancer susceptibility loci through a genome-wide association study. Nat. Genet. 2009, 41, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Carter, H.; Marty, R.; Hofree, M.; Gross, A.M.; Jensen, J.; Fisch, K.M.; Wu, X.Y.; DeBoever, C.; Van Nostrand, E.L.; Song, Y.; et al. Interaction landscape of inherited polymorphisms with somatic events in cancer. Cancer Discov. 2017, 7, 410–423. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Forer, L.; Schonherr, S.; Sidore, C.; Locke, A.E.; Kwong, A.; Vrieze, S.I.; Chew, E.Y.; Levy, S.; McGue, M.; et al. Next-generation genotype imputation service and methods. Nat. Genet. 2016, 48, 1284–1287. [Google Scholar] [CrossRef]
- Blokzijl, F.; Janssen, R.; van Boxtel, R.; Cuppen, E. Mutationalpatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 2018, 10, 33. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LogOR 1 (95% CI) | p | Cancer 2 | Race | Gender | Cases | Controls 3 |
|---|---|---|---|---|---|---|
| 1.8973 (1.6228–2.1920) | 4.89 × 10−28 | BRCA | Black | female | 163 | 342 |
| 1.1487 (0.8858–1.4336) | 5.28 × 10−12 | SKCM | Caucasian | male | 274 | 240 |
| 0.9630 (0.7327–1.2104) | 3.34 × 10−11 | OV | Caucasian | female | 369 | 263 |
| 1.4445 (1.0980–1.8303) | 8.83 × 10−11 | STAD | Caucasian | male | 153 | 240 |
| 1.1018 (0.8318–1.3940) | 1.17 × 10−10 | BRCA | Caucasian | female | 653 | 263 |
| 1.1224 (0.8393–1.4289) | 3.80 × 10−10 | HNSC | Caucasian | male | 300 | 240 |
| 1.2666 (0.9249–1.6396) | 5.68 × 10−9 | LUAD | Caucasian | female | 165 | 263 |
| 0.9607 (0.6844–1.2734) | 7.93 × 10−8 | HNSC | Caucasian | female | 120 | 263 |
| 0.9216 (0.6325–1.2450) | 7.44 × 10−7 | PRAD | Caucasian | male | 127 | 240 |
| 0.8458 (0.5814–1.1452) | 7.70 × 10−7 | COAD | Caucasian | male | 101 | 240 |
| 0.6438 (0.4208–0.8882) | 6.00 × 10−6 | SKCM | Caucasian | female | 168 | 263 |
| 0.4797 (0.2712–0.7050) | 2.71 × 10−4 | LUAD | Caucasian | male | 134 | 240 |
| 0.4689 (0.2634–0.6977) | 3.82 × 10−4 | LUSC | Caucasian | male | 166 | 240 |
| 0.4903 (0.2585–0.7537) | 1.12 × 10−3 | LIHC | Asian | male | 118 | 244 |
| LogOR 1 (95% CI) | p | Cancer 2 | Race | Gender | Cases | Controls 3 |
|---|---|---|---|---|---|---|
| 15.6383 (7.9048–23.5950) | 0.0010 | COAD | Caucasian | male | 101 | 240 |
| −0.2304 (−0.3658–−0.0969) | 0.0048 | OV | Caucasian | female | 369 | 263 |
| 5.3577 (2.2553–8.5218) | 0.0048 | PRAD | Caucasian | male | 127 | 240 |
| −0.2839 (−0.4606–−0.1132) | 0.0071 | LUAD | Caucasian | female | 165 | 263 |
| −0.1797 (−0.2989–−0.0623) | 0.0122 | BRCA | Caucasian | female | 653 | 263 |
| −0.2557 (−0.4285–−0.0882) | 0.0133 | SKCM | Caucasian | female | 168 | 263 |
| 8.1083 (2.2387–14.1885) | 0.0253 | HNSC | Caucasian | male | 300 | 240 |
| 0.2548 (0.0708–0.4519) | 0.0269 | LIHC | Asian | male | 118 | 244 |
| 7.1487 (1.0826–13.0103) | 0.0422 | LUSC | Caucasian | male | 166 | 240 |
| 5.0918 (0.6647–9.4886) | 0.0483 | STAD | Caucasian | male | 153 | 240 |
| −0.2022 (−0.3918–−0.0282) | 0.0670 | BRCA | Black | female | 163 | 342 |
| −0.0873 (−0.2877–0.0971) | 0.4534 | HNSC | Caucasian | female | 120 | 263 |
| 0.7471 (−0.0236–2.9058) | 0.5483 | SKCM | Caucasian | male | 274 | 240 |
| 0.0625 (−0.1152–0.2397) | 0.5614 | LUAD | Caucasian | male | 134 | 240 |
| Effect | Stderr 1 | Adusted p | Signature | Case | Predictor | Gender | Race | Cancer 2 |
|---|---|---|---|---|---|---|---|---|
| 0.2638 | 0.0387 | 4.62 × 10−8 | SBS44 | 116 | ROH | female | Caucasian | HNSC |
| 0.3571 | 0.0679 | 3.02 × 10−5 | SBS36 | 125 | ROH | male | Caucasian | PRAD |
| 0.8407 | 0.1970 | 0.0013 | SBS9 | 277 | HRNonRef | female | Caucasian | OV |
| 1.3245 | 0.3223 | 0.0013 | SBS18 | 277 | HRNonRef | female | Caucasian | OV |
| 1.0001 | 0.2359 | 0.0018 | SBS42 | 164 | ROH | male | Caucasian | LUSC |
| 0.6597 | 0.1891 | 0.0070 | SBS5 | 277 | HRNonRef | female | Caucasian | OV |
| 0.8939 | 0.2564 | 0.0070 | SBS7c | 277 | HRNonRef | female | Caucasian | OV |
| 2.9086 | 0.7533 | 0.0172 | SBS7b | 132 | ROH | male | Caucasian | LUAD |
| −0.0885 | 0.0234 | 0.0206 | SBS44 | 166 | HRNonRef | female | Caucasian | SKCM |
| 0.9714 | 0.3227 | 0.0280 | SBS22 | 277 | HRNonRef | female | Caucasian | OV |
| 0.2439 | 0.0718 | 0.0453 | SBS36 | 125 | HRNonRef | male | Caucasian | PRAD |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, L.; Guo, F.; Tang, J.; Leng, S.; Ness, S.; Ye, F.; Kang, H.; Samuels, D.C.; Guo, Y. Global Autozygosity Is Associated with Cancer Risk, Mutational Signature and Prognosis. Cancers 2020, 12, 3646. https://doi.org/10.3390/cancers12123646
Jiang L, Guo F, Tang J, Leng S, Ness S, Ye F, Kang H, Samuels DC, Guo Y. Global Autozygosity Is Associated with Cancer Risk, Mutational Signature and Prognosis. Cancers. 2020; 12(12):3646. https://doi.org/10.3390/cancers12123646
Chicago/Turabian StyleJiang, Limin, Fei Guo, Jijun Tang, Shuguan Leng, Scott Ness, Fei Ye, Huining Kang, David C. Samuels, and Yan Guo. 2020. "Global Autozygosity Is Associated with Cancer Risk, Mutational Signature and Prognosis" Cancers 12, no. 12: 3646. https://doi.org/10.3390/cancers12123646
APA StyleJiang, L., Guo, F., Tang, J., Leng, S., Ness, S., Ye, F., Kang, H., Samuels, D. C., & Guo, Y. (2020). Global Autozygosity Is Associated with Cancer Risk, Mutational Signature and Prognosis. Cancers, 12(12), 3646. https://doi.org/10.3390/cancers12123646