Pharmacogenetic Study of Trabectedin-Induced Severe Hepatotoxicity in Patients with Advanced Soft Tissue Sarcoma

 ,
,  ,
,  ,
,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Patients’ Characteristics and Toxicity Data
2.2. Evaluation of the NGS Data Quality and Building of the Genetic Database
2.3. Pharmacogenetic Association Studies
2.3.1. Primary Analysis Based on Literature-Selected SNPs
2.3.2. Exploratory Analysis
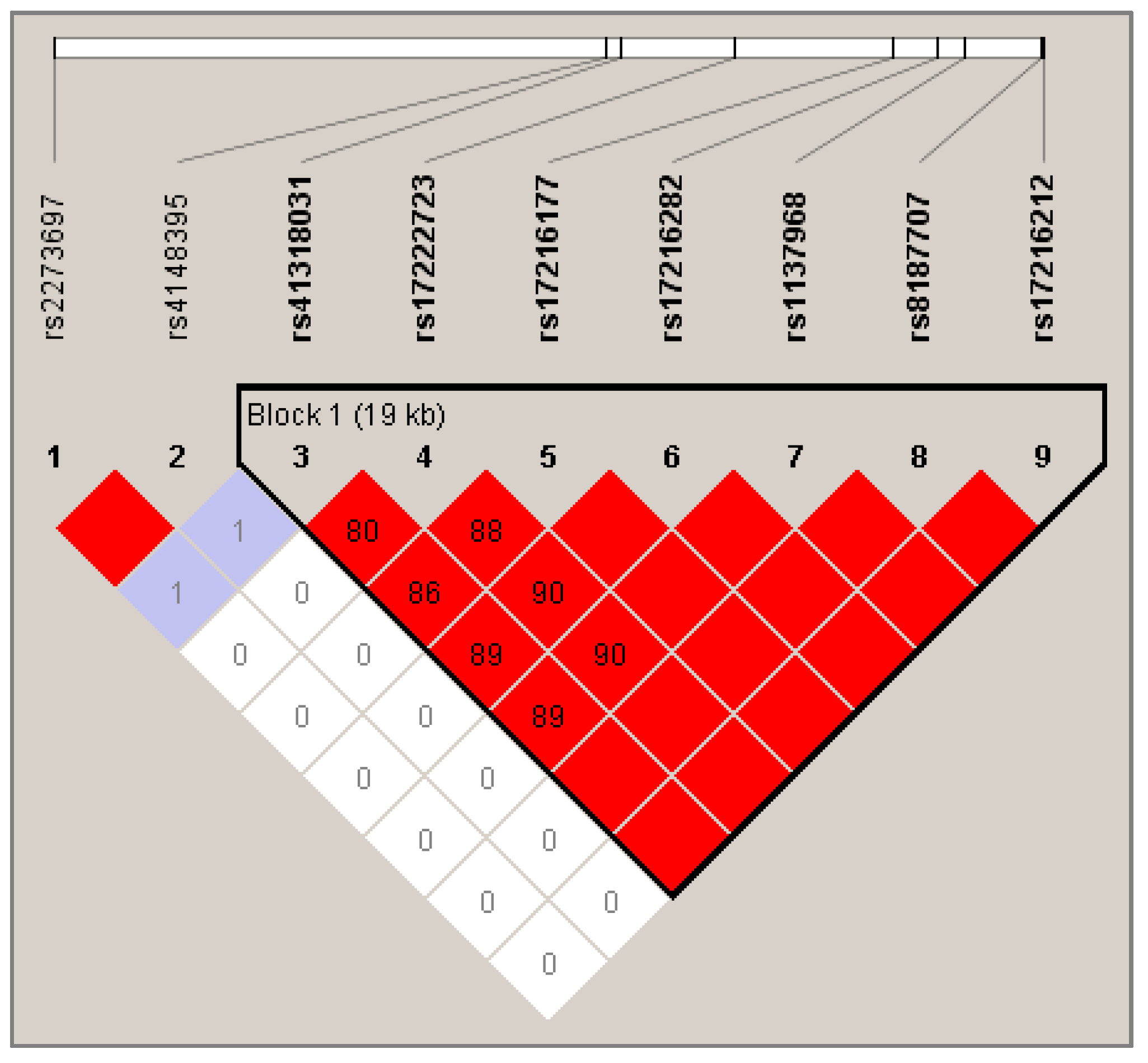
2.3.3. Haplotype Analysis
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. SNP Selection and Genotyping
4.2.1. SNP Selection
4.2.2. Customized Ion Ampliseq Panel for Gene Targeting
4.2.3. Libraries Preparation and Amplification for Next-Generation Sequencing
4.2.4. PGM Sequencing
4.3. Assessment of Liver Toxicity
4.4. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pommier, Y.; Kohlhagen, G.; Bailly, C.; Waring, M.; Mazumder, A.; Kohn, K.W. DNA Sequence- and Structure-Selective Alkylation of Guanine N2 in the DNA Minor Groove by Ecteinascidin 743, a Potent Antitumor Compound from the Caribbean Tunicate Ecteinascidia turbinata. Biochemistry 1996, 35, 13303–13309. [Google Scholar] [CrossRef] [PubMed]
- Herrero, A.B.; Martín-Castellanos, C.; Marco, E.; Gago, F.; Moreno, S. Cross-Talk between Nucleotide Excision and Homologous Recombination DNA Repair Pathways in the Mechanism of Action of Antitumor Trabectedin. Cancer Res. 2006, 66, 8155–8162. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Galmarini, C.M.; D’Incalci, M. Unique features of trabectedin mechanism of action. Cancer Chemother. Pharm. 2016, 77, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Izbicka, E.; Lawrence, R.; Raymond, E.; Eckhardt, G.; Faircloth, G.; Jimeno, J.; Clark, G.; Von Hoff, D.D. In vitro antitumor activity of the novel marine agent, ecteinascidin-743 (ET-743, NSC-648766) against human tumors explanted from patients. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1998, 9, 981–987. [Google Scholar] [CrossRef] [PubMed]
- EMEA Yondelis® INN-trabectedin—Summary of Product Characteristics. Available online: https://www.ema.europa.eu/en/documents/product-information/yondelis-epar-product-information_en.pdf (accessed on 21 December 2019).
- Le Cesne, A. Making the Best of Available Options for Optimal Sarcoma Treatment. Oncology 2018, 95, 11–20. [Google Scholar] [CrossRef]
- Caruso, C.; Garofalo, C. Pharmacogenomics Biomarkers of Soft Tissue Sarcoma Therapies. Front. Oncol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Laurand, A.; Laroche, A.; Casali, P.; Sanfilippo, R.; Cesne, A.L.; Judson, I.; Blay, J.-Y.; Ray-Coquard, I.; Bui, B.; et al. ERCC5/XPG, ERCC1, and BRCA1 gene status and clinical benefit of trabectedin in patients with soft tissue sarcoma. Cancer 2011, 117, 3445–3456. [Google Scholar] [CrossRef]
- Schöffski, P.; Taron, M.; Jimeno, J.; Grosso, F.; Sanfilipio, R.; Casali, P.G.; Le Cesne, A.; Jones, R.L.; Blay, J.-Y.; Poveda, A.; et al. Predictive impact of DNA repair functionality on clinical outcome of advanced sarcoma patients treated with trabectedin: A retrospective multicentric study. Eur. J. Cancer Oxf. Engl. 1990 2011, 47, 1006–1012. [Google Scholar] [CrossRef]
- Miolo, G.; Viel, A.; Canzonieri, V.; Baresic, T.; Buonadonna, A.; Santeufemia, D.A.; Lara, D.P.; Corona, G. Association of the germline BRCA2 missense variation Glu2663Lys with high sensitivity to trabectedin-based treatment in soft tissue sarcoma. Cancer Biol. 2016, 17, 1017–1021. [Google Scholar] [CrossRef][Green Version]
- Beumer, J.H.; Rademaker-Lakhai, J.M.; Rosing, H.; Hillebrand, M.J.X.; Bosch, T.M.; Lopez-Lazaro, L.; Schellens, J.H.M.; Beijnen, J.H. Metabolism of trabectedin (ET-743, YondelisTM) in patients with advanced cancer. Cancer Chemother. Pharm. 2007, 59, 825–837. [Google Scholar] [CrossRef]
- Sparidans, R.W.; Rosing, H.; Hillebrand, M.J.; López-Lázaro, L.; Jimeno, J.M.; Manzanares, I.; van Kesteren, C.; Cvitkovic, E.; van Oosterom, A.T.; Schellens, J.H.; et al. Search for metabolites of ecteinascidin 743, a novel, marine-derived, anti-cancer agent, in man. Anticancer Drugs 2001, 12, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Vermeir, M.; Hemeryck, A.; Cuyckens, F.; Francesch, A.; Bockx, M.; Van Houdt, J.; Steemans, K.; Mannens, G.; Avilés, P.; De Coster, R. In vitro studies on the metabolism of trabectedin (YONDELIS®) in monkey and man, including human CYP reaction phenotyping. Biochem. Pharm. 2009, 77, 1642–1654. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.H.; Rademaker-Lakhai, J.M.; Rosing, H.; Lopez-Lazaro, L.; Beijnen, J.H.; Schellens, J.H.M. Trabectedin (YondelisTM, formerly ET-743), a mass balance study in patients with advanced cancer. Investig. New Drugs 2005, 23, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Chawla, S.P.; von Mehren, M.; Ritch, P.; Baker, L.H.; Blay, J.Y.; Hande, K.R.; Keohan, M.L.; Samuels, B.L.; Schuetze, S.; et al. Efficacy and Safety of Trabectedin in Patients With Advanced or Metastatic Liposarcoma or Leiomyosarcoma After Failure of Prior Anthracyclines and Ifosfamide: Results of a Randomized Phase II Study of Two Different Schedules. J. Clin. Oncol. 2009, 27, 4188–4196. [Google Scholar] [CrossRef]
- Grosso, F.; D’Incalci, M.; Cartoafa, M.; Nieto, A.; Fernández-Teruel, C.; Alfaro, V.; Lardelli, P.; Roy, E.; Gómez, J.; Kahatt, C.; et al. A comprehensive safety analysis confirms rhabdomyolysis as an uncommon adverse reaction in patients treated with trabectedin. Cancer Chemother. Pharm. 2012, 69, 1557–1565. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taamma, A.; Misset, J.L.; Riofrio, M.; Guzman, C.; Brain, E.; Lopez Lazaro, L.; Rosing, H.; Jimeno, J.M.; Cvitkovic, E. Phase I and Pharmacokinetic Study of Ecteinascidin-743, a New Marine Compound, Administered as a 24-hour Continuous Infusion in Patients With Solid Tumors. J. Clin. Oncol. 2001, 19, 1256–1265. [Google Scholar] [CrossRef]
- Villalona-Calero, M.A.; Eckhardt, S.G.; Weiss, G.; Hidalgo, M.; Beijnen, J.H.; Kesteren C, v.a.n.; Rosing, H.; Campbell, E.; Kraynak, M.; Lopez-Lazaro, L.; et al. A Phase I and Pharmacokinetic Study of Ecteinascidin-743 on a Daily × 5 Schedule in Patients with Solid Malignancies. Clin. Cancer Res. 2002, 8, 75–85. [Google Scholar]
- Le Cesne, A.; Yovine, A.; Blay, J.-Y.; Delaloge, S.; Maki, R.G.; Misset, J.-L.; Frontelo, P.; Nieto, A.; Jiao, J.J.; Demetri, G.D. A retrospective pooled analysis of trabectedin safety in 1,132 patients with solid tumors treated in phase II clinical trials. Investig. New Drugs 2012, 30, 1193–1202. [Google Scholar] [CrossRef]
- D’Incalci, M.; Jimeno, J. Preclinical and clinical results with the natural marine product ET-743. Expert Opin. Investig. Drugs 2003, 12, 1843–1853. [Google Scholar] [CrossRef]
- Donald, S.; Verschoyle, R.D.; Edwards, R.; Judah, D.J.; Davies, R.; Riley, J.; Dinsdale, D.; Lazaro, L.L.; Smith, A.G.; Gant, T.W.; et al. Hepatobiliary Damage and Changes in Hepatic Gene Expression Caused by the Antitumor Drug Ecteinascidin-743 (ET-743) in the Female Rat. Cancer Res. 2002, 62, 4256–4262. [Google Scholar]
- Reid, J.M.; Kuffel, M.J.; Ruben, S.L.; Morales, J.J.; Rinehart, K.L.; Squillace, D.P.; Ames, M.M. Rat and Human Liver Cytochrome P-450 Isoform Metabolism of Ecteinascidin 743 Does Not Predict Gender-dependent Toxicity in Humans. Clin. Cancer Res. 2002, 8, 2952–2962. [Google Scholar] [PubMed]
- Brandon, E.F.A.; Sparidans, R.W.; Guijt, K.-J.; Löwenthal, S.; Meijerman, I.; Beijnen, J.H.; Schellens, J.H.M. In vitro characterization of the human biotransformation and CYP reaction phenotype of ET-743 (Yondelis®, Trabectedin), a novel marine anti-cancer drug. Investig. New Drugs 2006, 24, 3–14. [Google Scholar] [CrossRef] [PubMed]
- van Waterschoot, R.A.B.; Eman, R.M.; Wagenaar, E.; van der Kruijssen, C.M.M.; Rosing, H.; Beijnen, J.H.; Schinkel, A.H. ABCC2, ABCC3, and ABCB1, but not CYP3A, Protect against Trabectedin-Mediated Hepatotoxicity. Clin. Cancer Res. 2009, 15, 7616–7623. [Google Scholar] [CrossRef] [PubMed]
- Beumer, J.H.; Franke, N.E.; Tolboom, R.; Buckle, T.; Rosing, H.; Lopez-Lazaro, L.; Schellens, J.H.M.; Beijnen, J.H.; van Tellingen, O. Disposition and toxicity of trabectedin (ET-743) in wild-type and mdr1 gene (P-gp) knock-out mice. Investig. New Drugs 2010, 28, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Brandon, E.F.A.; Meijerman, I.; Klijn, J.S.; den Arend, D.; Sparidans, R.W.; Lázaro, L.L.; Beijnen, J.H.; Schellens, J.H.M. In-vitro cytotoxicity of ET-743 (Trabectedin, Yondelis), a marine anti-cancer drug, in the Hep G2 cell line: Influence of cytochrome P450 and phase II inhibition, and cytochrome P450 induction. Anticancer. Drugs 2005, 16, 935–943. [Google Scholar] [CrossRef]
- Donald, S.; Verschoyle, R.D.; Greaves, P.; Gant, T.W.; Colombo, T.; Zaffaroni, M.; Frapolli, R.; Zucchetti, M.; D’Incalci, M.; Meco, D.; et al. Complete Protection by High-Dose Dexamethasone against the Hepatotoxicity of the Novel Antitumor Drug Yondelis (ET-743) in the Rat. Cancer Res. 2003, 63, 5902–5908. [Google Scholar]
- Lee, J.K.; Leslie, E.M.; Zamek-Gliszczynski, M.J.; Brouwer, K.L.R. Modulation of trabectedin (ET-743) hepatobiliary disposition by multidrug resistance-associated proteins (Mrps) may prevent hepatotoxicity. Toxicol. Appl. Pharm. 2008, 228, 17–23. [Google Scholar] [CrossRef]
- Laurenty, A.-P.; Thomas, F.; Chatelut, E.; Bétrian, S.; Guellec, C.L.; Hennebelle, I.; Guellec, S.L.; Chevreau, C. Irreversible hepatotoxicity after administration of trabectedin to a pleiomorphic sarcoma patient with a rare ABCC2 polymorphism: A case report. Pharmacogenomics 2013, 14, 1389–1396. [Google Scholar] [CrossRef]
- Rathinamanickam, H. Trabectedin Induced Irreversible Hepatotoxicity: 1863. Am. J. Gastroenterol. 2016, 111, S893. [Google Scholar] [CrossRef]
- LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- EMEA Good Pharmacogenomic Practice. Available online: https://www.ema.europa.eu/en/good-pharmacogenomic-practice (accessed on 15 October 2020).
- Wen, X.; Joy, M.S.; Aleksunes, L.M. In Vitro Transport Activity and Trafficking of MRP2/ABCC2 Polymorphic Variants. Pharm. Res. 2017, 34, 1637–1647. [Google Scholar] [CrossRef]
- Meier, Y.; Pauli-Magnus, C.; Zanger, U.M.; Klein, K.; Schaeffeler, E.; Nussler, A.K.; Nussler, N.; Eichelbaum, M.; Meier, P.J.; Stieger, B. Interindividual variability of canalicular ATP-binding-cassette (ABC)–transporter expression in human liver. Hepatology 2006, 44, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Jedlitschky, G.; Hoffmann, U.; Kroemer, H.K. Structure and function of the MRP2 (ABCC2) protein and its role in drug disposition. Expert Opin. Drug Metab. Toxicol. 2006, 2, 351–366. [Google Scholar] [CrossRef] [PubMed]
- Boige, V.; Buffet, C. Diagnostic strategy in case of isolated increase in serum gamma-glutamyltranspeptidase. Gastroenterol. Clin. Biol. 1997, 21, 8. [Google Scholar]
- Megaraj, V.; Zhao, T.; Paumi, C.M.; Gerk, P.M.; Kim, R.; Vore, M. Functional Analysis of Non-synonymous Single Nucleotide Polymorphisms of Multidrug Resistance Protein 2 (MRP2; ABCC2). Pharm. Genom. 2011, 21, 506–515. [Google Scholar] [CrossRef]
- Deo, A.K.; Prasad, B.; Balogh, L.; Lai, Y.; Unadkat, J.D. Interindividual Variability in Hepatic Expression of the Multidrug Resistance-Associated Protein 2 (MRP2/ABCC2): Quantification by Liquid Chromatography/Tandem Mass Spectrometry. Drug Metab. Dispos. 2012, 40, 852–855. [Google Scholar] [CrossRef]
- Kim, W.-J.; Lee, J.H.; Yi, J.; Cho, Y.-J.; Heo, K.; Lee, S.H.; Kim, S.W.; Kim, M.-K.; Kim, K.H.; In Lee, B.; et al. A nonsynonymous variation in MRP2/ABCC2 is associated with neurological adverse drug reactions of carbamazepine in patients with epilepsy. Pharm. Genom. 2010, 20, 249–256. [Google Scholar] [CrossRef]
- Wolking, S.; Schaeffeler, E.; Lerche, H.; Schwab, M.; Nies, A.T. Impact of Genetic Polymorphisms of ABCB1 (MDR1, P-Glycoprotein) on Drug Disposition and Potential Clinical Implications: Update of the Literature. Clin. Pharm. 2015, 54, 709–735. [Google Scholar] [CrossRef]
- Beuselinck, B.; Lambrechts, D.; Van Brussel, T.; Wolter, P.; Cardinaels, N.; Joniau, S.; Lerut, E.; Karadimou, A.; Couchy, G.; Sebe, P.; et al. Efflux pump ABCB1 single nucleotide polymorphisms and dose reductions in patients with metastatic renal cell carcinoma treated with sunitinib. Acta Oncol. 2014, 53, 1413–1422. [Google Scholar] [CrossRef]
- Salama, N.N.; Yang, Z.; Bui, T.o.t.; Ho, R.J.Y. MDR1 haplotypes significantly minimize intracellular uptake and transcellular P-gp substrate transport in recombinant LLC-PK1 cells. J. Pharm. Sci. 2006, 95, 2293–2308. [Google Scholar] [CrossRef]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharm. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef]
- Bai, H.; Wu, T.; Jiao, L.; Wu, Q.; Zhao, Z.; Song, J.; Liu, T.; Lv, Y.; Lu, X.; Ying, B. Association of ABCC Gene Polymorphism With Susceptibility to Antituberculosis Drug–Induced Hepatotoxicity in Western Han Patients With Tuberculosis. J. Clin. Pharm. 2020, 60, 361–368. [Google Scholar] [CrossRef]
- Lopez-Lopez, E.; Ballesteros, J.; Piñan, M.; Toledo, J.S.; de Andoin, N.G.; de Garcia-Miguel, P.; Navajas, A.; Garcia-Orad, A. Polymorphisms in the methotrexate transport pathway: A new tool for MTX plasma level prediction in pediatric acute lymphoblastic leukemia. Pharm. Genom. 2013, 23, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Poonkuzhali, B.; Lamba, J.; Strom, S.; Sparreboom, A.; Thummel, K.; Watkins, P.; Schuetz, E. Association of Breast Cancer Resistance Protein/ABCG2 Phenotypes and Novel Promoter and Intron 1 Single Nucleotide Polymorphisms. Drug Metab. Dispos. 2008, 36, 780–795. [Google Scholar] [CrossRef] [PubMed]
- De Mattia, E.; Toffoli, G.; Polesel, J.; D’Andrea, M.; Corona, G.; Zagonel, V.; Buonadonna, A.; Dreussi, E.; Cecchin, E. Pharmacogenetics of ABC and SLC transporters in metastatic colorectal cancer patients receiving first-line FOLFIRI treatment. Pharm. Genom. 2013, 23, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef]
- Birdwell, K.; Decker, B.; Barbarino, J.; Peterson, J.; Stein, C.; Sadee, W.; Wang, D.; Vinks, A.; He, Y.; Swen, J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin. Pharm. 2015, 98, 19–24. [Google Scholar] [CrossRef]
- Court, M.H.; Peter, I.; Hazarika, S.; Vasiadi, M.; Greenblatt, D.J.; Lee, W.M. The Acute Liver Failure Study Group Candidate Gene Polymorphisms in Patients with Acetaminophen-Induced Acute Liver Failure. Drug Metab. Dispos. 2014, 42, 28–32. [Google Scholar] [CrossRef]
- Povo-Retana, A.; Mojena, M.; Stremtan, A.B.; Fernández-García, V.B.; Gómez-Sáez, A.; Nuevo-Tapioles, C.; Molina-Guijarro, J.M.; Avendaño-Ortiz, J.; Cuezva, J.M.; López-Collazo, E.; et al. Specific Effects of Trabectedin and Lurbinectedin on Human Macrophage Function and Fate—Novel Insights. Cancers 2020, 12, 3060. [Google Scholar] [CrossRef]
- Banerjee, P.; Zhang, R.; Ivan, C.; Galletti, G.; Clise-Dwyer, K.; Barbaglio, F.; Scarfò, L.; Aracil, M.; Klein, C.; Wierda, W.; et al. Trabectedin Reveals a Strategy of Immunomodulation in Chronic Lymphocytic Leukemia. Cancer Immunol. Res. 2019, 7, 2036–2051. [Google Scholar] [CrossRef]
- Mallick, P.; Taneja, G.; Moorthy, B.; Ghose, R. Regulation of drug-metabolizing enzymes in infectious and inflammatory disease: Implications for biologics–small molecule drug interactions. Expert Opin. Drug Metab. Toxicol. 2017, 13, 605–616. [Google Scholar] [CrossRef]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and Safety of Trabectedin or Dacarbazine for Metastatic Liposarcoma or Leiomyosarcoma After Failure of Conventional Chemotherapy: Results of a Phase III Randomized Multicenter Clinical Trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Yondelis. Avis de la Commission de Transparence–Haute Autorité de Santé. Available online: https://www.has-sante.fr/upload/docs/evamed/CT-16827_YONDELIS_sarcomes_PIC_REEV_Avis2_CT16827.pdf (accessed on 16 June 2020).
- Casado, A.; Callata, H.R.; Manzano, A.; Marquina, G.; Alonso, T.; Gajate, P.; Sotelo, M.; Cabezas, S.; Fernández, C.; Díaz-Rubio, E. Trabectedin for reversing platinum resistance and resensitization to platinum in patients with recurrent ovarian cancer. Future Oncol. Lond. Engl. 2019, 15, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Monk, B.J.; Herzog, T.J.; Wang, G.; Triantos, S.; Maul, S.; Knoblauch, R.; McGowan, T.; Shalaby, W.S.W.; Coleman, R.L. A phase 3 randomized, open-label, multicenter trial for safety and efficacy of combined trabectedin and pegylated liposomal doxorubicin therapy for recurrent ovarian cancer. Gynecol. Oncol. 2020, 156, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Le Cesne, A.; Blay, J.-Y.; Cupissol, D.; Italiano, A.; Delcambre, C.; Penel, N.; Isambert, N.; Chevreau, C.; Bompas, E.; Bertucci, F.; et al. Results of a prospective randomized phase III T-SAR trial comparing trabectedin (T) vs. best supportive care (BSC) in patients with pretreated advanced soft tissue sarcoma (ASTS): A French Sarcoma Group (FSG) trial. J. Clin. Oncol. 2018, 36, 11508. [Google Scholar] [CrossRef]
- Gonzalez, J.R.; Armengol, L.; Sole, X.; Guino, E.; Mercader, J.M.; Estivill, X.; Moreno, V. SNPassoc: An R package to perform whole genome association studies. Bioinformatics 2007, 23, 654–655. [Google Scholar] [CrossRef]
- Wigginton, J.E.; Cutler, D.J.; Abecasis, G.R. A note on exact tests of Hardy-Weinberg equilibrium. Am. J. Hum. Genet. 2005, 76, 887–893. [Google Scholar] [CrossRef]
- Minelli, C.; Thompson, J.R.; Abrams, K.R.; Thakkinstian, A.; Attia, J. The choice of a genetic model in the meta-analysis of molecular association studies. Int. J. Epidemiol. 2005, 34, 1319–1328. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Gabriel, S.B. The Structure of Haplotype Blocks in the Human Genome. Science 2002, 296, 2225–2229. [Google Scholar] [CrossRef]


{kind=link}
| Demographical Characteristics at Baseline | n (%) or (Range) | |
|---|---|---|
| Total patients included in the pharmacogenetic study | 63 | |
| Gender | Male | 31 (49.2%) |
| Female | 32 (50.8%) | |
| Age | Median (years) | 65 (21.5–82.3) |
| Initial arm before crossover | Trabectedin | 31 (49%) |
| Best supportive care | 32 (51%) | |
| Baseline ECOG performance status | 0 | 30 (47.6%) |
| 1 | 32 (50.8%) | |
| 2 | 1 (1.6%) | |
| Tumor characteristics at baseline | ||
| Histological subtype of soft tissue sarcoma | STS-L: liposarcoma or leiomyosarcoma | 35 (55.6%) |
| STS-non L: other subtype | 28 (44.4%) | |
| Hepatic metastasis at baseline | Yes | 13 (20.6%) |
| No | 50 (79.4%) | |
| Prior anticancer treatment before inclusion | ||
| Chemotherapy | Yes | 63 (100%) |
| No | 0 (0%) | |
| Neoadjuvant and/or adjuvant chemotherapy | Yes | 21 (33.3%) |
| No | 42 (66.7%) | |
| Lines of prior chemotherapy in advanced setting | Median | 1 (0; 3) |
| 0 | 7 (11.1%) | |
| 1 | 29 (46%) | |
| 2 | 22 (34.9%) | |
| 3 | 5 (7.9%) | |
| Common prior chemotherapies in advanced setting | Anthracyclines | 62 (98.4%) |
| Ifosfamide | 34 (54%) | |
| Gemcitabine | 17 (27%) | |
| Dacarbazine | 15 (23.8%) | |
| Pazopanib | 10 (15.9%) | |
| Cyclophosphamide | 5 (7.9%) | |
| Other | 8 (12.7%) | |
| Trabectedin treatment after inclusion | ||
| Number of cycles administered per patient | Median | 7 (1–33) |
| Total dose administered | Mean (mg) | 2.25 (1.35–3.24) |
| Treatment modification because of hepatotoxicity | ||
| Administration delay | Yes | 6 (10%) |
| No | 57 (90%) | |
| Dose reduction | Yes | 17 (27%) |
| No | 46 (73%) | |
| Stop of treatment | Yes | 2 (3%) |
| No | 61 (97%) | |
| Hepatic side effects recorded during the two first courses of trabectedin | ||
| Cytolysis | Grades 0, 1, 2 | 36 (57%) |
| Grades 3, 4 | 27 (43%) | |
| Cholestasis | Grades 0, 1, 2 | 60 (95%) |
| Grades 3, 4 | 3 (5%) | |
| Isolated elevation of γ-GT | Grades 0, 1, 2 | 56 (89%) |
| Grades 3, 4 | 7 (11%) | |
| Overall hepatotoxicity | Grades 0, 1, 2 | 27 (43%) |
| Grades 3, 4 | 36 (57%) | |
| Gene | SNP | Genotype (n) | Severe HAE | % Severe HAE per Genotype | Genotype Comparison 1 | p-Value | OR | 95% CI | FDR |
|---|---|---|---|---|---|---|---|---|---|
| ABCB1 | rs1128503 c.1236C > T | CC (19) CT (26) TT (14) | Cytolysis | 68.4/34.6/35.7 | CT-TT vs. CC | 0.015 | 0.25 | 0.08–0.80 | 0.147 |
| ABCB1 | rs2032582 c.2677G > T,A | GG (18) GT/A (34) TT (11) | Overall hepatotoxicity | 66.7/61.7/27.3 | TT vs. GG-GT | 0.027 | 0.22 | 0.05–0.91 | 0.705 |
| ABCC2 | rs17222723 c.3563T > A | TT (51) TA (9) AA (1) | Cytolysis | 50.9/11.1/0 | TA-AA vs. TT | 0.010 | 0.11 | 0.01–0.91 | 0.147 |
| ABCC2 | rs2273697 c.1249G > A | GG (40) GA (21) AA (1) | Cytolysis | 32.5/61.9/100 | GA-AA vs. GG | 0.018 | 3.63 | 1.22–10.83 | 0.147 |
| ABCC2 | rs2273697 c.1249G > A | GG (40) GA (21) AA (1) | Isolated elevation of γ-GT | 17.5/0/0 | GA-AA vs. GG | 0.044 | N/C | / | 0.811 |
| ABCC2 | rs8187707 c.4488C > T | CC (50) CT (8) TT (0) | Cytolysis | 48/12.5/0 | CT vs. CC | 0.045 | 0.15 | 0.02–1.35 | 0.371 |
| ABCC4 | rs9516519 c.*3261A > G | TT (42) TG (16) GG (1) | Overall hepatotoxicity | 69/43.8/0 | TG-GG vs. TT | 0.048 | 0.31 | 0.10–1.01 | 0.444 |
| ABCG2 | rs7699188 c.-15994C > T | CC (31) CT (19) TT (2) | Cytolysis | 32.3/63.2/50 | CT-TT vs. CC | 0.034 | 3.41 | 1.07–10.87 | 0.212 |
| CYP3A5 | rs776746 c.6986A > G | AA (2) AG (14) GG (47) | Overall hepatotoxicity | 0/85.7/51.1 | AA vs. GA vs. GG | 0.012 | 5.75 | 1.16–28.55 | 0.403 |
| Gene | SNP | Genotype (n) | Severe HAE | % Severe HAE per Genotype | Genotype Comparison 1 | p-Value | OR | 95% CI | FDR |
|---|---|---|---|---|---|---|---|---|---|
| ABCC2 | rs17216282 c.4146 + 11G > C | GG (50) GC (10) CC (0) | Cytolysis | 52/10/0 | GC vs. GG | 0.009 | 0.1 | 0.01–0.87 | 0.698 |
| ABCC3 | rs2072365 c.2714 + 29C > T | CC (28) CT (29) TT (6) | Overall hepatotoxicity | 39.2/79.3/33.3 | CT vs. CC vs. TT | 0.003 | 5.92 | 1.83–19.20 | 0.242 |
| ABCC3 | rs4148415 c.2600-123C > T | CC (27) CT (27) TT (5) | Overall hepatotoxicity | 40.7/85.2/40 | CT vs. CC vs. TT | 0.001 | 8.36 | 2.26–31 | 0.242 |
| ABCC4 | rs11568647 c.2213 + 108delC | CC (53) Cdel (6) DelDel (0) | Overall hepatotoxicity | 66/0/0 | Cdel vs. CC | 0.003 | N/C | - | 0.242 |
| ABCC4 | rs1751005 c.1727 + 91G > A | GG (43) GA (14) AA (2) | Cytolysis | 55.8/14.3/50 | GA-AA vs. GG | 0.009 | 0.18 | 0.05–0.74 | 0.385 |
| Overall hepatotoxicity | 72/28.6/50 | GA-AA vs. GG | 0.004 | 0.18 | 0.05–0.61 | 0.240 | |||
| ABCC4 | rs4148553 c.*694C > T | CC (23) CT (31) TT (6) | Cytolysis | 34.8/41.9/100 | TT vs. CC-CT | 0.006 | N/C | - | 0.644 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maillard, M.; Chevreau, C.; Le Louedec, F.; Cassou, M.; Delmas, C.; Gourdain, L.; Blay, J.-Y.; Cupissol, D.; Bompas, E.; Italiano, A.; et al. Pharmacogenetic Study of Trabectedin-Induced Severe Hepatotoxicity in Patients with Advanced Soft Tissue Sarcoma. Cancers 2020, 12, 3647. https://doi.org/10.3390/cancers12123647
Maillard M, Chevreau C, Le Louedec F, Cassou M, Delmas C, Gourdain L, Blay J-Y, Cupissol D, Bompas E, Italiano A, et al. Pharmacogenetic Study of Trabectedin-Induced Severe Hepatotoxicity in Patients with Advanced Soft Tissue Sarcoma. Cancers. 2020; 12(12):3647. https://doi.org/10.3390/cancers12123647
Chicago/Turabian StyleMaillard, Maud, Christine Chevreau, Félicien Le Louedec, Manon Cassou, Caroline Delmas, Laure Gourdain, Jean-Yves Blay, Didier Cupissol, Emmanuelle Bompas, Antoine Italiano, and et al. 2020. "Pharmacogenetic Study of Trabectedin-Induced Severe Hepatotoxicity in Patients with Advanced Soft Tissue Sarcoma" Cancers 12, no. 12: 3647. https://doi.org/10.3390/cancers12123647
APA StyleMaillard, M., Chevreau, C., Le Louedec, F., Cassou, M., Delmas, C., Gourdain, L., Blay, J.-Y., Cupissol, D., Bompas, E., Italiano, A., Isambert, N., Delcambre-Lair, C., Penel, N., Bertucci, F., Guillemet, C., Plenecassagnes, J., Foulon, S., Chatelut, É., Le Cesne, A., & Thomas, F. (2020). Pharmacogenetic Study of Trabectedin-Induced Severe Hepatotoxicity in Patients with Advanced Soft Tissue Sarcoma. Cancers, 12(12), 3647. https://doi.org/10.3390/cancers12123647