Growth Hormone Upregulates Mediators of Melanoma Drug Efflux and Epithelial-to-Mesenchymal Transition In Vitro and In Vivo

 , , , and
, , , and
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
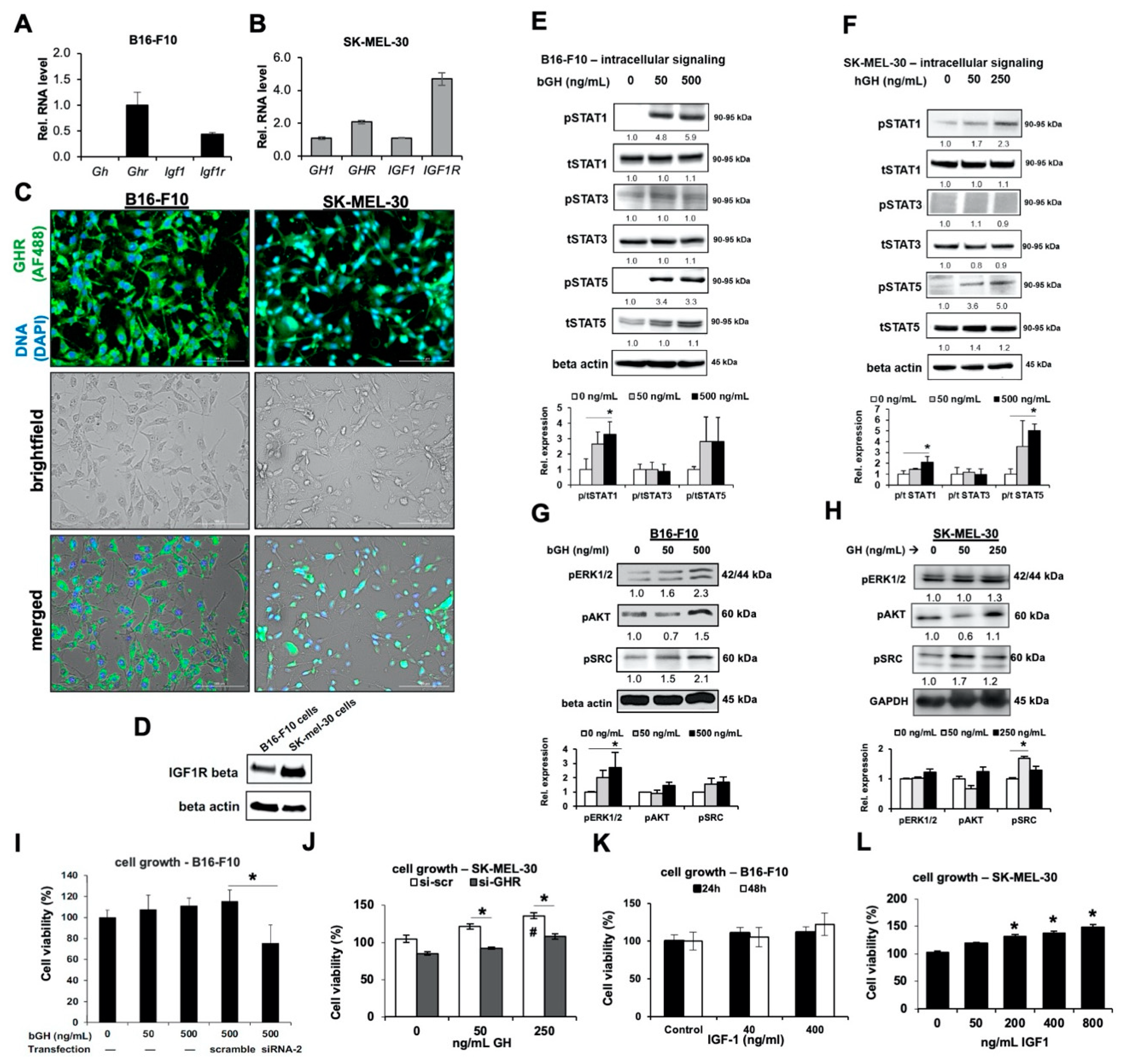
2.1. Melanoma Cells Are Responsive to GH Treatment
2.2. In Syngeneic Mouse Melanoma Models, Elevated GH or IGF-1 Did Not Affect Growth Rate of B16-F10 Tumors In Vivo
2.3. Elevated GH Drives Expression of ATP-Binding Cassette (ABC-Type) Multidrug Efflux Pumps in Melanoma Tumors In Vitro and In Vivo
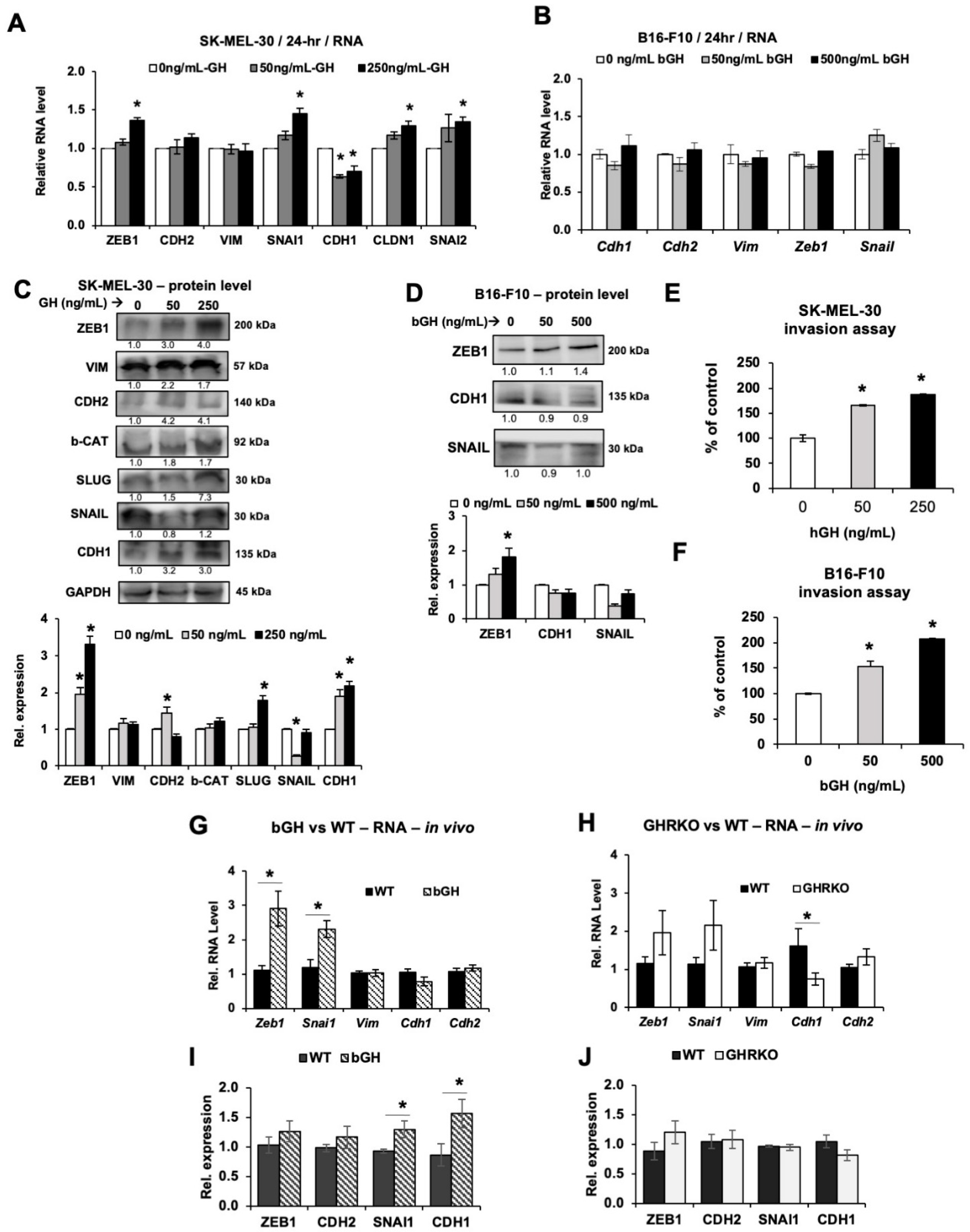
2.4. Elevated GH Drives Expression of Epithelial-to-Mesenchymal Transition (EMT) Markers In Vitro and In Vivo
3. Discussion
4. Materials and Methods
4.1. Cell Culture and GH
4.2. Real-Time RT-qPCR
4.3. Immunocytochemistry
4.4. Knockdown of GHR Expression by siRNA
4.5. Western Blotting
4.6. Cell Proliferation Assay
4.7. Cellular Metabolism
4.8. Mouse Models of Subcutaneous Melanoma
4.9. Enzyme-Linked Immunosorbent Assay (ELISA)
4.10. Drug Retention Assay
4.11. Invasion Assay
4.12. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ikeno, Y.; Hubbard, G.B.; Lee, S.; Cortez, L.A.; Lew, C.M.; Webb, C.R.; Berryman, D.E.; List, E.O.; Kopchick, J.J.; Bartke, A. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M.; Blouin, M.J.; Zhang, J.C.; Kopchick, J.J. Reduced mammary gland carcinogenesis in transgenic mice expressing a growth hormone antagonist. Br. J. Cancer 2001, 85, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Conway-Campbell, B.L.; Wooh, J.W.; Brooks, A.J.; Gordon, D.; Brown, R.J.; Lichanska, A.M.; Chin, H.S.; Barton, C.L.; Boyle, G.M.; Parsons, P.G.; et al. Nuclear targeting of the growth hormone receptor results in dysregulation of cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. USA 2007, 104, 13331–13336. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Lee, M.J.; Liou, H.C.; Liou, H.H.; Fu, W.M. Growth hormone is increased in the lungs and enhances experimental lung metastasis of melanoma in DJ-1 KO mice. BMC Cancer 2016, 16. [Google Scholar] [CrossRef]
- Van der Lely, A.J.; Muller, A.; Janssen, J.A.; Davis, R.J.; Zib, K.A.; Scarlett, J.A.; Lamberts, S.W. Control of tumor size and disease activity during cotreatment with octreotide and the growth hormone receptor antagonist pegvisomant in an acromegalic patient. J. Clin. Endocrinol. Metab. 2001, 86, 478–481. [Google Scholar] [CrossRef]
- Boguszewski, C.L.; Boguszewski, M. Growth Hormone’s Links to Cancer. Endocr. Rev. 2019, 40, 558–574. [Google Scholar] [CrossRef]
- Boguszewski, C.L.; Boguszewski, M.C.; Kopchick, J.J. Growth hormone, insulin-like growth factor system and carcinogenesis. Endokrynol. Pol. 2016, 67, 414–426. [Google Scholar] [CrossRef]
- Gebre-Medhin, M.; Kindblom, L.G.; Wennbo, H.; Tornell, J.; Meis-Kindblom, J.M. Growth hormone receptor is expressed in human breast cancer. Am. J. Pathol. 2001, 158, 1217–1222. [Google Scholar] [CrossRef]
- Felice, D.L.; El-Shennawy, L.; Zhao, S.; Lantvit, D.L.; Shen, Q.; Unterman, T.G.; Swanson, S.M.; Frasor, J. Growth hormone potentiates 17beta-estradiol-dependent breast cancer cell proliferation independently of IGF-I receptor signaling. Endocrinology 2013, 154, 3219–3227. [Google Scholar] [CrossRef][Green Version]
- Minoia, M.; Gentilin, E.; Mole, D.; Rossi, M.; Filieri, C.; Tagliati, F.; Baroni, A.; Ambrosio, M.R.; degli Uberti, E.; Zatelli, M.C. Growth hormone receptor blockade inhibits growth hormone-induced chemoresistance by restoring cytotoxic-induced apoptosis in breast cancer cells independently of estrogen receptor expression. J. Clin. Endocrinol. Metab. 2012, 97, E907–E916. [Google Scholar] [CrossRef]
- Chen, Y.J.; Zhang, X.; Wu, Z.S.; Wang, J.J.; Lau, A.Y.; Zhu, T.; Lobie, P.E. Autocrine human growth hormone stimulates the tumor initiating capacity and metastasis of estrogen receptor-negative mammary carcinoma cells. Cancer Lett. 2015, 365, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Divisova, J.; Kuiatse, I.; Lazard, Z.; Weiss, H.; Vreeland, F.; Hadsell, D.L.; Schiff, R.; Osborne, C.K.; Lee, A.V. The growth hormone receptor antagonist pegvisomant blocks both mammary gland development and MCF-7 breast cancer xenograft growth. Breast Cancer Res. Treat. 2006, 98, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Emerman, J.T.; Leahy, M.; Gout, P.W.; Bruchovsky, N. Elevated growth hormone levels in sera from breast cancer patients. Horm. Metab. Res. 1985, 17, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mehta, R.G.; Lantvit, D.D.; Coschigano, K.T.; Kopchick, J.J.; Green, J.E.; Hedayat, S.; Christov, K.T.; Ray, V.H.; Unterman, T.G.; et al. Inhibition of estrogen-independent mammary carcinogenesis by disruption of growth hormone signaling. Carcinogenesis 2007, 28, 143–150. [Google Scholar] [CrossRef]
- Waters, M.J.; Conway-Campbell, B.L. The oncogenic potential of autocrine human growth hormone in breast cancer. Proc. Natl. Acad. Sci. USA 2004, 101, 14992–14993. [Google Scholar] [CrossRef]
- Wang, Z.; Prins, G.S.; Coschigano, K.T.; Kopchick, J.J.; Green, J.E.; Ray, V.H.; Hedayat, S.; Christov, K.T.; Unterman, T.G.; Swanson, S.M. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/T antigen mouse. Endocrinology 2005, 146, 5188–5196. [Google Scholar] [CrossRef]
- Weiss-Messer, E.; Merom, O.; Adi, A.; Karry, R.; Bidosee, M.; Ber, R.; Kaploun, A.; Stein, A.; Barkey, R.J. Growth hormone (GH) receptors in prostate cancer: Gene expression in human tissues and cell lines and characterization, GH signaling and androgen receptor regulation in LNCaP cells. Mol. Cell. Endocrinol. 2004, 220, 109–123. [Google Scholar] [CrossRef]
- Wu, X.; Liu, F.; Yao, X.; Li, W.; Chen, C. Growth hormone receptor expression is up-regulated during tumorigenesis of human colorectal cancer. J. Surg. Res. 2007, 143, 294–299. [Google Scholar] [CrossRef]
- Yang, X.; Liu, F.; Xu, Z.; Chen, C.; Li, G.; Wu, X.; Li, J. Growth hormone receptor expression in human colorectal cancer. Dig. Dis. Sci. 2004, 49, 1493–1498. [Google Scholar] [CrossRef]
- Lea, R.W.; Dawson, T.; Martinez-Moreno, C.G.; El-Abry, N.; Harvey, S. Growth hormone and cancer: GH production and action in glioma? Gen. Comp. Endocrinol. 2015, 220, 119–123. [Google Scholar] [CrossRef]
- Subramani, R.; Lopez-Valdez, R.; Salcido, A.; Boopalan, T.; Arumugam, A.; Nandy, S.; Lakshmanaswamy, R. Growth hormone receptor inhibition decreases the growth and metastasis of pancreatic ductal adenocarcinoma. Exp. Mol. Med. 2014, 46, e117. [Google Scholar] [CrossRef] [PubMed]
- Dal, J.; Leisner, M.Z.; Hermansen, K.; Farkas, D.K.; Bengtsen, M.; Kistorp, C.; Nielsen, E.H.; Andersen, M.; Feldt-Rasmussen, U.; Dekkers, O.M.; et al. Cancer Incidence in Patients With Acromegaly: A Cohort Study and Meta-Analysis of the Literature. J. Clin. Endocrinol. Metab. 2018, 103, 2182–2188. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; You, M.L.; Chong, Q.Y.; Pandey, V.; Zhuang, Q.S.; Liu, D.X.; Ma, L.; Zhu, T.; Lobie, P.E. Autocrine Human Growth Hormone Promotes Invasive and Cancer Stem Cell-Like Behavior of Hepatocellular Carcinoma Cells by STAT3 Dependent Inhibition of CLAUDIN-1 Expression. Int. J. Mol. Sci. 2017, 18, 1274. [Google Scholar] [CrossRef] [PubMed]
- Pandey, V.; Perry, J.K.; Mohankumar, K.M.; Kong, X.J.; Liu, S.M.; Wu, Z.S.; Mitchell, M.D.; Zhu, T.; Lobie, P.E. Autocrine human growth hormone stimulates oncogenicity of endometrial carcinoma cells. Endocrinology 2008, 149, 3909–3919. [Google Scholar] [CrossRef]
- Tang, J.Z.; Kong, X.J.; Banerjee, A.; Muniraj, N.; Pandey, V.; Steiner, M.; Perry, J.K.; Zhu, T.; Liu, D.X.; Lobie, P.E. STAT3alpha is oncogenic for endometrial carcinoma cells and mediates the oncogenic effects of autocrine human growth hormone. Endocrinology 2010, 151, 4133–4145. [Google Scholar] [CrossRef]
- Sakharova, A.A.; Horowitz, J.F.; Surya, S.; Goldenberg, N.; Harber, M.P.; Symons, K.; Barkan, A. Role of growth hormone in regulating lipolysis, proteolysis, and hepatic glucose production during fasting. J. Clin. Endocrinol. Metab. 2008, 93, 2755–2759. [Google Scholar] [CrossRef]
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The somatomedin hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef]
- Zhu, T.; Goh, E.L.; Graichen, R.; Ling, L.; Lobie, P.E. Signal transduction via the growth hormone receptor. Cell Signal 2001, 13, 599–616. [Google Scholar] [CrossRef]
- Dehkhoda, F.; Lee, C.M.M.; Medina, J.; Brooks, A.J. The Growth Hormone Receptor: Mechanism of Receptor Activation, Cell Signaling, and Physiological Aspects. Front. Endocrinol. 2018, 9, 35. [Google Scholar] [CrossRef]
- Barclay, J.L.; Kerr, L.M.; Arthur, L.; Rowland, J.E.; Nelson, C.N.; Ishikawa, M.; d’Aniello, E.M.; White, M.; Noakes, P.G.; Waters, M.J. In vivo targeting of the growth hormone receptor (GHR) Box1 sequence demonstrates that the GHR does not signal exclusively through JAK2. Mol. Endocrinol. 2010, 24, 204–217. [Google Scholar] [CrossRef]
- Arnaldez, F.I.; Helman, L.J. Targeting the insulin growth factor receptor 1. Hematol. Oncol. Clin. N. Am. 2012, 26, 527–542, vii–viii. [Google Scholar] [CrossRef] [PubMed]
- Junnila, R.K.; List, E.O.; Berryman, D.E.; Murrey, J.W.; Kopchick, J.J. The GH/IGF-1 axis in ageing and longevity. Nat. Rev. Endocrinol. 2013, 9, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yee, D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef]
- Clark, R.G.; Mortensen, D.L.; Carlsson, L.M. Insulin-like growth factor-1 and growth hormone (GH) have distinct and overlapping anabolic effects in GH-deficient rats. Endocrine 1995, 3, 297–304. [Google Scholar] [CrossRef]
- Palmer, A.J.; Chung, M.Y.; List, E.O.; Walker, J.; Okada, S.; Kopchick, J.J.; Berryman, D.E. Age-related changes in body composition of bovine growth hormone transgenic mice. Endocrinology 2009, 150, 1353–1360. [Google Scholar] [CrossRef]
- Naar, E.M.; Bartke, A.; Majumdar, S.S.; Buonomo, F.C.; Yun, J.S.; Wagner, T.E. Fertility of transgenic female mice expressing bovine growth hormone or human growth hormone variant genes. Biol. Reprod. 1991, 45, 178–187. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef]
- Basu, R.; Qian, Y.; Kopchick, J.J. Mechanisms In Endocrinology: Lessons from growth hormone receptor gene-disrupted mice: Are there benefits of endocrine defects? Eur. J. Endocrinol. 2018, 178, R155–R181. [Google Scholar] [CrossRef]
- Basu, R.; Kopchick, J.J. The effects of growth hormone on therapy resistance in cancer. Cancer Drug Resist. 2019, 2, 827–846. [Google Scholar] [CrossRef]
- Wu, Z.S.; Yang, K.; Wan, Y.; Qian, P.X.; Perry, J.K.; Chiesa, J.; Mertani, H.C.; Zhu, T.; Lobie, P.E. Tumor expression of human growth hormone and human prolactin predict a worse survival outcome in patients with mammary or endometrial carcinoma. J. Clin. Endocrinol. Metab. 2011, 96, E1619–E1629. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, V.; Zonis, S.; Barrett, R.J.; Gleeson, J.P.; Melmed, S. Growth Hormone Induces Colon DNA Damage Independent of IGF-1. Endocrinology 2019, 160, 1439–1447. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, V.; Melmed, S. Growth hormone in the tumor microenvironment. Arch. Endocrinol. Metab. 2019, 63, 568–575. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, V.; Zonis, S.; Barrett, R.; Kameda, H.; Wawrowsky, K.; Ben-Shlomo, A.; Yamamoto, M.; Gleeson, J.; Bresee, C.; Gorbunova, V.; et al. Excess growth hormone suppresses DNA damage repair in epithelial cells. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Chesnokova, V.; Zonis, S.; Zhou, C.; Recouvreux, M.V.; Ben-Shlomo, A.; Araki, T.; Barrett, R.; Workman, M.; Wawrowsky, K.; Ljubimov, V.A.; et al. Growth hormone is permissive for neoplastic colon growth. Proc. Natl. Acad. Sci. USA 2016, 113, E3250–E3259. [Google Scholar] [CrossRef]
- Chiesa, J.; Ferrer, C.; Arnould, C.; Vouyovitch, C.M.; Diaz, J.J.; Gonzalez, S.; Mares, P.; Morel, G.; Wu, Z.S.; Zhu, T.; et al. Autocrine proliferative effects of hGH are maintained in primary cultures of human mammary carcinoma cells. J. Clin. Endocrinol. Metab. 2011, 96, E1418–E1426. [Google Scholar] [CrossRef][Green Version]
- Bougen, N.M.; Steiner, M.; Pertziger, M.; Banerjee, A.; Brunet-Dunand, S.E.; Zhu, T.; Lobie, P.E.; Perry, J.K. Autocrine human GH promotes radioresistance in mammary and endometrial carcinoma cells. Endocr. Relat. Cancer 2012, 19, 625–644. [Google Scholar] [CrossRef][Green Version]
- Bougen, N.M.; Yang, T.; Chen, H.; Lobie, P.E.; Perry, J.K. Autocrine human growth hormone reduces mammary and endometrial carcinoma cell sensitivity to mitomycin C. Oncol Rep 2011, 26, 487–493. [Google Scholar] [CrossRef]
- Perry, J.K.; Mohankumar, K.M.; Emerald, B.S.; Mertani, H.C.; Lobie, P.E. The contribution of growth hormone to mammary neoplasia. J. Mammary Gland Biol. Neoplasia 2008, 13, 131–145. [Google Scholar] [CrossRef]
- Perry, J.K.; Wu, Z.S.; Mertani, H.C.; Zhu, T.; Lobie, P.E. Tumour-Derived Human Growth Hormone As a Therapeutic Target in Oncology. Trends Endocrinol. Metab. 2017, 28, 587–596. [Google Scholar] [CrossRef]
- Zhu, T.; Starling-Emerald, B.; Zhang, X.; Lee, K.O.; Gluckman, P.D.; Mertani, H.C.; Lobie, P.E. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res. 2005, 65, 317–324. [Google Scholar] [PubMed]
- Zhu, Z.; Mukhina, S.; Zhu, T.; Mertani, H.C.; Lee, K.O.; Lobie, P.E. p44/42 MAP kinase-dependent regulation of catalase by autocrine human growth hormone protects human mammary carcinoma cells from oxidative stress-induced apoptosis. Oncogene 2005, 24, 3774–3785. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.Q.; Emerald, B.S.; Goh, E.L.; Kannan, N.; Miller, L.D.; Gluckman, P.D.; Liu, E.T.; Lobie, P.E. Gene expression profiling to identify oncogenic determinants of autocrine human growth hormone in human mammary carcinoma. J. Biol. Chem. 2005, 280, 23987–24003. [Google Scholar] [CrossRef] [PubMed]
- Kaulsay, K.K.; Mertani, H.C.; Tornell, J.; Morel, G.; Lee, K.O.; Lobie, P.E. Autocrine stimulation of human mammary carcinoma cell proliferation by human growth hormone. Exp. Cell Res. 1999, 250, 35–50. [Google Scholar] [CrossRef] [PubMed]
- American Cancer Society. Cancer Facts & Figures; The Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Sustarsic, E.G.; Junnila, R.K.; Kopchick, J.J. Human metastatic melanoma cell lines express high levels of growth hormone receptor and respond to GH treatment. Biochem. Biophys. Res. Commun. 2013, 441, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Wu, S.; Kopchick, J.J. Targeting growth hormone receptor in human melanoma cells attenuates tumor progression and epithelial mesenchymal transition via suppression of multiple oncogenic pathways. Oncotarget 2017, 8, 21579–21598. [Google Scholar] [CrossRef]
- Basu, R.; Baumgaertel, N.; Wu, S.; Kopchick, J.J. Growth Hormone Receptor Knockdown Sensitizes Human Melanoma Cells to Chemotherapy by Attenuating Expression of ABC Drug Efflux Pumps. Horm. Cancer 2017, 8, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Kulkarni, P.; Qian, Y.; Walsh, C.; Arora, P.; Davis, E.; Duran-Ortiz, S.; Funk, K.; Ibarra, D.; Kruse, C.; et al. Growth Hormone Upregulates Melanocyte-Inducing Transcription Factor Expression and Activity via JAK2-STAT5 and SRC Signaling in GH Receptor-Positive Human Melanoma. Cancers 2019, 11, 1352. [Google Scholar] [CrossRef]
- Le Coz, V.; Zhu, C.; Devocelle, A.; Vazquez, A.; Boucheix, C.; Azzi, S.; Gallerne, C.; Eid, P.; Lecourt, S.; Giron-Michel, J. IGF-1 contributes to the expansion of melanoma-initiating cells through an epithelial-mesenchymal transition process. Oncotarget 2016, 7, 82511–82527. [Google Scholar] [CrossRef]
- Zhao, K.; Wei, L.; Hui, H.; Dai, Q.; You, Q.D.; Guo, Q.L.; Lu, N. Wogonin suppresses melanoma cell B16-F10 invasion and migration by inhibiting Ras-medicated pathways. PLoS ONE 2014, 9, e106458. [Google Scholar] [CrossRef][Green Version]
- Carter-Su, C.; Schwartz, J.; Argetsinger, L.S. Growth hormone signaling pathways. Growth Horm. IGF Res. 2016, 28, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Park, M.J. Effects of growth hormone on glucose metabolism and insulin resistance in human. Ann. Pediatr. Endocrinol. Metab. 2017, 22, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Sag, D.; Cekic, C.; Wu, R.; Linden, J.; Hedrick, C.C. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat. Commun. 2015, 6, 6354. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.P.; Ambudkar, S.V. The pharmacological impact of ATP-binding cassette drug transporters on vemurafenib-based therapy. Acta Pharm. Sin. B 2014, 4, 105–111. [Google Scholar] [CrossRef]
- Landreville, S.; Agapova, O.A.; Kneass, Z.T.; Salesse, C.; Harbour, J.W. ABCB1 identifies a subpopulation of uveal melanoma cells with high metastatic propensity. Pigm. Cell Melanoma R 2011, 24, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.M.; Ai-Hajj, M.A. ABCB8 Mediates Doxorubicin Resistance in Melanoma Cells by Protecting the Mitochondrial Genome. Mol. Cancer Res. 2009, 7, 79–87. [Google Scholar] [CrossRef]
- Monzani, E.; Facchetti, F.; Galmozzi, E.; Corsini, E.; Benetti, A.; Cavazzin, C.; Gritti, A.; Piccinini, A.; Porro, D.; Santinami, M.; et al. Melanoma contains CD133 and ABCG2 positive cells with enhanced tumourigenic potential. Eur. J. Cancer 2007, 43, 935–946. [Google Scholar] [CrossRef]
- Szakacs, G.; Annereau, J.P.; Lababidi, S.; Shankavaram, U.; Arciello, A.; Bussey, K.J.; Reinhold, W.; Guo, Y.; Kruh, G.D.; Reimers, M.; et al. Predicting drug sensitivity and resistance: Profiling ABC transporter genes in cancer cells. Cancer Cell 2004, 6, 129–137. [Google Scholar] [CrossRef]
- Liedert, B.; Materna, V.; Schadendorf, D.; Thomale, J.; Lage, H. Overexpression of cMOAT (MRP2/ABCC2) is associated with decreased formation of platinum-DNA adducts and decreased G2-arrest in melanoma cells resistant to cisplatin. J. Investig. Dermatol. 2003, 121, 172–176. [Google Scholar] [CrossRef]
- Arumugam, A.; Subramani, R.; Nandy, S.B.; Terreros, D.; Dwivedi, A.K.; Saltzstein, E.; Lakshmanaswamy, R. Silencing growth hormone receptor inhibits estrogen receptor negative breast cancer through ATP-binding cassette sub-family G member 2. Exp. Mol. Med. 2019, 51, 2. [Google Scholar] [CrossRef]
- Du, J.; Shi, H.R.; Ren, F.; Wang, J.L.; Wu, Q.H.; Li, X.; Zhang, R.T. Inhibition of the IGF signaling pathway reverses cisplatin resistance in ovarian cancer cells. BMC Cancer 2017, 17, 851. [Google Scholar] [CrossRef] [PubMed]
- Benabbou, N.; Mirshahi, P.; Cadillon, M.; Soria, J.; Therwath, A.; Mirshahi, M. Hospicells promote upregulation of the ATP-binding cassette genes by insulin-like growth factor-I via the JAK2/STAT3 signaling pathway in an ovarian cancer cell line. Int. J. Oncol. 2013, 43, 685–694. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shen, K.; Cui, D.; Sun, L.; Lu, Y.; Han, M.; Liu, J. Inhibition of IGF-IR increases chemosensitivity in human colorectal cancer cells through MRP-2 promoter suppression. J. Cell Biochem. 2012, 113, 2086–2097. [Google Scholar] [CrossRef] [PubMed]
- Benabbou, N.; Mirshahi, P.; Bordu, C.; Faussat, A.M.; Tang, R.; Therwath, A.; Soria, J.; Marie, J.P.; Mirshahi, M. A subset of bone marrow stromal cells regulate ATP-binding cassette gene expression via insulin-like growth factor-I in a leukemia cell line. Int. J. Oncol. 2014, 45, 1372–1380. [Google Scholar] [CrossRef]
- Brittain, A.L.; Basu, R.; Qian, Y.; Kopchick, J.J. Growth Hormone and the Epithelial-to-Mesenchymal Transition. J. Clin. Endocrinol. Metab. 2017. [Google Scholar] [CrossRef]
- Mukhina, S.; Mertani, H.C.; Guo, K.; Lee, K.O.; Gluckman, P.D.; Lobie, P.E. Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc. Natl. Acad. Sci. USA 2004, 101, 15166–15171. [Google Scholar] [CrossRef]
- Zhang, W.; Qian, P.; Zhang, X.; Zhang, M.; Wang, H.; Wu, M.; Kong, X.; Tan, S.; Ding, K.; Perry, J.K.; et al. Autocrine/Paracrine Human Growth Hormone-stimulated MicroRNA 96-182-183 Cluster Promotes Epithelial-Mesenchymal Transition and Invasion in Breast Cancer. J. Biol. Chem. 2015, 290, 13812–13829. [Google Scholar] [CrossRef]
- Wang, J.J.; Chong, Q.Y.; Sun, X.B.; You, M.L.; Pandey, V.; Chen, Y.J.; Zhuang, Q.S.; Liu, D.X.; Ma, L.; Wu, Z.S.; et al. Autocrine hGH stimulates oncogenicity, epithelial-mesenchymal transition and cancer stem cell-like behavior in human colorectal carcinoma. Oncotarget 2017, 8, 103900–103918. [Google Scholar] [CrossRef]
- Liao, G.; Wang, M.; Ou, Y.; Zhao, Y. IGF-1-induced epithelial-mesenchymal transition in MCF-7 cells is mediated by MUC1. Cell Signal. 2014, 26, 2131–2137. [Google Scholar] [CrossRef]
- Ford, N.A.; Nunez, N.P.; Holcomb, V.B.; Hursting, S.D. IGF1 dependence of dietary energy balance effects on murine Met1 mammary tumor progression, epithelial-to-mesenchymal transition, and chemokine expression. Endocr. Relat. Cancer 2013, 20, 39–51. [Google Scholar] [CrossRef]
- Yao, C.; Su, L.; Shan, J.; Zhu, C.; Liu, L.; Liu, C.; Xu, Y.; Yang, Z.; Bian, X.; Shao, J.; et al. IGF/STAT3/NANOG/Slug Signaling Axis Simultaneously Controls Epithelial-Mesenchymal Transition and Stemness Maintenance in Colorectal Cancer. Stem Cells 2016, 34, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Park, G.B.; Kim, D. Insulin-like growth factor-1 activates different catalytic subunits p110 of PI3K in a cell-type-dependent manner to induce lipogenesis-dependent epithelial-mesenchymal transition through the regulation of ADAM10 and ADAM17. Mol. Cell. Biochem. 2018, 439, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Kobayashi, I.; Murakami, A.; Kato, M.; Minakata, K.; Nara, T.; Hashimoto, M.; Yagishita, S.; Baskoro, H.; et al. Treatment with insulin-like growth factor 1 receptor inhibitor reverses hypoxia-induced epithelial-mesenchymal transition in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2014, 455, 332–338. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, J.; Zeng, Y.; Zhang, X.; Hu, Q.; Zheng, J.; Chen, B.; Xie, B.; Zhang, W.M. Implication of epithelial-mesenchymal transition in IGF1R-induced resistance to EGFR-TKIs in advanced non-small cell lung cancer. Oncotarget 2015, 6, 44332–44345. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, J.; Wu, D.; Han, G. The involvement of survivin in insulin-like growth factor 1-induced epithelial-mesenchymal transition in gastric cancer. Tumour Biol. 2016, 37, 1091–1096. [Google Scholar] [CrossRef]
- Li, H.; Xu, L.; Zhao, L.; Ma, Y.; Zhu, Z.; Liu, Y.; Qu, X. Insulin-like growth factor-I induces epithelial to mesenchymal transition via GSK-3beta and ZEB2 in the BGC-823 gastric cancer cell line. Oncol. Lett. 2015, 9, 143–148. [Google Scholar] [CrossRef]
- Li, H.; Xu, L.; Li, C.; Zhao, L.; Ma, Y.; Zheng, H.; Li, Z.; Zhang, Y.; Wang, R.; Liu, Y.; et al. Ubiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cells. Mol. Cancer 2014, 13, 136. [Google Scholar] [CrossRef]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef]
- Zhau, H.E.; Odero-Marah, V.; Lue, H.W.; Nomura, T.; Wang, R.; Chu, G.; Liu, Z.R.; Zhou, B.P.; Huang, W.C.; Chung, L.W. Epithelial to mesenchymal transition (EMT) in human prostate cancer: Lessons learned from ARCaP model. Clin. Exp. Metastasis 2008, 25, 601–610. [Google Scholar] [CrossRef][Green Version]
- Liu, F.; Sun, Y.; Liu, B.; Lu, J.; Li, H.; Zhu, H.; Gao, H.; Zhou, X.; Chang, H. Insulin-like growth factor-1 induces epithelial-mesenchymal transition in hepatocellular carcinoma by activating survivin. Oncol. Rep. 2018, 40, 952–958. [Google Scholar] [CrossRef]
- Lin, Y.C.; Lin, J.C.; Hung, C.M.; Chen, Y.; Liu, L.C.; Chang, T.C.; Kao, J.Y.; Ho, C.T.; Way, T.D. Osthole inhibits insulin-like growth factor-1-induced epithelial to mesenchymal transition via the inhibition of PI3K/Akt signaling pathway in human brain cancer cells. J. Agric. Food Chem. 2014, 62, 5061–5071. [Google Scholar] [CrossRef] [PubMed]
- Alonso, S.R.; Tracey, L.; Ortiz, P.; Perez-Gomez, B.; Palacios, J.; Pollan, M.; Linares, J.; Serrano, S.; Saez-Castillo, A.I.; Sanchez, L.; et al. A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 2007, 67, 3450–3460. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Z.; Dhillon, A.S.; Anderson, R.L.; McArthur, G.; Ferrao, P.T. Phenotype switching in melanoma: Implications for progression and therapy. Front. Oncol. 2015, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A Switch in the Expression of Embryonic EMT-Inducers Drives the Development of Malignant Melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, X.; Montoya-Durango, D.E.; Liu, Y.H.; Dean, K.C.; Darling, D.S.; Kaplan, H.J.; Dean, D.C.; Gao, L.; Liu, Y. ZEB1 Regulates Multiple Oncogenic Components Involved in Uveal Melanoma Progression. Sci Rep 2017, 7, 45. [Google Scholar] [CrossRef]
- Wauthier, V.; Waxman, D.J. Sex-specific early growth hormone response genes in rat liver. Mol. Endocrinol. 2008, 22, 1962–1974. [Google Scholar] [CrossRef]
- Waxman, D.J.; O’Connor, C. Growth hormone regulation of sex-dependent liver gene expression. Mol. Endocrinol. 2006, 20, 2613–2629. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Green, H.; Morikawa, M.; Nixon, T. A dual effector theory of growth-hormone action. Differentiation 1985, 29, 195–198. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Flanagan, J.U.; Langley, R.J.; Hay, M.P.; Perry, J.K. Targeting growth hormone function: Strategies and therapeutic applications. Signal Transduct. Target Ther. 2019, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Friend, K.E. Cancer and the potential place for growth hormone receptor antagonist therapy. Growth Horm. IGF Res. 2001, 11 (Suppl. A), S121–S123. [Google Scholar] [CrossRef]
- Holly, J.; Perks, C. Growth hormone and cancer: Are we asking the right questions? Clin. Endocrinol. 2006, 64, 122–124. [Google Scholar] [CrossRef]
- Holly, J.M.; Gunnell, D.J.; Davey Smith, G. Growth hormone, IGF-I and cancer. Less intervention to avoid cancer? More intervention to prevent cancer? J. Endocrinol. 1999, 162, 321–330. [Google Scholar] [CrossRef][Green Version]
- Critical evaluation of the safety of recombinant human growth hormone administration: Statement from the Growth Hormone Research Society. J. Clin. Endocrinol. Metab. 2001, 86, 1868–1870. [CrossRef]
- Stochholm, K.; Johannsson, G. Reviewing the safety of GH replacement therapy in adults. Growth Horm. IGF Res. 2015, 25, 149–157. [Google Scholar] [CrossRef]
- Raman, S.; Grimberg, A.; Waguespack, S.G.; Miller, B.S.; Sklar, C.A.; Meacham, L.R.; Patterson, B.C. Risk of Neoplasia in Pediatric Patients Receiving Growth Hormone Therapy-A Report From the Pediatric Endocrine Society Drug and Therapeutics Committee. J. Clin. Endocrinol. Metab. 2015, 100, 2192–2203. [Google Scholar] [CrossRef]
- Sklar, C.A.; Mertens, A.C.; Mitby, P.; Occhiogrosso, G.; Qin, J.; Heller, G.; Yasui, Y.; Robison, L.L. Risk of disease recurrence and second neoplasms in survivors of childhood cancer treated with growth hormone: A report from the Childhood Cancer Survivor Study. J. Clin. Endocrinol. Metab. 2002, 87, 3136–3141. [Google Scholar] [CrossRef]
- Cao, Y.; Nimptsch, K.; Shui, I.M.; Platz, E.A.; Wu, K.; Pollak, M.N.; Kenfield, S.A.; Stampfer, M.J.; Giovannucci, E.L. Prediagnostic plasma IGFBP-1, IGF-1 and risk of prostate cancer. Int. J. Cancer 2015, 136, 2418–2426. [Google Scholar] [CrossRef]
- Gao, Y.; Katki, H.; Graubard, B.; Pollak, M.; Martin, M.; Tao, Y.; Schoen, R.E.; Church, T.; Hayes, R.B.; Greene, M.H.; et al. Serum IGF1, IGF2 and IGFBP3 and risk of advanced colorectal adenoma. Int. J. Cancer 2012, 131, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, D.R. Modifying IGF1 activity: An approach to treat endocrine disorders, atherosclerosis and cancer. Nat. Rev. Drug Discov. 2007, 6, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.E.; Suarez, J.A.; Brandhorst, S.; Balasubramanian, P.; Cheng, C.W.; Madia, F.; Fontana, L.; Mirisola, M.G.; Guevara-Aguirre, J.; Wan, J.; et al. Low protein intake is associated with a major reduction in IGF-1, cancer, and overall mortality in the 65 and younger but not older population. Cell Metab. 2014, 19, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Milman, S.; Atzmon, G.; Huffman, D.M.; Wan, J.; Crandall, J.P.; Cohen, P.; Barzilai, N. Low insulin-like growth factor-1 level predicts survival in humans with exceptional longevity. Aging Cell 2014, 13, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2008, 8, 915–928. [Google Scholar] [CrossRef]
- Beckwith, H.; Yee, D. Minireview: Were the IGF Signaling Inhibitors All Bad? Mol. Endocrinol. 2015, 29, 1549–1557. [Google Scholar] [CrossRef]
- Chhabra, Y.; Nelson, C.N.; Plescher, M.; Barclay, J.L.; Smith, A.G.; Andrikopoulos, S.; Mangiafico, S.; Waxman, D.J.; Brooks, A.J.; Waters, M.J. Loss of growth hormone-mediated signal transducer and activator of transcription 5 (STAT5) signaling in mice results in insulin sensitivity with obesity. FASEB J. 2019, 33, 6412–6430. [Google Scholar] [CrossRef]
- Adamson, U. On the diabetogenic effect of growth hormone in man: Effects of growth hormone of glucagon and insulin secretion. Eur. J. Clin. Investig. 1981, 11, 115–119. [Google Scholar] [CrossRef]
- Bornstein, J.; Armstrong, J.M.; Gould, M.K.; Harcourt, J.A.; Jones, M.D. Mechanism of the diabetogenic action of growth hormone. I. Effect of polypeptides derived from growth hormone on glycolysis in muscle. Biochim. Biophys. Acta 1969, 192, 265–270. [Google Scholar] [CrossRef]
- Jara, A.; Liu, X.; Sim, D.; Benner, C.M.; Duran-Ortiz, S.; Qian, Y.; List, E.O.; Berryman, D.E.; Kim, J.K.; Kopchick, J.J. Cardiac-Specific Disruption of GH Receptor Alters Glucose Homeostasis While Maintaining Normal Cardiac Performance in Adult Male Mice. Endocrinology 2016, 157, 1929–1941. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Wang, X.; Liu, Y.; Li, Y.; Colvin, R.A.; Tong, L.; Wu, S.; Chen, X. Extracellular ATP is internalized by macropinocytosis and induces intracellular ATP increase and drug resistance in cancer cells. Cancer Lett 2014, 351, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Vonk, A.G.; Wieland, C.W.; Versteegen, M.; Verschueren, I.C.; Netea, M.G.; Joosten, L.A.B.; Verweij, P.E.; Kullberg, B.J. Influence of endogenous pro-inflammatory cytokines on neutrophil-mediated damage of Candida albicans pseudohyphae, quantified in a modified tetrazolium dye assay. Med. Mycol. 2005, 43, 551–557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Qian, Y.; Wang, X.; Li, Y.; Cao, Y.; Chen, X. Extracellular ATP a New Player in Cancer Metabolism: NSCLC Cells Internalize ATP In Vitro and In Vivo Using Multiple Endocytic Mechanisms. Mol. Cancer Res. 2016, 14, 1087–1096. [Google Scholar] [CrossRef]
- Liu, Y.; Cao, Y.; Zhang, W.; Bergmeier, S.; Qian, Y.; Akbar, H.; Colvin, R.; Ding, J.; Tong, L.; Wu, S.; et al. A small-molecule inhibitor of glucose transporter 1 downregulates glycolysis, induces cell-cycle arrest, and inhibits cancer cell growth in vitro and in vivo. Mol. Cancer Ther. 2012, 11, 1672–1682. [Google Scholar] [CrossRef]
- Wang, J.; Yu, F.; Jia, X.; Iwanowycz, S.; Wang, Y.; Huang, S.; Ai, W.; Fan, D. MicroRNA-155 deficiency enhances the recruitment and functions of myeloid-derived suppressor cells in tumor microenvironment and promotes solid tumor growth. Int. J. Cancer 2015, 136, E602–E613. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Effects of GH | Observed Effects | Refs |
| Cancer Type | ||
| Breast Cancer (In vivo) | Increased ABCG2 | [71] |
| Human Melanoma (In vitro) | Increased ABCB1, ABCB5, ABCB8, ABCC1, ABCC2, ABCG1, ABCG2 | [58] Current study |
| Mouse Melanoma (In vitro) | Increased Abcb1a, Abcg1, Abcg2 | Current study |
| Mouse Melanoma (In vivo) | Increased Abcb1a, Abcb8, Abcg1, Abcg2 | Current study |
| Effects of IGF-1 | Observed Effects | Refs |
| Cancer Type | ||
| Ovarian cancer (In vitro and vivo) | Increased ABCB1, ABCC1, ABCC2, ABCC3, ABCC5, ABCG2 | [72,73] |
| Colorectal cancer (In vitro) | Increased ABCC2 | [74] |
| Leukemia (In vitro) | Increased ABCB1, ABCC1, ABCC2, ABCC3, ABCG2 | [75] |
| Mouse Melanoma (In vivo) | Increased Abcc2, Abcc4 | Current study |
| Effects of GH | Observed Effects | Refs |
| Cancer Type | ||
| Breast Cancer (In vitro and in vivo) | Increased SNAIL, SLUG, VIM, CDH2, FN1 Decreased CDH1, OCLN, CTNNA1, JUP | [77,78] |
| Endometrial adenocarcinoma (In vitro) | Increased FN1 Decreased CTNNA1 | [24] |
| Colorectal cancer (In vitro and in vivo) | Increased SNAIL, TWIST2, FN1 Decreased CDH1, OCLN | [45,79] |
| Pancreatic ductal adenocarcinoma (In vitro) | Increased SNAIL, SLUG, VIM, ZEB1, CDH2, CTNNB2 Decreased CDH1 | [21] |
| Human Melanoma (In vitro) | Increased SNAIL, SLUG, VIM, ZEB1, CDH2, CLDN1 Decreased CDH1 | [57] Current study |
| Mouse Melanoma (In vivo) | Increased Zeb1, Snai1 Decreased Cdh1 | Current study |
| Effects of IGF-1 | Observed Effects | Refs |
| Cancer Type | ||
| Breast Cancer (In vitro and in vivo) | Increased SNAIL, VIM, ZEB1, CDH2, TWIST1, FN1 Decreased CDH1, OCLN | [80,81] |
| Colorectal cancer (In vitro and in vivo) | Increased SNAIL, VIM, CDH2 Decreased CDH1, ZO-1 | [82,83] |
| Lung cancer (In vitro) | Increased SNAIL, VIM, CDH2, FN1, CTNNB1 Decreased CDH1 | [84,85] |
| Gastric cancer (In vitro) | Increased SNAIL, ZEB2, CDH2 | [86,87,88] |
| Prostate cancer (In vitro) | Increased ZEB1, VIM, CDH2, FN1, CTNNB1 Decreased CDH1 | [89,90] |
| Liver cancer (In vitro) | Increased SNAIL, VIM, CDH2 Decreased CDH1 | [91] |
| Glioblastoma (In vitro) | Increased VIM, CDH2 Decreased CDH1, ZO-1 | [92] |
| Human Melanoma (In vitro) | Increased ZEB1, CDH2 Decreased CDH1 | [60] |
| Mouse Melanoma (In vitro) | Increased Zeb1 | Current study |
| Mouse Melanoma (In vivo) | Increased Snai1, Zeb1 Decreased Cdh1 (m) | Current study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, Y.; Basu, R.; Mathes, S.C.; Arnett, N.A.; Duran-Ortiz, S.; Funk, K.R.; Brittain, A.L.; Kulkarni, P.; Terry, J.C.; Davis, E.; et al. Growth Hormone Upregulates Mediators of Melanoma Drug Efflux and Epithelial-to-Mesenchymal Transition In Vitro and In Vivo. Cancers 2020, 12, 3640. https://doi.org/10.3390/cancers12123640
Qian Y, Basu R, Mathes SC, Arnett NA, Duran-Ortiz S, Funk KR, Brittain AL, Kulkarni P, Terry JC, Davis E, et al. Growth Hormone Upregulates Mediators of Melanoma Drug Efflux and Epithelial-to-Mesenchymal Transition In Vitro and In Vivo. Cancers. 2020; 12(12):3640. https://doi.org/10.3390/cancers12123640
Chicago/Turabian StyleQian, Yanrong, Reetobrata Basu, Samuel C. Mathes, Nathan A. Arnett, Silvana Duran-Ortiz, Kevin R. Funk, Alison L. Brittain, Prateek Kulkarni, Joseph C. Terry, Emily Davis, and et al. 2020. "Growth Hormone Upregulates Mediators of Melanoma Drug Efflux and Epithelial-to-Mesenchymal Transition In Vitro and In Vivo" Cancers 12, no. 12: 3640. https://doi.org/10.3390/cancers12123640
APA StyleQian, Y., Basu, R., Mathes, S. C., Arnett, N. A., Duran-Ortiz, S., Funk, K. R., Brittain, A. L., Kulkarni, P., Terry, J. C., Davis, E., Singerman, J. T., Henry, B. E., List, E. O., Berryman, D. E., & Kopchick, J. J. (2020). Growth Hormone Upregulates Mediators of Melanoma Drug Efflux and Epithelial-to-Mesenchymal Transition In Vitro and In Vivo. Cancers, 12(12), 3640. https://doi.org/10.3390/cancers12123640