Silencing of VEGFR2 by RGD-Modified Lipid Nanoparticles Enhanced the Efficacy of Anti-PD-1 Antibody by Accelerating Vascular Normalization and Infiltration of T Cells in Tumors

 ,
, 
Abstract
:Simple Summary
Abstract
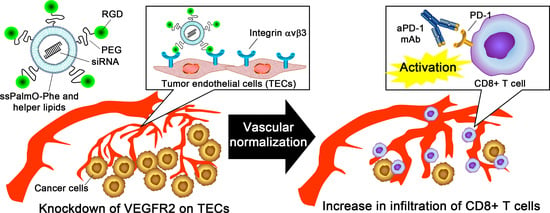
1. Introduction
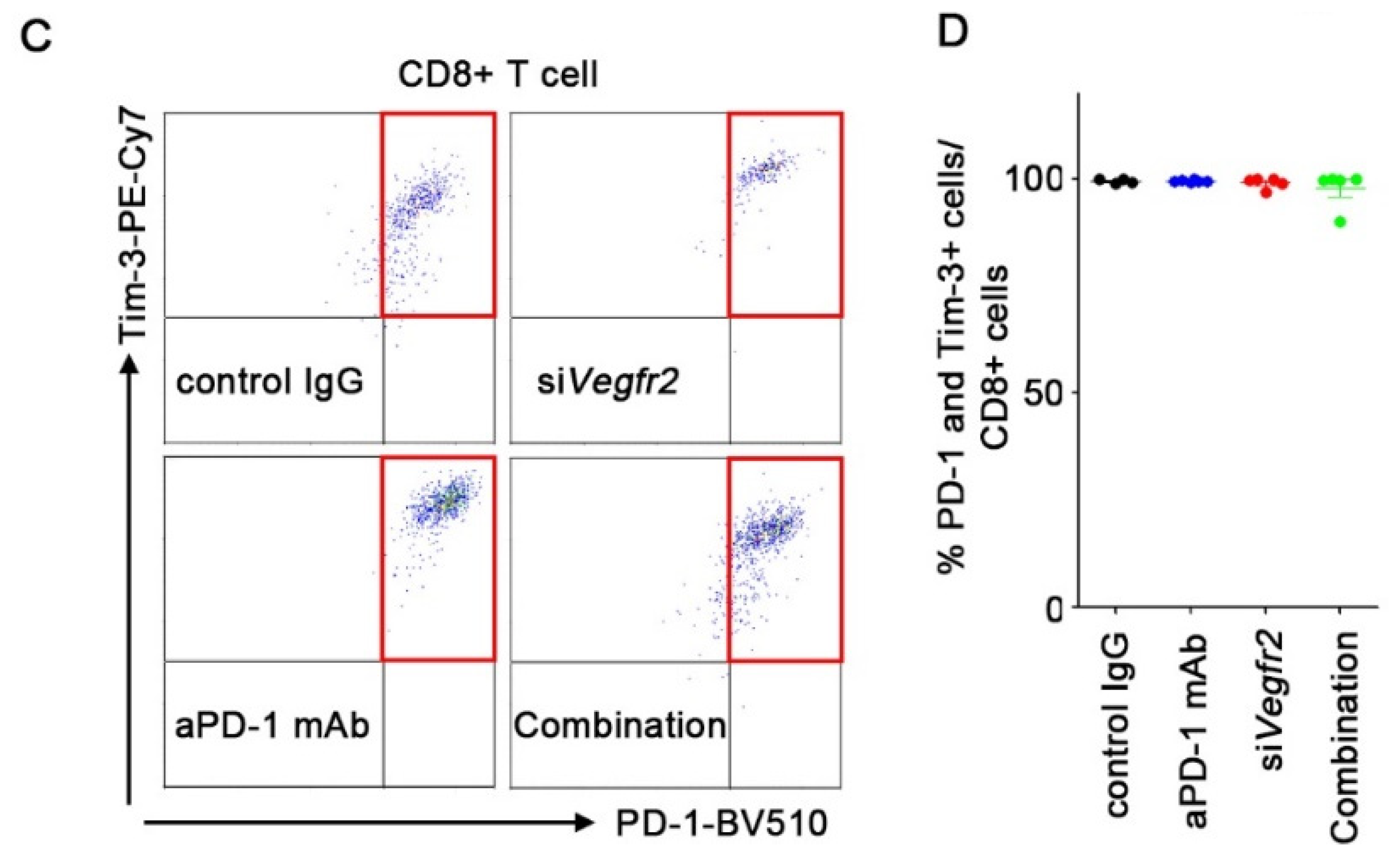
2. Results
2.1. Characterization of LNPs
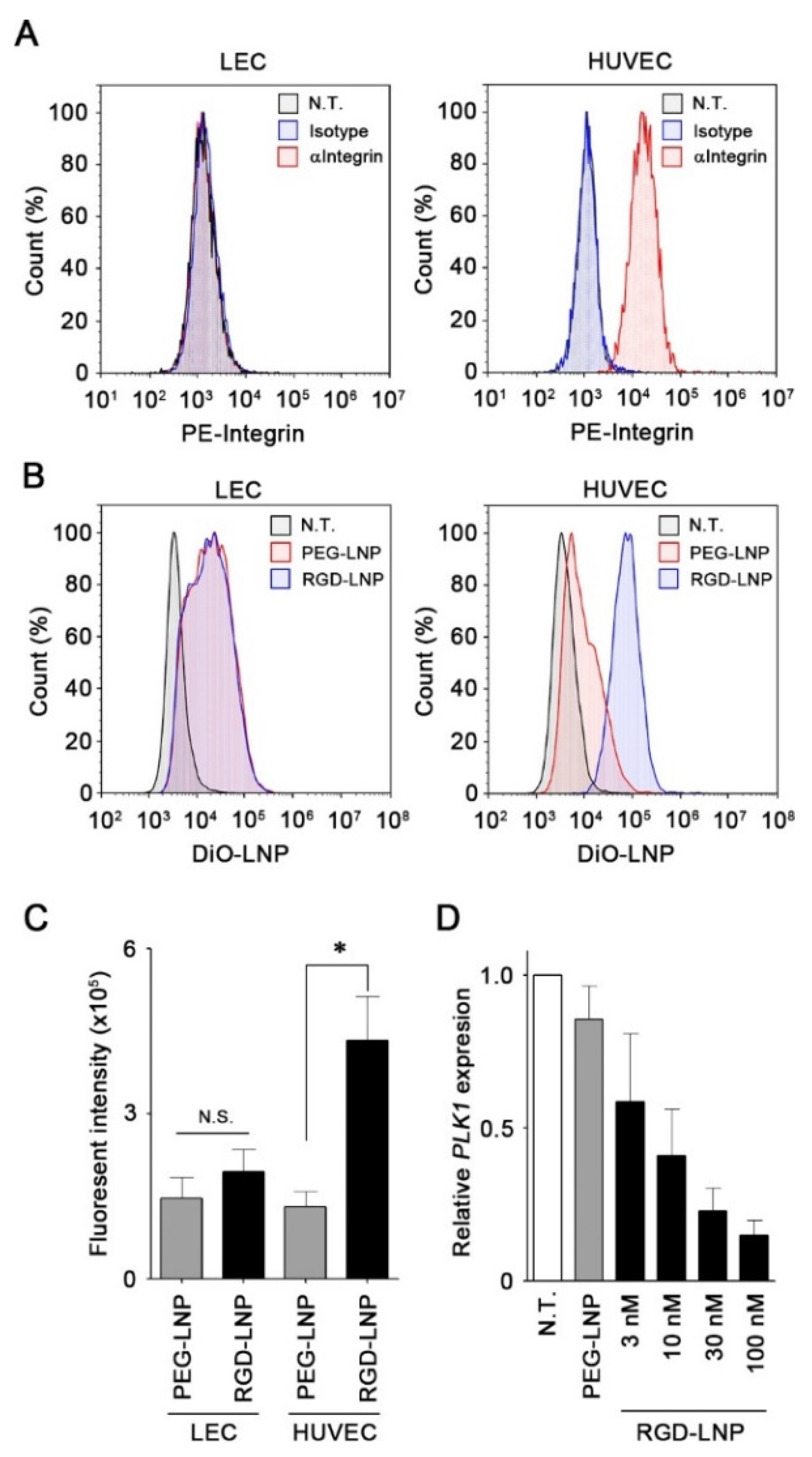
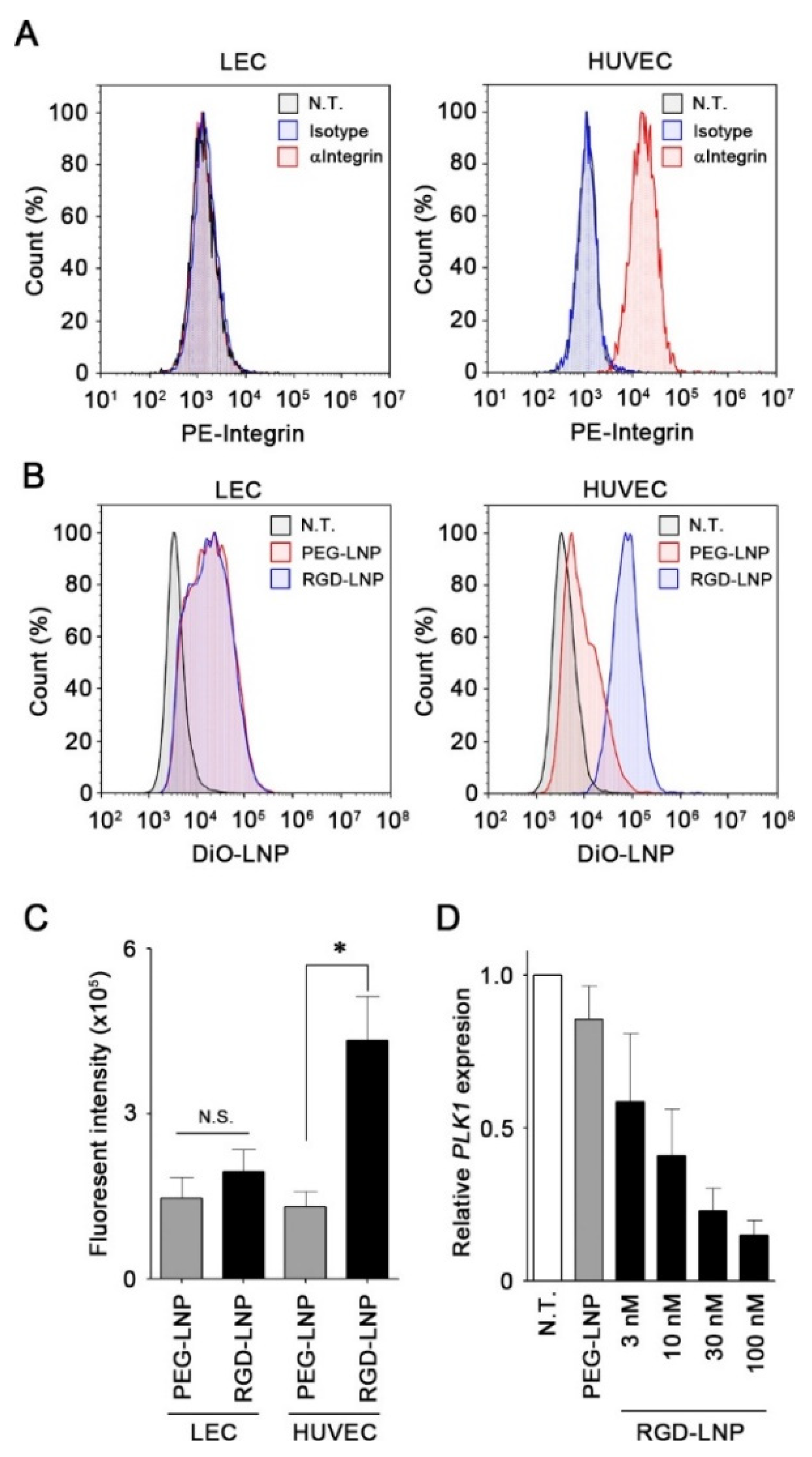
2.2. In Vitro Cellular Uptake and Knockdown by LNPs in Endothelial Cells
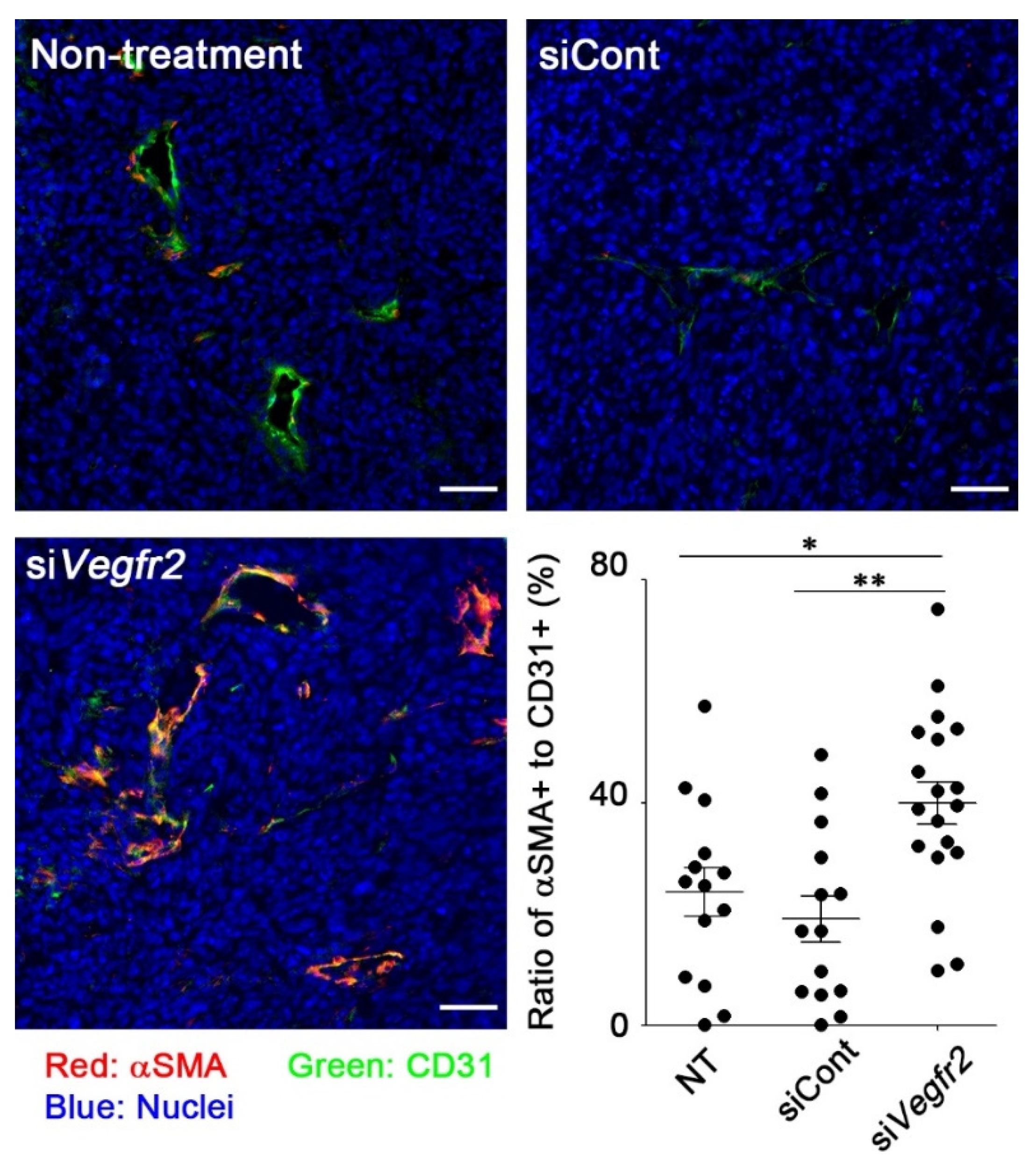
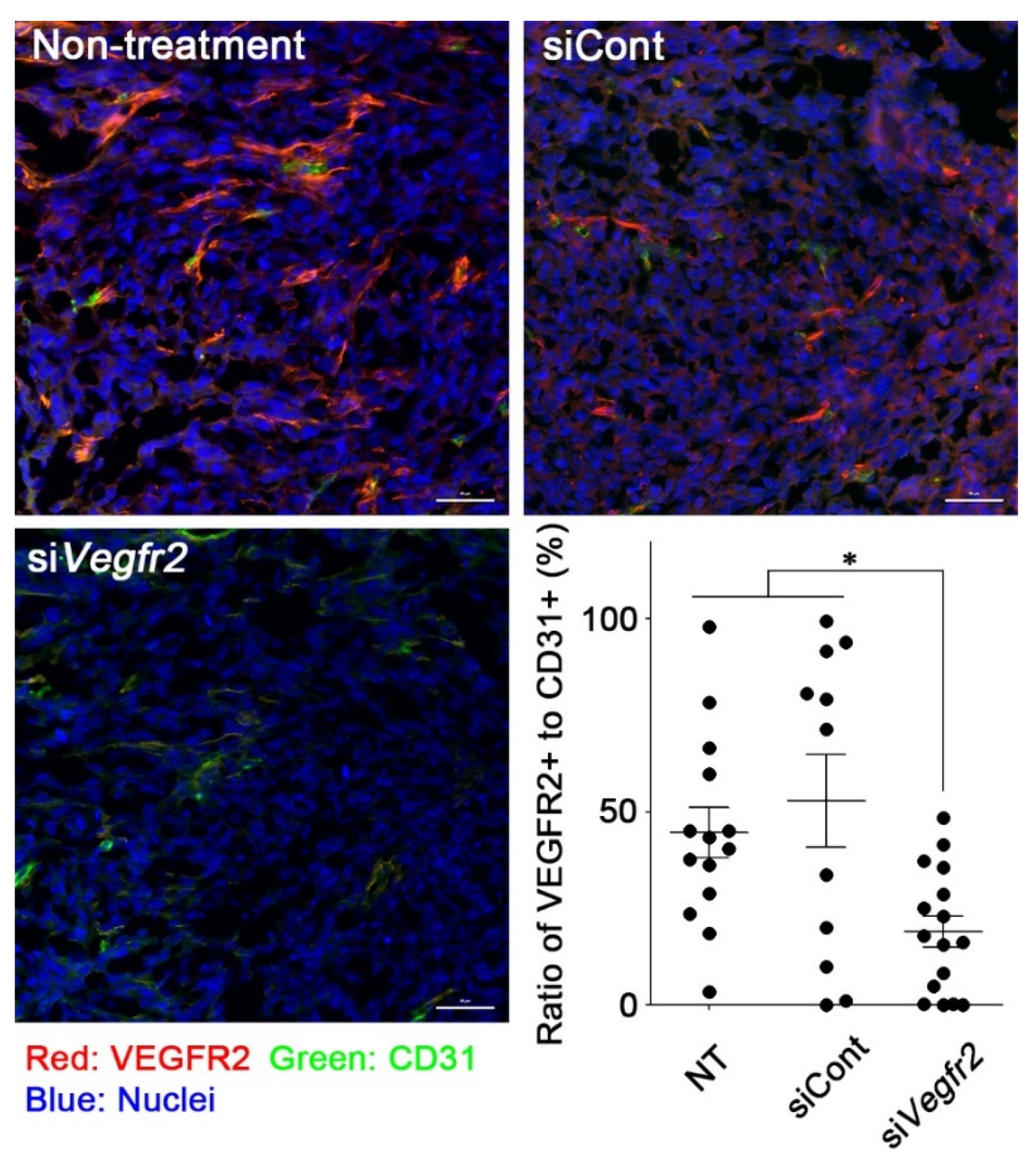
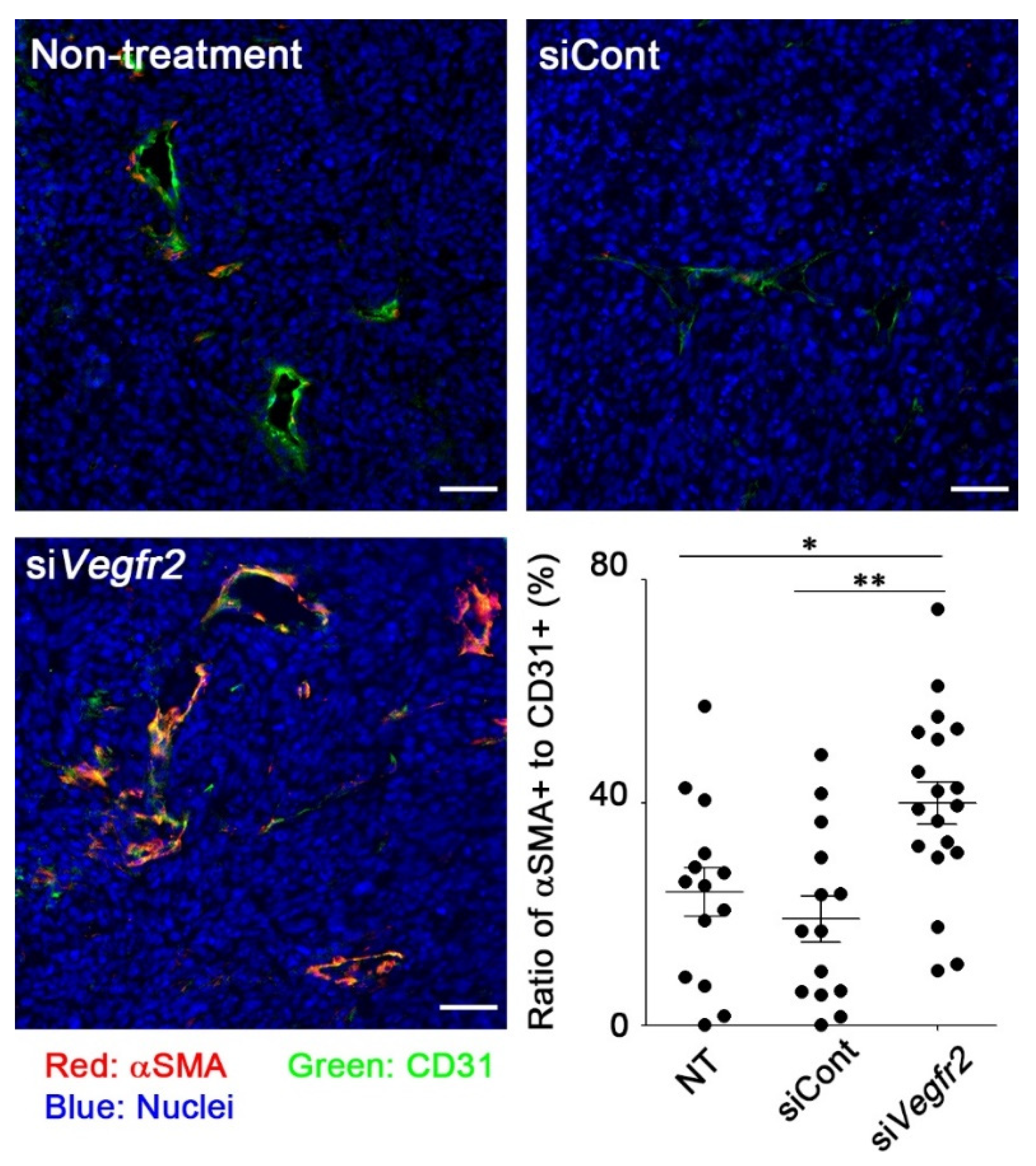
2.3. In Vivo Knockdown of VEGFR2 on TECs and Vascular Normalization
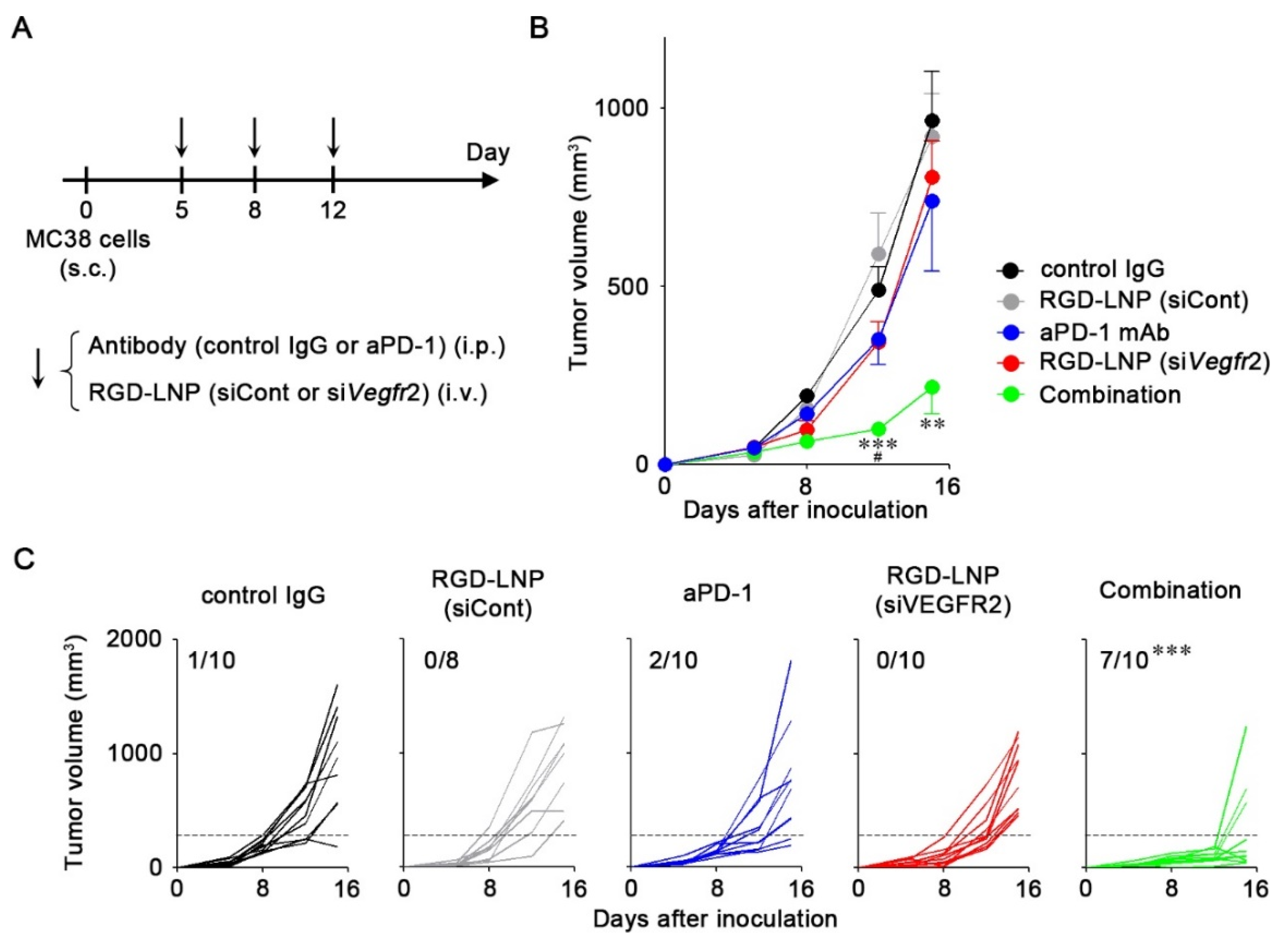
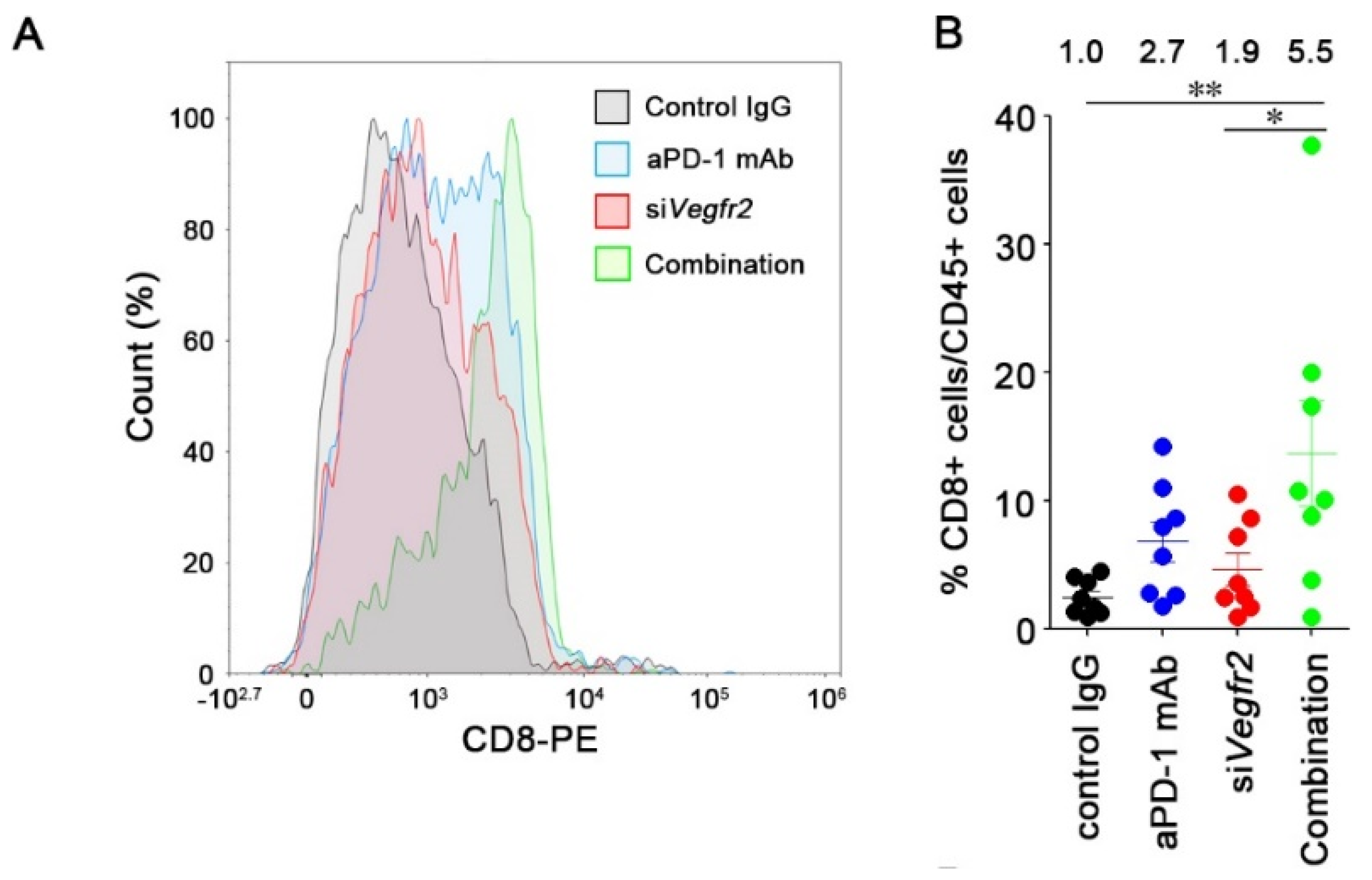
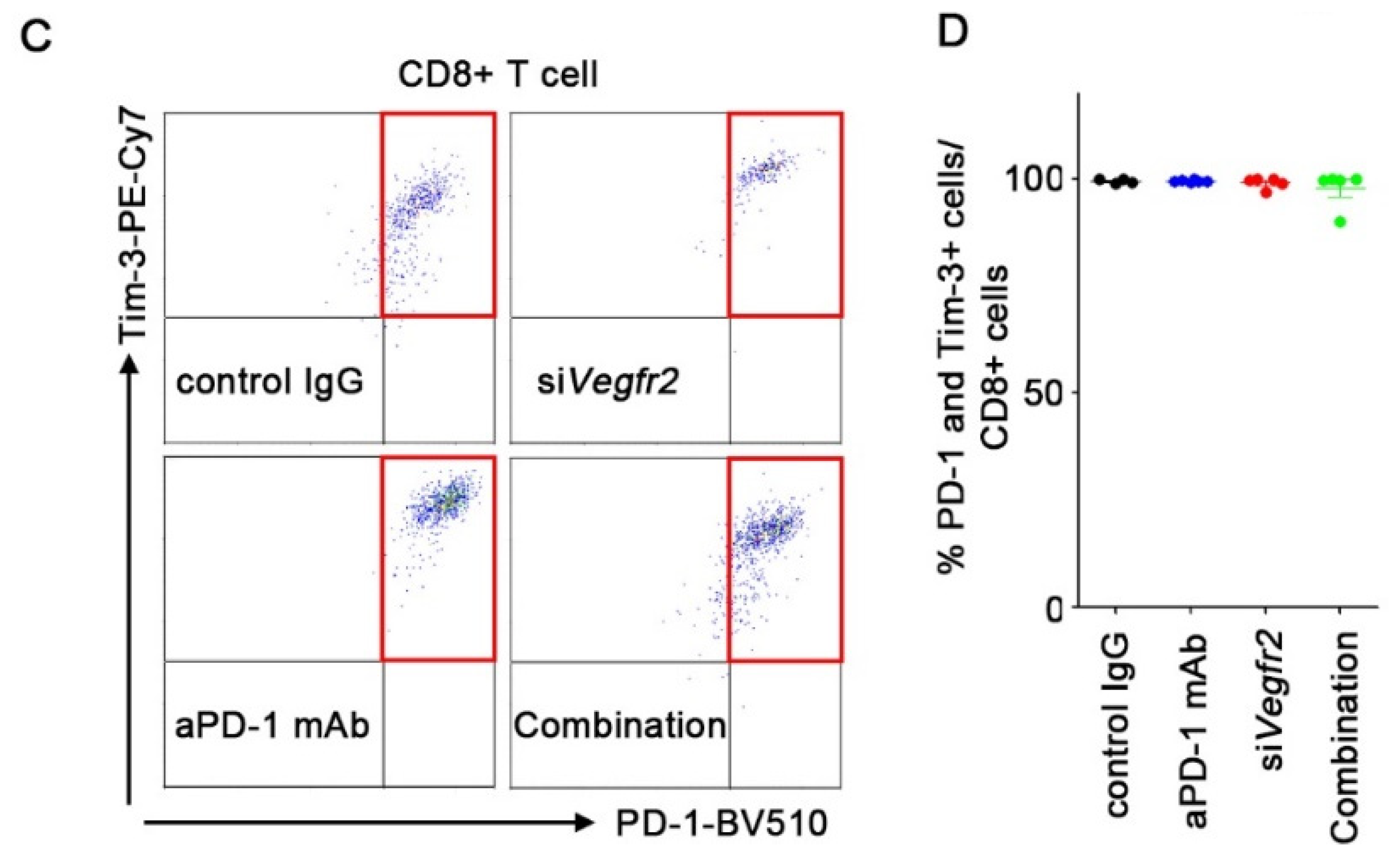
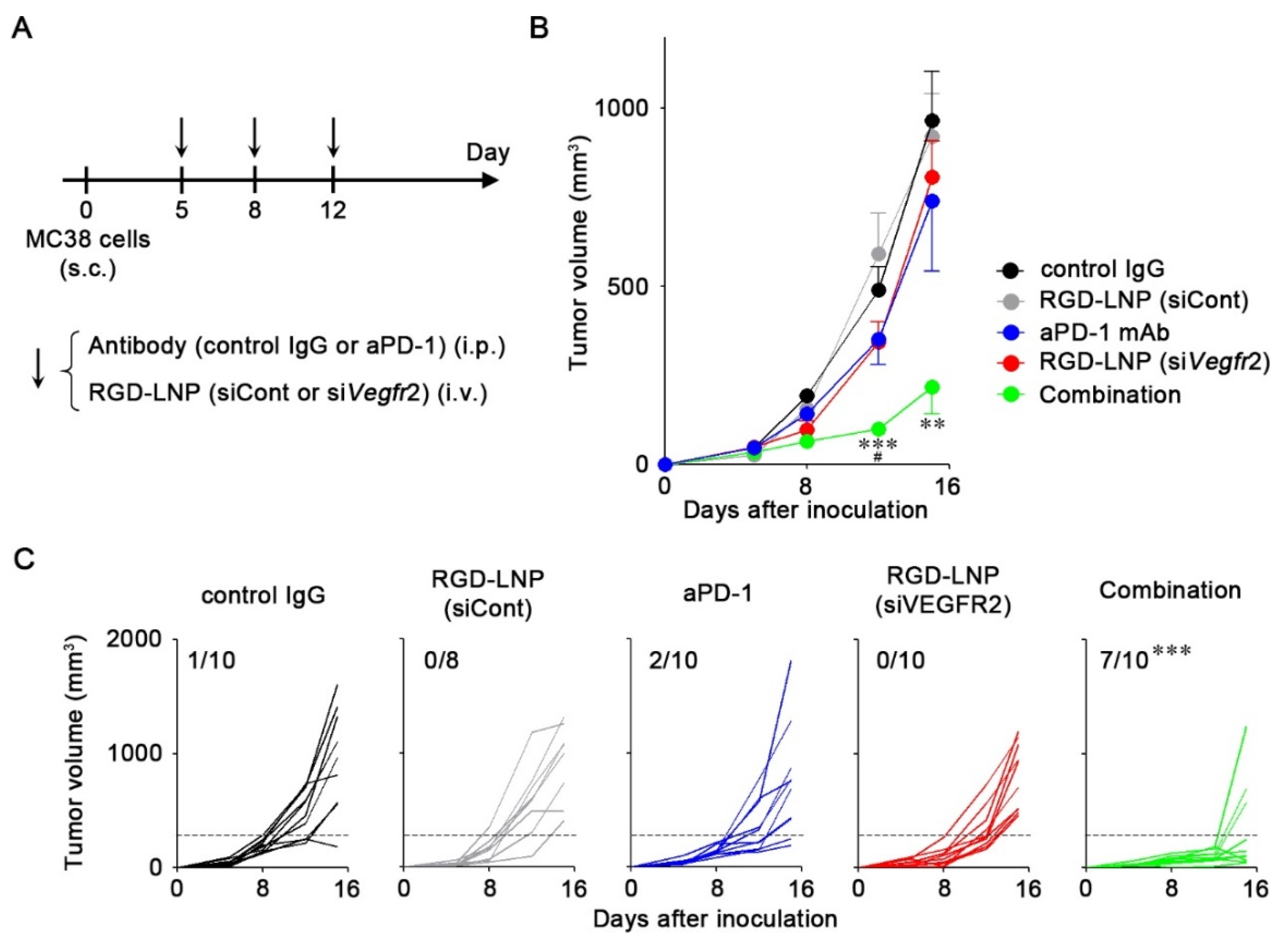
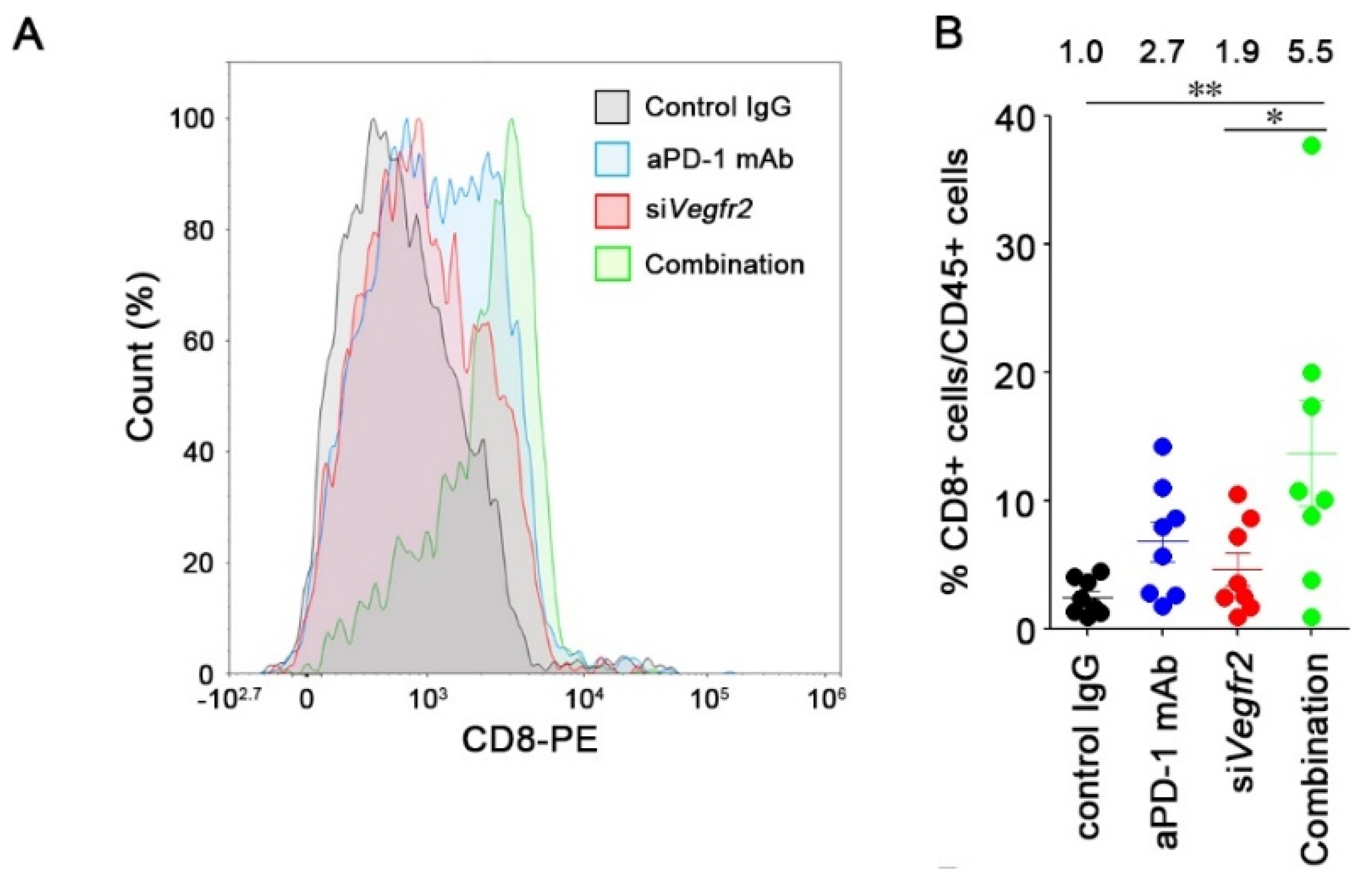
2.4. In Vivo Anti-Tumor Efficacy
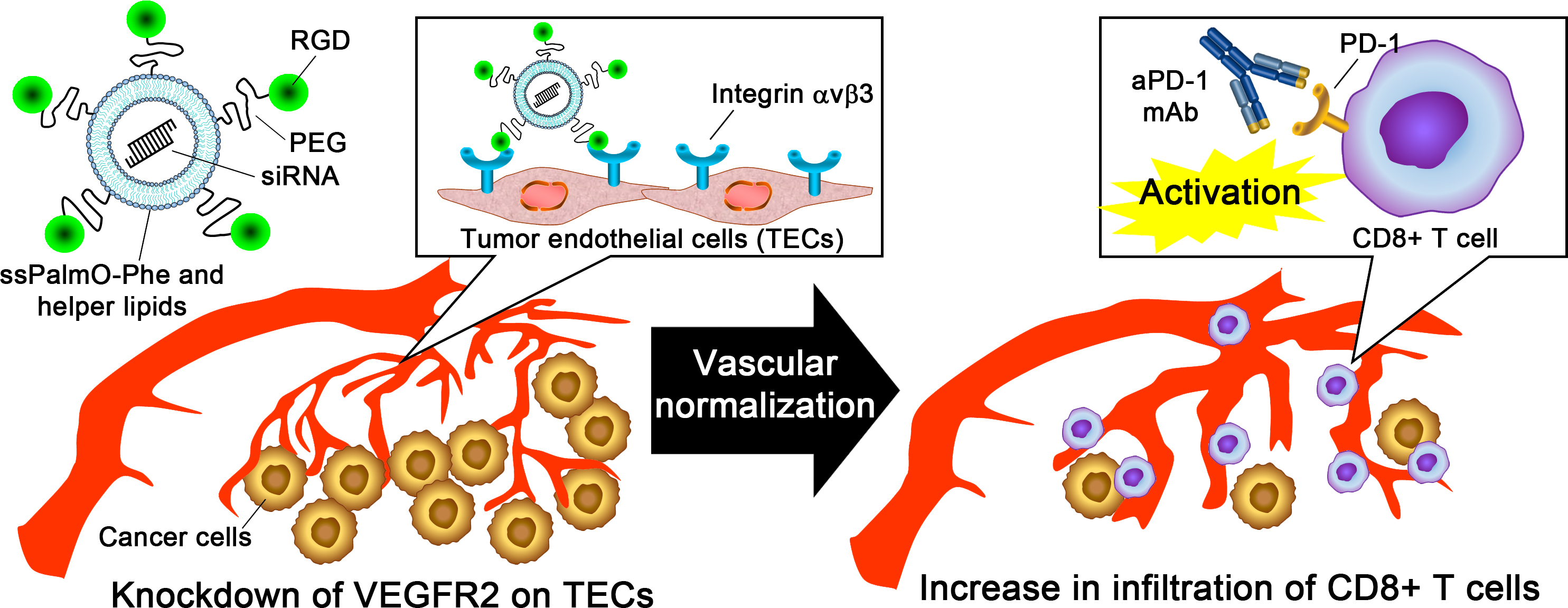
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Preparation and Characterization of LNPs
4.4. Flow Cytometry
4.5. Cellular Uptake of PEG-LNP and RGD-LNP
4.6. In Vitro Knockdown Studies
4.7. Tumor Inoculation
4.8. Immunofluorescence Analysis for In Vivo Knockdown of Vegfr2
4.9. Tumor Inoculation
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor Mutational Burden and Response Rate to PD-1 Inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef]
- Yi, M.; Qin, S.; Zhao, W.; Yu, S.; Chu, Q.; Wu, K. The role of neoantigen in immune checkpoint blockade therapy. Exp. Hematol. Oncol. 2018, 7, 28. [Google Scholar] [CrossRef]
- Fukumura, D.; Kloepper, J.; Amoozgar, Z.; Duda, D.G.; Jain, R.K. Enhancing cancer immunotherapy using antiangiogenics: Opportunities and challenges. Nat. Rev. Clin. Oncol. 2018, 15, 325–340. [Google Scholar] [CrossRef] [PubMed]
- Ramjiawan, R.R.; Griffioen, A.W.; Duda, D.G. Anti-angiogenesis for cancer revisited: Is there a role for combinations with immunotherapy? Angiogenesis 2017, 20, 185–204. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Molecular regulation of vessel maturation. Nat. Med. 2003, 9, 685–693. [Google Scholar] [CrossRef]
- Cooke, V.G.; LeBleu, V.S.; Keskin, D.; Khan, Z.; O’Connell, J.T.; Teng, Y.; Duncan, M.B.; Xie, L.; Maeda, G.; Vong, S.; et al. Pericyte depletion results in hypoxia-associated epithelial-to-mesenchymal transition and metastasis mediated by met signaling pathway. Cancer Cell 2012, 21, 66–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcon, B.L.; O’Clair, B.; McClure, D.; Evans, G.F.; Stewart, J.; Swearingen, M.L.; Chen, Y.; Allard, K.; Lee, L.N.; Neote, K.; et al. Development and characterization of a high-throughput in vitro cord formation model insensitive to VEGF inhibition. J. Hematol. Oncol. 2013, 6, 31. [Google Scholar] [CrossRef] [Green Version]
- Basagiannis, D.; Zografou, S.; Murphy, C.; Fotsis, T.; Morbidelli, L.; Ziche, M.; Bleck, C.; Mercer, J.; Christoforidis, S. VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J. Cell Sci. 2016, 129, 4091–4104. [Google Scholar] [CrossRef] [Green Version]
- Keating, G.M. Bevacizumab: A review of its use in advanced cancer. Drugs 2014, 74, 1891–1925. [Google Scholar] [CrossRef]
- Vasudev, N.S.; Reynolds, A.R. Anti-angiogenic therapy for cancer: Current progress, unresolved questions and future directions. Angiogenesis 2014, 17, 471–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kibria, G.; Hatakeyama, H.; Ohga, N.; Hida, K.; Harashima, H. The effect of liposomal size on the targeted delivery of doxorubicin to Integrin alphavbeta3-expressing tumor endothelial cells. Biomaterials 2013, 34, 5617–5627. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Hyodo, M.; Akita, H.; Ohga, N.; Hida, K.; Harashima, H. RNAi-mediated gene knockdown and anti-angiogenic therapy of RCCs using a cyclic RGD-modified liposomal-siRNA system. J. Control. Release 2014, 173, 110–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, S.; Kato, A.; Sakurai, Y.; Hada, T.; Harashima, H. Modality of tumor endothelial VEGFR2 silencing-mediated improvement in intratumoral distribution of lipid nanoparticles. J. Control. Release 2017, 251, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Hada, T.; Kato, A.; Hagino, Y.; Mizumura, W.; Harashima, H. Effective Therapy Using a Liposomal siRNA that Targets the Tumor Vasculature in a Model Murine Breast Cancer with Lung Metastasis. Mol. Ther. Oncolytics 2018, 11, 102–108. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Mizumura, W.; Ito, K.; Iwasaki, K.; Katoh, T.; Goto, Y.; Suga, H.; Harashima, H. Improved Stability of siRNA-Loaded Lipid Nanoparticles Prepared with a PEG-Monoacyl Fatty Acid Facilitates Ligand-Mediated siRNA Delivery. Mol. Pharm. 2020, 17, 1397–1404. [Google Scholar] [CrossRef]
- Akita, H.; Ishiba, R.; Hatakeyama, H.; Tanaka, H.; Sato, Y.; Tange, K.; Arai, M.; Kubo, K.; Harashima, H. A neutral envelope-type nanoparticle containing pH-responsive and SS-cleavable lipid-like material as a carrier for plasmid DNA. Adv. Healthc. Mater. 2013, 2, 1120–1125. [Google Scholar] [CrossRef]
- Tanaka, H.; Nakatani, T.; Furihata, T.; Tange, K.; Nakai, Y.; Yoshioka, H.; Harashima, H.; Akita, H. In Vivo Introduction of mRNA Encapsulated in Lipid Nanoparticles to Brain Neuronal Cells and Astrocytes via Intracerebroventricular Administration. Mol. Pharm. 2018, 15, 2060–2067. [Google Scholar] [CrossRef]
- Tanaka, H.; Watanabe, A.; Konishi, M.; Nakai, Y.; Yoshioka, H.; Ohkawara, T.; Takeda, H.; Harashima, H.; Akita, H. The delivery of mRNA to colon inflammatory lesions by lipid-nano-particles containing environmentally-sensitive lipid-like materials with oleic acid scaffolds. Heliyon 2018, 4, e00959. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, H.; Takahashi, T.; Konishi, M.; Takata, N.; Gomi, M.; Shirane, D.; Miyama, R.; Hagiwara, S.; Yamasaki, Y.; Sakurai, Y.; et al. Self-degradable Lipid-like Materials based on “Hydrolysis accelerated by the intra-Particle Enrichment of Reactant (HyPER)” for Messenger RNA Delivery. Adv. Funct. Mater. 2020, 30, 1910575. [Google Scholar] [CrossRef]
- Wu, J.; Tracey, L.; Davidoff, A.M. Assessing interactions for fixed-dose drug combinations in tumor xenograft studies. J. Biopharm. Stat. 2012, 22, 535–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotte, A. Combination of CTLA-4 and PD-1 blockers for treatment of cancer. J. Exp. Clin. Cancer Res. 2019, 38, 255. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, S.; Sho, M.; Yamato, I.; Yoshiji, H.; Wakatsuki, K.; Nishiwada, S.; Yagita, H.; Nakajima, Y. Simultaneous blockade of programmed death 1 and vascular endothelial growth factor receptor 2 (VEGFR2) induces synergistic anti-tumour effect in vivo. Clin. Exp. Immunol. 2013, 172, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Voron, T.; Colussi, O.; Marcheteau, E.; Pernot, S.; Nizard, M.; Pointet, A.L.; Latreche, S.; Bergaya, S.; Benhamouda, N.; Tanchot, C.; et al. VEGF-A modulates expression of inhibitory checkpoints on CD8+ T cells in tumors. J. Exp. Med. 2015, 212, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.T.H.; Xu, P.; Chow, A.; Man, S.; Kruger, J.; Khan, K.A.; Paez-Ribes, M.; Pham, E.; Kerbel, R.S. Pre- and post-operative anti-PD-L1 plus anti-angiogenic therapies in mouse breast or renal cancer models of micro- or macro-metastatic disease. Br. J. Cancer 2019, 120, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Jabouille, A.; Rivera, L.B.; Lodewijckx, I.; Missiaen, R.; Steri, V.; Feyen, K.; Tawney, J.; Hanahan, D.; Michael, I.P.; et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci. Transl. Med. 2017, 9, eaak9679. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Ren, S.; Jiang, T.; Zhu, B.; Li, X.; Zhao, C.; Jia, Y.; Shi, J.; Zhang, L.; Liu, X.; et al. Low-Dose Apatinib Optimizes Tumor Microenvironment and Potentiates Antitumor Effect of PD-1/PD-L1 Blockade in Lung Cancer. Cancer Immunol. Res. 2019, 7, 630–643. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Jia, W.; Deng, H.; Li, G.; Deng, W.; Chen, J.; Kim, B.Y.S.; Jiang, W.; Liu, Q.; et al. Low-Dose Anti-Angiogenic Therapy Sensitizes Breast Cancer to PD-1 Blockade. Clin. Cancer Res. 2020, 26, 1712–1724. [Google Scholar] [CrossRef] [Green Version]
- Shigeta, K.; Datta, M.; Hato, T.; Kitahara, S.; Chen, I.X.; Matsui, A.; Kikuchi, H.; Mamessier, E.; Aoki, S.; Ramijiawan, R.R.; et al. Dual Programmed Death Receptor-1 and Vascular Endothelial Growth Factor Receptor-2 Blockade Promotes Vascular Normalization and Enhances Antitumor Immune Responses in Hepatocellular Carcinoma. Hepatology 2020, 71, 1247–1261. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Bendell, J.C.; Funke, R.; Sznol, M.; Korski, K.; Jones, S.; Hernandez, G.; Mier, J.; He, X.; Hodi, F.S.; et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat. Commun. 2016, 7, 12624. [Google Scholar] [CrossRef] [PubMed]
- Ciciola, P.; Cascetta, P.; Bianco, C.; Formisano, L.; Bianco, R. Combining immune checkpoint inhibitors with anti-angiogenic agents. J. Clin. Med. 2020, 9, 675. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y.; Jia, R.; Yue, C.; Chang, L.; Liu, R.; Zhang, G.; Zhao, C.; Zhang, Y.; Chen, C.; et al. Anti-PD-1 Antibody SHR-1210 Combined with Apatinib for Advanced Hepatocellular Carcinoma, Gastric, or Esophagogastric Junction Cancer: An Open-label, Dose Escalation and Expansion Study. Clin. Cancer Res. 2019, 25, 515–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Arkenau, H.T.; Santana-Davila, R.; Calvo, E.; Paz-Ares, L.; Cassier, P.A.; Bendell, J.; Penel, N.; Krebs, M.G.; Martin-Liberal, J.; et al. Ramucirumab plus pembrolizumab in patients with previously treated advanced non-small-cell lung cancer, gastro-oesophageal cancer, or urothelial carcinomas (JVDF): A multicohort, non-randomised, open-label, phase 1a/b trial. Lancet Oncol. 2019, 20, 1109–1123. [Google Scholar] [CrossRef]
- Makker, V.; Rasco, D.; Vogelzang, N.J.; Brose, M.S.; Cohn, A.L.; Mier, J.; Simone, C.D.; Hyman, D.M.; Stepan, D.E.; Dutcus, C.E.; et al. Lenvatinib plus pembrolizumab in patients with advanced endometrial cancer: An interim analysis of a multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 711–718. [Google Scholar] [CrossRef]
- Sakurai, Y.; Akita, H.; Harashima, H. Targeting tumor endothelial cells with nanoparticles. Int. J. Mol. Sci. 2019, 20, 5819. [Google Scholar] [CrossRef] [Green Version]
- Danhier, F.; Le Breton, A.; Preat, V. RGD-based strategies to target alpha(v) beta(3) integrin in cancer therapy and diagnosis. Mol. Pharm. 2012, 9, 2961–2973. [Google Scholar] [CrossRef]
- Schiffelers, R.M.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Akita, H.; Harashima, H. A multifunctional envelope type nano device (MEND) for gene delivery to tumours based on the EPR effect: A strategy for overcoming the PEG dilemma. Adv. Drug Deliv. Rev. 2011, 63, 152–160. [Google Scholar] [CrossRef]
- Takara, K.; Hatakeyama, H.; Kibria, G.; Ohga, N.; Hida, K.; Harashima, H. Size-controlled, dual-ligand modified liposomes that target the tumor vasculature show promise for use in drug-resistant cancer therapy. J. Control. Release 2012, 162, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Goldstein, A.; Wang, H.; Ching Lo, H.; Sun Kim, I.; Welte, T.; Sheng, K.; Dobrolecki, L.E.; Zhang, X.; Putluri, N.; et al. Mutual regulation of tumour vessel normalization and immunostimulatory reprogramming. Nature 2017, 544, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Sweeny, L.; Prince, A.; Patel, N.; Moore, L.S.; Rosenthal, E.L.; Hughley, B.B.; Warram, J.M. Antiangiogenic antibody improves melanoma detection by fluorescently labeled therapeutic antibodies. Laryngoscope 2016, 126, e387–e395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, R.T.; Boucher, Y.; Kozin, S.V.; Winkler, F.; Hicklin, D.J.; Jain, R.K. Vascular normalization by vascular endothelial growth factor receptor 2 blockade induces a pressure gradient across the vasculature and improves drug penetration in tumors. Cancer Res. 2004, 64, 3731–3736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Brown, J.; Petkova, V.; Liu, F.; Li, L.; Boussiotis, V.A. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci. Signal. 2012, 5, ra46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlman, J.E.; Barnes, C.; Khan, O.; Thiriot, A.; Jhunjunwala, S.; Shaw, T.E.; Xing, Y.; Sager, H.B.; Sahay, G.; Speciner, L.; et al. In vivo endothelial siRNA delivery using polymeric nanoparticles with low molecular weight. Nat. Nanotechnol. 2014, 9, 648–655. [Google Scholar] [CrossRef]
- Sakurai, Y.; Hatakeyama, H.; Akita, H.; Harashima, H. Improvement of doxorubicin efficacy using liposomal anti-polo-like kinase 1 siRNA in human renal cell carcinomas. Mol. Pharm. 2014, 11, 2713–2719. [Google Scholar] [CrossRef]
- Sato, Y.; Hatakeyama, H.; Sakurai, Y.; Hyodo, M.; Akita, H.; Harashima, H. A pH-sensitive cationic lipid facilitates the delivery of liposomal siRNA and gene silencing activity in vitro and in vivo. J. Control. Release 2012, 163, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Judge, A.D.; Robbins, M.; Tavakoli, I.; Levi, J.; Hu, L.; Fronda, A.; Ambegia, E.; McClintock, K.; MacLachlan, I. Confirming the RNAi-mediated mechanism of action of siRNA-based cancer therapeutics in mice. J. Clin. Investig. 2009, 119, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Sakurai, Y.; Hatakeyama, H.; Sato, Y.; Hyodo, M.; Akita, H.; Harashima, H. Gene silencing via RNAi and siRNA quantification in tumor tissue using MEND, a liposomal siRNA delivery system. Mol. Ther. 2013, 21, 1195–1203. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Diameter (nm) | PDI | ξ-Potential (mV) | EE (%) | RR (%) |
|---|---|---|---|---|---|
| PEG-LNP | 157 ± 4 | 0.21 ± 0.03 | −9 ± 4 | 95.2 ± 1.6 | 67.5 ± 1.6 |
| RGD-LNP | 167 ± 6 | 0.22 ± 0.01 | −11 ± 4 | 91.9 ± 2.6 | 58.1 ± 10.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, R.; Sakurai, Y.; Jones, H.S.; Akita, H.; Hisaka, A.; Hatakeyama, H. Silencing of VEGFR2 by RGD-Modified Lipid Nanoparticles Enhanced the Efficacy of Anti-PD-1 Antibody by Accelerating Vascular Normalization and Infiltration of T Cells in Tumors. Cancers 2020, 12, 3630. https://doi.org/10.3390/cancers12123630
Cho R, Sakurai Y, Jones HS, Akita H, Hisaka A, Hatakeyama H. Silencing of VEGFR2 by RGD-Modified Lipid Nanoparticles Enhanced the Efficacy of Anti-PD-1 Antibody by Accelerating Vascular Normalization and Infiltration of T Cells in Tumors. Cancers. 2020; 12(12):3630. https://doi.org/10.3390/cancers12123630
Chicago/Turabian StyleCho, Riki, Yu Sakurai, Haleigh Sakura Jones, Hidetaka Akita, Akihiro Hisaka, and Hiroto Hatakeyama. 2020. "Silencing of VEGFR2 by RGD-Modified Lipid Nanoparticles Enhanced the Efficacy of Anti-PD-1 Antibody by Accelerating Vascular Normalization and Infiltration of T Cells in Tumors" Cancers 12, no. 12: 3630. https://doi.org/10.3390/cancers12123630
APA StyleCho, R., Sakurai, Y., Jones, H. S., Akita, H., Hisaka, A., & Hatakeyama, H. (2020). Silencing of VEGFR2 by RGD-Modified Lipid Nanoparticles Enhanced the Efficacy of Anti-PD-1 Antibody by Accelerating Vascular Normalization and Infiltration of T Cells in Tumors. Cancers, 12(12), 3630. https://doi.org/10.3390/cancers12123630