Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus

 , ,
, ,  and
and 
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Setup of a Cellular System to Dissect Cancer Cell-Resident STING Pathway In Vitro and In Vivo
2.2. STING Restricts the Replicative Potential of HSV-1 in Cancer Cell Lines
2.3. Sting_KO-Dependent Improvements in Oncolytic Viral Replication and Cytotoxicity Do Not Correlate with Tumor Clearance Efficacy In Vivo
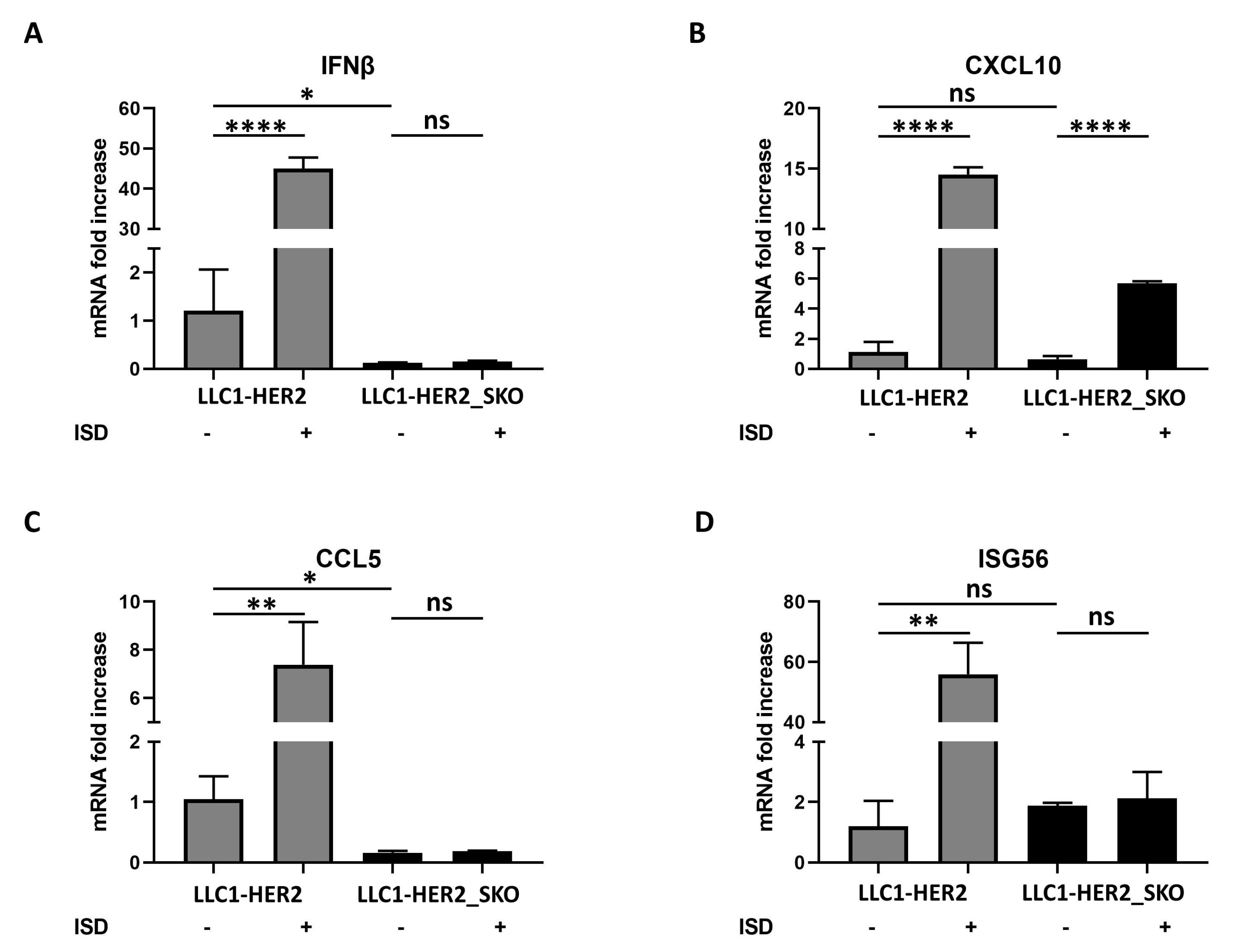
2.4. STING-Deficient Tumor Cells Do Not Trigger Type I IFN Cascade and Show Impaired Immunogenic Cell Death Responses
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Manipulation and Characterization
4.2. Cytotoxicity Assay
4.3. Virus Production, Titration and Real Time PCR Analysis
4.4. In Vivo Studies and Ex Vivo Genome Copies Analysis
4.5. NanoString Data
4.6. In Vitro mRNA Dosage
4.7. Immunogenic Cell Death
4.8. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic Viruses: A New Class of Immunotherapy Drugs. Nat. Rev. Drug Discov. 2015, 14, 642–662, Erratum in 2016, 15, 660. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Dummer, R.; Puzanov, I.; Vander Walde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanksi, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef] [PubMed]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol. Ther. Oncolytics 2019, 13, 93–106. [Google Scholar] [CrossRef]
- Russell, L.; Peng, K.W.; Russell, S.J.; Diaz, R.M. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs 2019, 33, 485–501. [Google Scholar] [CrossRef] [PubMed]
- Twumasi-Boateng, K.; Pettigrew, J.L.; Kwok, Y.Y.E.; Bell, J.C.; Nelson, B.H. Oncolytic Viruses as Engineering Platforms for Combination Immunotherapy. Nat. Rev. Cancer 2018, 18, 419–432. [Google Scholar] [CrossRef]
- Workenhe, S.T.; Simmons, G.; Pol, J.G.; Lichty, B.D.; Halford, W.P.; Mossman, K.L. Immunogenic HSV-Mediated Oncolysis Shapes the Antitumor Immune Response and Contributes to Therapeutic Efficacy. Mol. Ther. 2014, 22, 123–131. [Google Scholar] [CrossRef]
- Bezu, L.; Gomes-De-Silva, L.C.; Dewitte, H.; Breckpot, K.; Fucikova, J.; Spisek, R.; Galluzzi, L.; Kepp, O.; Kroemer, G. Combinatorial Strategies for the Induction of Immunogenic Cell Death. Front. Immunol. 2015, 6, 187. [Google Scholar] [CrossRef]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type I Interferon-Dependent Innate Immunity. Nat. Cell Biol. 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell Intrinsic Immunity Spreads to Bystander Cells via the Intercellular Transfer of cGAMP. Nat. Cell Biol. 2013, 503, 530–534. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Extracellular Vesicles Released by Herpes Simplex Virus 1-Infected Cells Block Virus Replication in Recipient Cells in a STING-Dependent Manner. J. Virol. 2018, 92, e01102-18. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Du, T.; Roizman, B. Cells Infected with Herpes Simplex Virus 1 Export to Uninfected Cells Exosomes Containing STING, Viral MRNAs, and MicroRNAs. Proc. Natl. Acad. Sci. USA 2014, 111, E4991–E4996. [Google Scholar] [CrossRef]
- Ni, G.; Ma, Z.; Damania, B. CGAS and STING: At the Intersection of DNA and RNA Virus-Sensing Networks. PLoS Pathog. 2018, 14, e1007148. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Damania, B. The CGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Stempel, M.; Chan, B.; Brinkmann, M.M. Coevolution Pays Off: Herpesviruses Have the License to Escape the DNA Sensing Pathway. Med. Microbiol. Immunol. 2019, 208, 495–512. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Liu, X.; Ma, Y.; Cao, Y.; He, B. Herpes Simplex Virus 1 γ134.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 92, e01015-18. [Google Scholar] [CrossRef]
- Wang, H.; Hu, S.; Chen, X.; Shi, H.; Chen, C.; Sun, L.; Chen, Z.J. CGAS is Essential for the Antitumor Effect of Immune Checkpoint Blockade. Proc. Natl. Acad. Sci. USA 2017, 114, 1637–1642. [Google Scholar] [CrossRef]
- Woo, S.-R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.K.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic CGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248.e7. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. CGAS in Action: Expanding Roles in Immunity and Inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef]
- Corrales, L.; Glickman, L.H.; McWhirter, S.M.; Kanne, D.B.; Sivick, K.E.; Katibah, G.E.; Woo, S.-R.; Lemmens, E.; Banda, T.; Leong, J.J.; et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015, 11, 1018–1030. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Xia, T.; Capote, A.R.; Betancourt, D.; Barber, G.N. Extrinsic Phagocyte-Dependent STING Signaling Dictates the Immunogenicity of Dying Cells. Cancer Cell 2018, 33, 862–873.e5. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Zhu, Y.; Zheng, T.; Wang, G.; Zhang, M.; Li, J.; Ji, H.; Li, S.; Yang, S.; Xu, D.; et al. An Analysis of the Expression and Association with Immune Cell Infiltration of the cGAS/STING Pathway in Pan-Cancer. Mol. Ther. Nucleic Acids 2019, 14, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.J.; Barber, G.N. Suppression of STING Signaling through Epigenetic Silencing and Missense Mutation Impedes DNA Damage Mediated Cytokine Production. Oncogene 2018, 37, 2037–2051. [Google Scholar] [CrossRef]
- Xia, T.; Konno, H.; Barber, G.N. Recurrent Loss of STING Signaling in Melanoma Correlates with Susceptibility to Viral Oncolysis. Cancer Res. 2016, 76, 6747–6759. [Google Scholar] [CrossRef]
- De Queiroz, N.M.G.P.; Xia, T.; Konno, H.; Barber, G.N. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Mol. Cancer Res. 2018, 17, 974–986. [Google Scholar] [CrossRef]
- Lee, J.M.; Ghonime, M.G.; Cassady, K.A. STING Restricts OHSV Replication and Spread in Resistant MPNSTs but Is Dispensable for Basal IFN-Stimulated Gene Upregulation. Mol. Ther. Oncolytics 2019, 15, 91–100. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Zloza, A.; Rabkin, S.D.; Kaufman, H.L. Oncolytic Virus Immunotherapy Induces Immunogenic Cell Death and Overcomes STING Deficiency in Melanoma. OncoImmunology 2019, 8, e1591875. [Google Scholar] [CrossRef]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a Fully Retargeted Herpes Simplex Virus 1 Recombinant Capable of Entering Cells Solely via Human Epidermal Growth Factor Receptor 2. J. Virol. 2008, 82, 10153–10161. [Google Scholar] [CrossRef]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of Murine Solid Tumor Models as a Defining Feature of In Vivo Behavior and Response to Immunotherapy. J. Immunother. 2013, 36, 477–489. [Google Scholar] [CrossRef]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.-L.; Casiraghi, C.; et al. A Fully-Virulent Retargeted Oncolytic HSV Armed with IL-12 Elicits Local Immunity and Vaccine Therapy Towards Distant Tumors. PLoS Pathog. 2018, 14, e1007209. [Google Scholar] [CrossRef] [PubMed]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and Multi-Cytokine Armed Herpes Virus is a Potent Cancer Endovaccine for Local and Systemic Anti-Tumor Treatment in Combination with Anti-PD1. Mol. Ther. Oncolytics 2020. [Google Scholar] [CrossRef]
- Sasso, E.; Latino, D.; Froechlich, G.; Succoio, M.; Passariello, M.; De Lorenzo, C.; Nicosia, A.; Zambrano, N. A Long Non-Coding SINEUP RNA Boosts Semi-Stable Production of Fully Human Monoclonal Antibodies in HEK293E Cells. mAbs 2018, 10, 730–737. [Google Scholar] [CrossRef]
- Chamoto, K.; Chowdhury, P.S.; Kumar, A.; Sonomura, K.; Matsuda, F.; Fagarasan, S.; Honjo, T. Mitochondrial Activation Chemicals Synergize with Surface Receptor PD-1 Blockade for T Cell-Dependent Antitumor Activity. Proc. Natl. Acad. Sci. USA 2017, 114, E761–E770. [Google Scholar] [CrossRef] [PubMed]
- Sasso, E.; Froechlich, G.; Cotugno, G.; D’Alise, A.M.; Gentile, C.; Bignone, V.; De Lucia, M.; Petrovic, B.; Campadelli-Fiume, G.; Scarselli, E.; et al. Replicative Conditioning of Herpes Simplex Type 1 Virus by Survivin Promoter, Combined to ERBB2 Retargeting, Improves Tumour Cell-Restricted Oncolysis. Sci. Rep. 2020, 10, 4307–4312. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Ricca, J.M.; Sadekova, S.; Oseledchyk, A.; Yu, Y.; Blumenschein, W.M.; Wong, J.; Gigoux, M.; Merghoub, T.; Wolchok, J. PD-L1 in Tumor Microenvironment Mediates Resistance to Oncolytic Immunotherapy. J. Clin. Investig. 2018, 128, 1413–1428. [Google Scholar] [CrossRef] [PubMed]
- Nirmal, A.J.; Regan, T.; Shih, B.; Hume, D.A.; Sims, A.H.; Freeman, T.C. Immune Cell Gene Signatures for Profiling the Microenvironment of Solid Tumors. Cancer Immunol. Res. 2018, 6, 1388–1400. [Google Scholar] [CrossRef]
- Pellecchia, A.; Pescucci, C.; De Lorenzo, E.; Luceri, C.; Passaro, N.; Sica, M.; Notaro, R.; De Angioletti, M. Overexpression of ETV4 is Oncogenic in Prostate Cells through Promotion of Both Cell Proliferation and Epithelial to Mesenchymal Transition. Oncogenesis 2012, 1, e20. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiao, H.; James, S.J. DUSP4 Regulates STING- and RIG-I-Mediated Signalling in Response to Virus Infection. J. Immunol. 2018, 200, 169.21. [Google Scholar]
- Schilling, M.; Bulli, L.; Weigang, S.; Graf, L.; Naumann, S.; Patzina, C.; Wagner, V.; Bauersfeld, L.; Goujon, C.; Hengel, H.; et al. Human MxB Protein Is a Pan-Herpesvirus Restriction Factor. J. Virol. 2018, 92, e01056-18. [Google Scholar] [CrossRef]
- Chee, A.V.; Roizman, B. Herpes Simplex Virus 1 Gene Products Occlude the Interferon Signaling Pathway at Multiple Sites. J. Virol. 2004, 78, 4185–4196. [Google Scholar] [CrossRef] [PubMed]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes Simplex Virus 1 Counteracts Tetherin Restriction via Its Virion Host Shutoff Activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Xie, X.; Pedro, J.M.B.-S.; Zitvogel, L.; White, E.; Kroemer, G. An Autophagy-Dependent Anticancer Immune Response Determines the Efficacy of Melanoma Chemotherapy. OncoImmunology 2014, 3, e944047. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients with Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef]
- Vile, R.G. The Immune System in Oncolytic Immunovirotherapy: Gospel, Schism and Heresy. Mol. Ther. 2018, 26, 942–946. [Google Scholar] [CrossRef]
- Davola, M.E.; Mossman, K.L. Oncolytic Viruses: How “Lytic” Must They be for Therapeutic Efficacy? Oncoimmunology 2019, 8, e1581528. [Google Scholar] [CrossRef]
- Dai, P.; Wang, W.; Yang, N.; Serna-Tamayo, C.; Ricca, J.M.; Zamarin, D.; Shuman, S.; Merghoub, T.; Wolchok, J.D.; Deng, L. Intratumoral Delivery of Inactivated Modified Vaccinia Virus Ankara (IMVA) Induces Systemic Antitumor Immunity via STING and Batf3-Dependent Dendritic Cells. Sci. Immunol. 2017, 2, eaal1713. [Google Scholar] [CrossRef]
- Lee, J.; Ghonime, M.G.; Wang, R.; Cassady, K.A. The Antiviral Apparatus: STING and Oncolytic Virus Restriction. Mol. Ther. Oncolytics 2019, 13, 7–13. [Google Scholar] [CrossRef]
- Sasso, E.; Vitale, M.; Monteleone, F.; Boffo, F.L.; Santoriello, M.; Sarnataro, D.; Garbi, C.; Sabatella, M.; Crifo, B.; Paolella, L.A.; et al. Binding of Carbonic Anhydrase IX to 45S RDNA Genes Is Prevented by Exportin-1 in Hypoxic Cells. Bio. Med. Res. Int. 2015, 2015, 1–10. [Google Scholar] [CrossRef]
- Sasso, E.; D’Avino, C.; Passariello, M.; D’Alise, A.M.; Siciliano, D.; Esposito, M.L.; Froechlich, G.; Cortese, R.; Scarselli, E.; Zambrano, N.; et al. Massive Parallel Screening of Phage Libraries for the Generation of Repertoires of Human Immunomodulatory Monoclonal Antibodies. mAbs 2018, 10, 1060–1072. [Google Scholar] [CrossRef]
- Falahat, R.; Perez-Villarroel, P.; Mailloux, A.W.; Zhu, G.; Pilon-Thomas, S.; Barber, G.N.; Mulé, J.J. STING Signaling in Melanoma Cells Shapes Antigenicity and Can Promote Antitumor T-cell Activity. Cancer Immunol. Res. 2019, 7, 1837–1848. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Oligonucleotide Sequences |
|---|---|
| Taqman_DNApol_Fwd | 5′-catcaccgacccggagagggac-3′ |
| Taqman_DNApol_Rev | 5′-gggccaggcgcttgttggtgta-3′ |
| Taqman Probe | FAM-ccgccgaactgagcagacacccgcgc-Tamra |
| CCL5_RT_Fwd | 5′-cctcaccatatggctcggac-3′ |
| CCL5_RT_Rev | 5′tcttctctgggttggcacac-3′ |
| CXCL10_RT_ Fwd | 5′-gccgtcattttctgcctcatc-3′ |
| CXCL10_RT_ Rev | 5′-taggctcgcagggatgatttc-3′ |
| IFIT/ISG56_RT_Fwd | 5′-tccgtaggaaacatcgcgtag-3′ |
| IFIT/ISG56_RT_Rev | 5′-tcttgcacattgtcctgcct-3′ |
| IFNβ1_RT_Fwd | 5′-atttctccagcactgggtgg-3′ |
| IFNβ1_RT_Rev | 5′-aggtacctttgcaccctcca-3′ |
| CAS9_Fwd | 5′-gctctttgatgccctcttcg-3′ |
| CAS9_Rev | 5′-gctgaccctgacactgtttg-3′ |
| GFP_Fwd | 5′-cacgacttcttcaagtccgc-3′ |
| GFP_Rev | 5′-ggtgttctgctggtagtggt-3′ |
| GuideRNA_1 | 5′-gaggtcaccgctccaaatat-3′ |
| GuideRNA_2 | 5′-cacctagcctcgcacgaact-3′ |
| GuideRNA_3 | 5′-gggatgccccatccactgta-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; De Lucia, M.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers 2020, 12, 3407. https://doi.org/10.3390/cancers12113407
Froechlich G, Caiazza C, Gentile C, D’Alise AM, De Lucia M, Langone F, Leoni G, Cotugno G, Scisciola V, Nicosia A, et al. Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers. 2020; 12(11):3407. https://doi.org/10.3390/cancers12113407
Chicago/Turabian StyleFroechlich, Guendalina, Carmen Caiazza, Chiara Gentile, Anna Morena D’Alise, Maria De Lucia, Francesca Langone, Guido Leoni, Gabriella Cotugno, Vittorio Scisciola, Alfredo Nicosia, and et al. 2020. "Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus" Cancers 12, no. 11: 3407. https://doi.org/10.3390/cancers12113407
APA StyleFroechlich, G., Caiazza, C., Gentile, C., D’Alise, A. M., De Lucia, M., Langone, F., Leoni, G., Cotugno, G., Scisciola, V., Nicosia, A., Scarselli, E., Mallardo, M., Sasso, E., & Zambrano, N. (2020). Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers, 12(11), 3407. https://doi.org/10.3390/cancers12113407