Epigenetics in Breast Cancer Therapy—New Strategies and Future Nanomedicine Perspectives

 , , , , , ,
, , , , , ,  , and
, and 
Abstract
Simple Summary
Abstract

1. Introduction
2. Molecular Pathology of Breast Cancer
2.1. Role of Epigenetics in BC Pathogenesis
2.1.1. DNA Methylation
2.1.2. Histone Modifications
2.1.3. Epigenetic Regulation by Non-Coding RNA
2.2. Epigenetics in BC Progression
3. Breast Cancer Therapeutic Opportunities
3.1. The Biomarker-Directed Approach in BC Treatment
3.2. Precision Medicine Concept
- Oncotype DX provides prognostic information in terms of 10-year distant recurrence. It predicts the likelihood of adjuvant chemotherapy benefit in ER+ BC patients, based on the expression of a panel of 21 genes (16 cancer-related and five reference genes) [102].
- Breast Cancer Index assesses the expression of 7 genes to predict the benefit from extended, adjuvant, endocrine therapy (Tamoxifen) in HR+ patients. It is a gene expression signature comprising two functional biomarker panels, the molecular grade index (MGI) and the two-gene ratio HOXB13/IL17BR (H/I), that evaluate tumor proliferation and estrogen signaling, respectively [103]. MGI is a gene expression assay, measuring the expression of five genes (BUB1B, CENPA, NEK2, RACGAP1, RRM2) related to histological grade and tumor progression, which recapitulates tumor grade and can predict the clinical outcome with high performance [104].
- EndoPredict (Myriad Genetics, Inc., Salt Lake City, UT, USA) is a genomic test for people newly diagnosed with early-stage, ER+, HER2-negative BC (node-negative). It assesses the expression of 12 genes (8 target genes, 3 normalization genes, and 1 control gene) to predict response to chemotherapy [105].
- MammaPrint (Agendia, Irvine, CA, USA) is a 70-gene signature test that predicts the clinical outcome/response to chemotherapy in ER+ early-stage BC [106].
- Prosigna Breast Cancer Prognostic Gene Signature Assay (Nanostring, Seattle, WA, USA), formerly PAM50, assesses Tamoxifen response for HR+ BC patients based on the expression of 58 genes after 5 years of hormonal therapy treatment in postmenopausal women [107].
3.3. Potential of Epigenetic Therapy
Epigenetic Therapy in BC
4. Nanomedicine as a Tool to Overcome the Current Limitations of Epigenetic Therapy
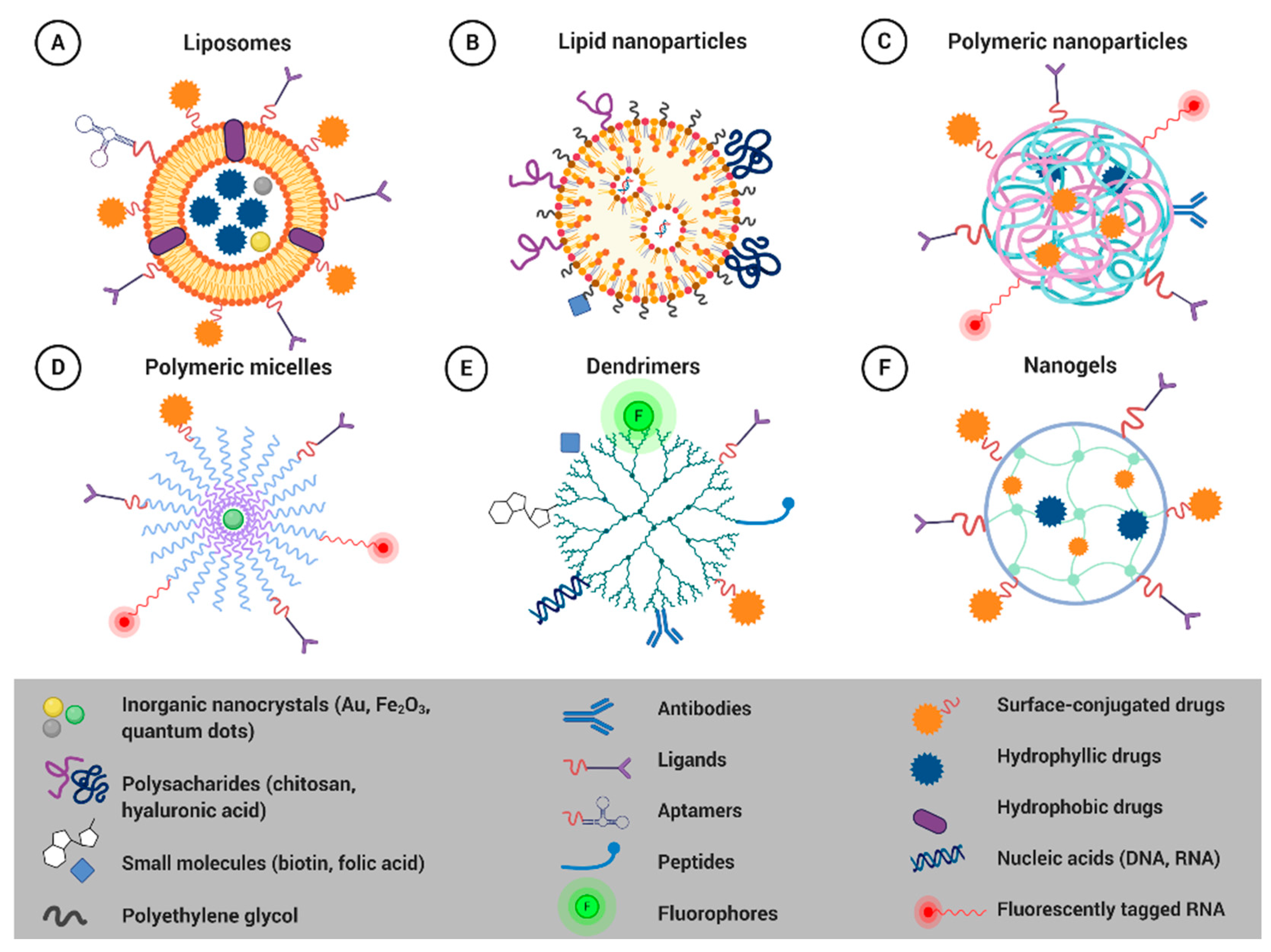
4.1. Smart Nanoformulations for Drug Delivery Applications
4.1.1. Liposomes
4.1.2. Solid Lipid Nanoparticles
4.1.3. Polymeric Nanoparticles
4.1.4. Polymeric Micelles
4.1.5. Dendrimers
4.1.6. Nanogels
4.2. Nanoplatforms for Combination Therapy
5. Biosafety of Soft Nanocarriers
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Dworkin, A.M.; Huang, T.H.M.; Toland, A.E. Epigenetic alterations in the breast: Implications for breast cancer detection, prognosis and treatment. Semin. Cancer Biol. 2009, 19, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Roche, J. The Epithelial-to-Mesenchymal Transition in Cancer. Cancers 2018, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Fang, J. Epigenetic regulation of epithelial-mesenchymal transition. Cell. Mol. Life Sci. 2016, 73, 4493–4515. [Google Scholar] [CrossRef] [PubMed]
- Moo, T.A.; Sanford, R.; Dang, C.; Morrow, M. Overview of Breast Cancer Therapy. PET Clin. 2018, 13, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Li, S.; Wang, N.; Feng, Y. Epigenetic regulation in human cancer: The potential role of epi-drug in cancer therapy. Mol. Cancer 2020, 19, 79. [Google Scholar] [CrossRef] [PubMed]
- Morel, D.; Jeffery, D.; Aspeslagh, S.; Almouzni, G.; Postel-Vinay, S. Combining epigenetic drugs with other therapies for solid tumours—Past lessons and future promise. Nat. Rev. Clin. Oncol. 2020, 17, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Magro, M.; Venerando, A.; Macone, A.; Canettieri, G.; Agostinelli, E.; Vianello, F. Nanotechnology-Based Strategies to Develop New Anticancer Therapies. Biomolecules 2020, 10, 735. [Google Scholar] [CrossRef] [PubMed]
- Jain, V.; Kumar, H.; Anod, H.V.; Chand, P.; Gupta, N.V.; Dey, S.; Kesharwani, S.S. A review of nanotechnology-based approaches for breast cancer and triple-negative breast cancer. J. Control. Release 2020, 326, 628–647. [Google Scholar] [CrossRef] [PubMed]
- Thakur, V.; Kutty, R.V. Recent advances in nanotheranostics for triple negative breast cancer treatment. J. Exp. Clin. Cancer Res. 2019, 38, 430. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.K.; Monsky, W.L.; Yuan, F.; Roberts, W.G.; Griffith, L.; Torchilin, V.P.; Jain, R.K. Regulation of transport pathways in tumor vessels: Role of tumor type and microenvironment. Proc. Natl. Acad. Sci. USA 1998, 95, 4607–4612. [Google Scholar] [CrossRef] [PubMed]
- Ernsting, M.J.; Murakami, M.; Roy, A.; Li, S.D. Factors controlling the pharmacokinetics, biodistribution and intratumoral penetration of nanoparticles. J. Control. Release 2013, 172, 782–794. [Google Scholar] [CrossRef]
- Navya, P.N.; Kaphle, A.; Srinivas, S.P.; Bhargava, S.K.; Rotello, V.M.; Daima, H.K. Current trends and challenges in cancer management and therapy using designer nanomaterials. Nano Converg. 2019, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Tang, M.; Leung, E.; Svirskis, D.; Shelling, A.; Wu, Z. Dual or multiple drug loaded nanoparticles to target breast cancer stem cells. RSC Adv. 2020, 10, 19089–19105. [Google Scholar] [CrossRef]
- Harbeck, N.; Penault-Llorca, F.; Cortes, J.; Gnant, M.; Houssami, N.; Poortmans, P.; Ruddy, K.; Tsang, J.; Cardoso, F. Breast cancer. Nat. Rev. Dis. Primers 2019, 5, 66. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.; Krapcho, M. SEER Cancer Statistics Review, 1975–2014, Based on November 2016 SEER Data Submission; National Cancer Institute: Bethesda, MD, USA, 2017. [Google Scholar]
- Goncalves, H., Jr.; Guerra, M.R.; Duarte Cintra, J.R.; Fayer, V.A.; Brum, I.V.; Bustamante Teixeira, M.T. Survival Study of Triple-Negative and Non-Triple-Negative Breast Cancer in a Brazilian Cohort. Clin. Med. Insights Oncol. 2018, 12. [Google Scholar] [CrossRef]
- Hinohara, K.; Polyak, K. Intratumoral heterogeneity: More than just mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Bentires-Alj, M. Breast Tumor Heterogeneity: Source of Fitness, Hurdle for Therapy. Mol. Cell 2015, 60, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Turashvili, G.; Brogi, E. Tumor Heterogeneity in Breast Cancer. Front. Med. 2017, 4, 227. [Google Scholar] [CrossRef]
- Taylor, A.; Brady, A.F.; Frayling, I.M.; Hanson, H.; Tischkowitz, M.; Turnbull, C.; Side, L. Consensus for genes to be included on cancer panel tests offered by UK genetics services: Guidelines of the UK Cancer Genetics Group. J. Med. Genet. 2018, 55, 372–377. [Google Scholar] [CrossRef]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Desmedt, C.; Yates, L.; Kulka, J. Catalog of genetic progression of human cancers: Breast cancer. Cancer Metastasis Rev. 2016, 35, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, R.; Gupta, S. Epigenetic modifications in cancer. Clin. Genet. 2012, 81, 303–311. [Google Scholar] [CrossRef]
- Veeck, J.; Esteller, M. Breast cancer epigenetics: From DNA methylation to microRNAs. J. Mammary Gland Biol. Neoplasia 2010, 15, 5–17. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA methylation and its basic function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef]
- Laird, P.W. The power and the promise of DNA methylation markers. Nat. Rev. Cancer 2003, 3, 253–266. [Google Scholar] [CrossRef]
- Guo, M.; Peng, Y.; Gao, A.; Du, C.; Herman, J.G. Epigenetic heterogeneity in cancer. Biomark Res. 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- De Almeida, B.P.; Apolónio, J.D.; Binnie, A.; Castelo-Branco, P. Roadmap of DNA methylation in breast cancer identifies novel prognostic biomarkers. BMC Cancer 2019, 19, 219. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Hinshelwood, R.A.; Clark, S.J. Breast cancer epigenetics: Normal human mammary epithelial cells as a model system. J. Mol. Med. 2008, 86, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, J.; Rønneberg, J.A.; Tost, J.; Kristensen, V. The epigenetics of breast cancer. Mol. Oncol. 2010, 4, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Pasculli, B.; Barbano, R.; Parrella, P. Epigenetics of breast cancer: Biology and clinical implication in the era of precision medicine. Semin. Cancer Biol. 2018, 51, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, X.; Clark, E.; Mulcahey, M.; Huang, S.; Shi, Y.G. TET1 is a DNA-binding protein that modulates DNA methylation and gene transcription via hydroxylation of 5-methylcytosine. Cell Res. 2010, 20, 1390–1393. [Google Scholar] [CrossRef]
- Huang, Y.; Rao, A. Connections between TET proteins and aberrant DNA modification in cancer. Trends Genet. 2014, 30, 464–474. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Chervona, Y.; Costa, M. Histone modifications and cancer: Biomarkers of prognosis? Am. J. Cancer Res. 2012, 2, 589–597. [Google Scholar] [PubMed]
- Kurdistani, S.K. Histone modifications as markers of cancer prognosis: A cellular view. Br. J. Cancer 2007, 97, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Stoccoro, A.; Karlsson, H.L.; Coppedè, F.; Migliore, L. Epigenetic effects of nano-sized materials. Toxicology 2013, 313, 3–14. [Google Scholar] [CrossRef]
- Horn, P.J.; Peterson, C.L. Heterochromatin assembly: A new twist on an old model. Chromosome Res. 2006, 14, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Herman, J.G.; Guo, M. Epigenome-based personalized medicine in human cancer. Epigenomics 2016, 8, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef]
- Kapoor-Vazirani, P.; Kagey, J.D.; Powell, D.R.; Vertino, P.M. Role of hMOF-dependent histone H4 lysine 16 acetylation in the maintenance of TMS1/ASC gene activity. Cancer Res. 2008, 68, 6810–6821. [Google Scholar] [CrossRef]
- Karsli-Ceppioglu, S.; Dagdemir, A.; Judes, G.; Ngollo, M.; Penault-Llorca, F.; Pajon, A.; Bignon, Y.J.; Bernard-Gallon, D. Epigenetic mechanisms of breast cancer: An update of the current knowledge. Epigenomics 2014, 6, 651–664. [Google Scholar] [CrossRef]
- Marmorstein, R.; Zhou, M.M. Writers and readers of histone acetylation: Structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014, 6, a018762. [Google Scholar] [CrossRef]
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef] [PubMed]
- You, J.S.; Jones, P.A. Cancer genetics and epigenetics: Two sides of the same coin? Cancer Cell 2012, 22, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, A.; Minucci, S. Chapter 6—Alterations of histone modifications in cancer. In Epigenetics in Human Disease, 2nd ed.; Tollefsbol, T.O., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 6, pp. 141–217. [Google Scholar]
- Yang, X.; Karuturi, R.K.; Sun, F.; Aau, M.; Yu, K.; Shao, R.; Miller, L.D.; Tan, P.B.; Yu, Q. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS ONE 2009, 4, e5011. [Google Scholar] [CrossRef]
- Shi, L.; Sun, L.; Li, Q.; Liang, J.; Yu, W.; Yi, X.; Yang, X.; Li, Y.; Han, X.; Zhang, Y.; et al. Histone demethylase JMJD2B coordinates H3K4/H3K9 methylation and promotes hormonally responsive breast carcinogenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 7541–7546. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, H.; Chen, Y.; Sun, Y.; Yang, F.; Yu, W.; Liang, J.; Sun, L.; Yang, X.; Shi, L.; et al. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 2009, 138, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef]
- Peschansky, V.J.; Wahlestedt, C. Non-coding RNAs as direct and indirect modulators of epigenetic regulation. Epigenetics 2014, 9, 3–12. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Esteller, M. Non-coding RNAs, epigenetics, and cancer: Tying it all together. Cancer Metastasis Rev. 2018, 37, 55–73. [Google Scholar] [CrossRef]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, J.; Wang, G. The role of microRNAs in human breast cancer progression. Tumor Biol. 2014, 35, 6235–6244. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Slack, F.J. Epigenetics and genetics. MicroRNAs en route to the clinic: Progress in validating and targeting microRNAs for cancer therapy. Nat. Rev. Cancer 2011, 11, 849–864. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, G.; Cava, C.; Castiglioni, I. MicroRNAs: New Biomarkers for Diagnosis, Prognosis, Therapy Prediction and Therapeutic Tools for Breast Cancer. Theranostics 2015, 5, 1122–1143. [Google Scholar] [CrossRef] [PubMed]
- Van Roosbroeck, K.; Calin, G.A. Cancer Hallmarks and MicroRNAs: The Therapeutic Connection. Adv. Cancer Res. 2017, 135, 119–149. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Brane, A.C.; Tollefsbol, T.O. MicroRNAs and Epigenetics Strategies to Reverse Breast Cancer. Cells 2019, 8, 1214. [Google Scholar] [CrossRef]
- Søkilde, R.; Persson, H.; Ehinger, A.; Pirona, A.C.; Fernö, M.; Hegardt, C.; Larsson, C.; Loman, N.; Malmberg, M.; Rydén, L.; et al. Refinement of breast cancer molecular classification by miRNA expression profiles. BMC Genom. 2019, 20, 503. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.; Liaw, C.S.; Ji, S.M.; Tan, H.H.; Wong, C.Y.; Thike, A.A.; Tan, P.H.; Ho, G.H.; Lee, A.S. Identification of circulating microRNA signatures for breast cancer detection. Clin. Cancer Res. 2013, 19, 4477–4487. [Google Scholar] [CrossRef]
- Martinez-Gutierrez, A.D.; Cantú de León, D.; Millan-Catalan, O.; Coronel-Hernandez, J.; Campos-Parra, A.D.; Porras-Reyes, F.; Exayana-Alderete, A.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Ramos-Payan, R. Identification of miRNA Master Regulators in Breast Cancer. Cells 2020, 9, 1610. [Google Scholar] [CrossRef]
- Markou, A.; Yousef, G.M.; Stathopoulos, E.; Georgoulias, V.; Lianidou, E. Prognostic significance of metastasis-related microRNAs in early breast cancer patients with a long follow-up. Clin. Chem. 2014, 60, 197–205. [Google Scholar] [CrossRef]
- Tormo, E.; Ballester, S.; Adam-Artigues, A.; Burgués, O.; Alonso, E.; Bermejo, B.; Menéndez, S.; Zazo, S.; Madoz-Gúrpide, J.; Rovira, A.; et al. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci. Rep. 2019, 9, 5316. [Google Scholar] [CrossRef]
- Lavin, D.P.; Tiwari, V.K. Unresolved Complexity in the Gene Regulatory Network Underlying EMT. Front. Oncol. 2020, 10, 554. [Google Scholar] [CrossRef]
- Felipe Lima, J.; Nofech-Mozes, S.; Bayani, J.; Bartlett, J.M. EMT in Breast Carcinoma—A Review. J. Clin. Med. 2016, 5, 65. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef] [PubMed]
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; André, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Rashidian, M.; Reinhardt, F.; Bagnato, A.; Keckesova, Z.; Ploegh, H.L.; Weinberg, R.A. Epithelial-to-Mesenchymal Transition Contributes to Immunosuppression in Breast Carcinomas. Cancer Res. 2017, 77, 3982–3989. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Bueno, G.; Portillo, F.; Cano, A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene 2008, 27, 6958–6969. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Cheung, K.J.; Padmanaban, V.; Silvestri, V.; Schipper, K.; Cohen, J.D.; Fairchild, A.N.; Gorin, M.A.; Verdone, J.E.; Pienta, K.J.; Bader, J.S.; et al. Polyclonal breast cancer metastases arise from collective dissemination of keratin 14-expressing tumor cell clusters. Proc. Natl. Acad. Sci. USA 2016, 113, E854–E863. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Ye, X.; Brabletz, T.; Kang, Y.; Longmore, G.D.; Nieto, M.A.; Stanger, B.Z.; Yang, J.; Weinberg, R.A. Upholding a role for EMT in breast cancer metastasis. Nature 2017, 547, E1–E3. [Google Scholar] [CrossRef]
- Brabletz, T. To differentiate or not—Routes towards metastasis. Nat. Rev. Cancer 2012, 12, 425–436. [Google Scholar] [CrossRef]
- Beerling, E.; Seinstra, D.; de Wit, E.; Kester, L.; van der Velden, D.; Maynard, C.; Schäfer, R.; van Diest, P.; Voest, E.; van Oudenaarden, A.; et al. Plasticity between Epithelial and Mesenchymal States Unlinks EMT from Metastasis-Enhancing Stem Cell Capacity. Cell Rep. 2016, 14, 2281–2288. [Google Scholar] [CrossRef] [PubMed]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massagué, J.; Benezra, R. TGF-β-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef] [PubMed]
- Bedi, U.; Mishra, V.K.; Wasilewski, D.; Scheel, C.; Johnsen, S.A. Epigenetic plasticity: A central regulator of epithelial-to-mesenchymal transition in cancer. Oncotarget 2014, 5, 2016–2029. [Google Scholar] [CrossRef] [PubMed]
- Casciello, F.; Windloch, K.; Gannon, F.; Lee, J.S. Functional Role of G9a Histone Methyltransferase in Cancer. Front. Immunol. 2015, 6, 487. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Wu, Y.; Yao, J.; Wang, Y.; Yu, Y.; Rychahou, P.G.; Evers, B.M.; Zhou, B.P. G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer. J. Clin. Investig. 2012, 122, 1469–1486. [Google Scholar] [CrossRef] [PubMed]
- Skrypek, N.; Bruneel, K.; Vandewalle, C.; De Smedt, E.; Soen, B.; Loret, N.; Taminau, J.; Goossens, S.; Vandamme, N.; Berx, G. ZEB2 stably represses RAB25 expression through epigenetic regulation by SIRT1 and DNMTs during epithelial-to-mesenchymal transition. Epigenetics Chromatin 2018, 11, 70. [Google Scholar] [CrossRef]
- Lin, Y.T.; Wu, K.J. Epigenetic regulation of epithelial-mesenchymal transition: Focusing on hypoxia and TGF-β signaling. J. Biomed. Sci. 2020, 27, 39. [Google Scholar] [CrossRef]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 cooperatively determine the mammary stem cell state. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef]
- Lim, S.; Becker, A.; Zimmer, A.; Lu, J.; Buettner, R.; Kirfel, J. SNAI1-mediated epithelial-mesenchymal transition confers chemoresistance and cellular plasticity by regulating genes involved in cell death and stem cell maintenance. PLoS ONE 2013, 8, e66558. [Google Scholar] [CrossRef]
- Barneh, F.; Mirzaie, M.; Nickchi, P.; Tan, T.Z.; Thiery, J.P.; Piran, M.; Salimi, M.; Goshadrou, F.; Aref, A.R.; Jafari, M. Integrated use of bioinformatic resources reveals that co-targeting of histone deacetylases, IKBK and SRC inhibits epithelial-mesenchymal transition in cancer. Brief. Bioinform. 2019, 20, 717–731. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Li, L.T.; Jiang, G.; Chen, Q.; Zheng, J.N. Ki67 is a promising molecular target in the diagnosis of cancer. Mol. Med. Rep. 2015, 11, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.K. Adjuvant therapy for breast cancer: Who should get what? West. J. Med. 2001, 174, 284–287. [Google Scholar] [CrossRef]
- Cardoso, F.; Kyriakides, S.; Ohno, S.; Penault-Llorca, F.; Poortmans, P.; Rubio, I.T.; Zackrisson, S.; Senkus, E. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2019, 30, 1674. [Google Scholar] [CrossRef]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; André, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)†. Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. New Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef]
- Bartlett, J.; Sgroi, D.; Treuner, K.; Zhang, Y.; Ahmed, I.; Piper, T.; Salunga, R.; Brachtel, E.; Pirrie, S.; Schnabel, C. Breast Cancer Index and prediction of benefit from extended endocrine therapy in breast cancer patients treated in the Adjuvant Tamoxifen—To Offer More?(aTTom) trial. Ann. Oncol. 2019, 30, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-J.; Salunga, R.; Dahiya, S.; Wang, W.; Carney, E.; Durbecq, V.; Harris, A.; Goss, P.; Sotiriou, C.; Erlander, M. A five-gene molecular grade index and HOXB13: IL17BR are complementary prognostic factors in early stage breast cancer. Clin. Cancer Res. 2008, 14, 2601–2608. [Google Scholar] [CrossRef] [PubMed]
- Simon, R.M.; Paik, S.; Hayes, D.F. Use of archived specimens in evaluation of prognostic and predictive biomarkers. J. Natl. Cancer Inst. 2009, 101, 1446–1452. [Google Scholar] [CrossRef]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.-Y.; Brain, E.; Causeret, S.; DeLorenzi, M. 70-gene signature as an aid to treatment decisions in early-stage breast cancer. New Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.-B.; Lænkholm, A.-V.; Nielsen, T.O.; Eriksen, J.O.; Wehn, P.; Hood, T.; Ram, N.; Buckingham, W.; Ferree, S.; Ejlertsen, B. The Prosigna gene expression assay and responsiveness to adjuvant cyclophosphamide-based chemotherapy in premenopausal high-risk patients with breast cancer. Breast Cancer Res. 2018, 20, 79. [Google Scholar] [CrossRef]
- Bae, Y.K.; Brown, A.; Garrett, E.; Bornman, D.; Fackler, M.J.; Sukumar, S.; Herman, J.G.; Gabrielson, E. Hypermethylation in histologically distinct classes of breast cancer. Clin. Cancer Res. 2004, 10, 5998–6005. [Google Scholar] [CrossRef]
- De Oca, R.M.; Gurard-Levin, Z.A.; Berger, F.; Rehman, H.; Martel, E.; Corpet, A.; de Koning, L.; Vassias, I.; Wilson, L.O.; Meseure, D. The histone chaperone HJURP is a new independent prognostic marker for luminal A breast carcinoma. Mol. Oncol. 2015, 9, 657–674. [Google Scholar] [CrossRef]
- Roessler, J.; Ammerpohl, O.; Gutwein, J.; Steinemann, D.; Schlegelberger, B.; Weyer, V.; Sariyar, M.; Geffers, R.; Arnold, N.; Schmutzler, R. The CpG island methylator phenotype in breast cancer is associated with the lobular subtype. Epigenomics 2015, 7, 187–199. [Google Scholar] [CrossRef]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.-F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214. [Google Scholar] [CrossRef]
- Braicu, C.; Raduly, L.; Morar-Bolba, G.; Cojocneanu, R.; Jurj, A.; Pop, L.-A.; Pileczki, V.; Ciocan, C.; Moldovan, A.; Irimie, A. Aberrant miRNAs expressed in HER-2 negative breast cancers patient. J. Exp. Clin. Cancer Res. 2018, 37, 257. [Google Scholar] [CrossRef]
- Qi, P.; Du, X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod. Pathol. 2013, 26, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Valdespino, V.; Valdespino, P.M. Potential of epigenetic therapies in the management of solid tumors. Cancer Manag. Res. 2015, 7, 241–251. [Google Scholar] [CrossRef]
- Jones, P.A.; Issa, J.P.; Baylin, S. Targeting the cancer epigenome for therapy. Nat. Rev. Genet. 2016, 17, 630–641. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Makita, S.; Tobinai, K. Development of new agents for peripheral T-cell lymphoma. Expert Opin. Biol. Ther. 2019, 19, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Roberti, A.; Valdes, A.F.; Torrecillas, R.; Fraga, M.F.; Fernandez, A.F. Epigenetics in cancer therapy and nanomedicine. Clin. Epigenetics 2019, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Ient, J.; Göttgens, E.L.; Krieg, A.J.; Hammond, E.M. Epigenetic Therapy for Solid Tumors: Highlighting the Impact of Tumor Hypoxia. Genes 2015, 6, 935–956. [Google Scholar] [CrossRef]
- Ganesan, A.; Arimondo, P.B.; Rots, M.G.; Jeronimo, C.; Berdasco, M. The timeline of epigenetic drug discovery: From reality to dreams. Clin. Epigenetics 2019, 11, 174. [Google Scholar] [CrossRef]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar]
- Bohl, S.R.; Bullinger, L.; Rücker, F.G. Epigenetic therapy: Azacytidine and decitabine in acute myeloid leukemia. Expert Rev. Hematol. 2018, 11, 361–371. [Google Scholar] [CrossRef]
- Roche, J.; Bertrand, P. Inside HDACs with more selective HDAC inhibitors. Eur. J. Med. Chem. 2016, 121, 451–483. [Google Scholar] [CrossRef]
- Ghasemi, S. Cancer’s epigenetic drugs: Where are they in the cancer medicines? Pharm. J. 2020, 20, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Filippova, E.V.; Zemaitaitis, B.; Aung, T.; Wolfe, A.J.; Anderson, W.F. Structural Basis for DNA Recognition by the Two-Component Response Regulator RcsB. mBio 2018, 9, e01993-17. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.A.; Dokmanovic, M. Histone deacetylase inhibitors: Discovery and development as anticancer agents. Expert Opin. Investig. Drugs 2005, 14, 1497–1511. [Google Scholar] [CrossRef] [PubMed]
- Montalvo-Casimiro, M.; González-Barrios, R.; Meraz-Rodriguez, M.A.; Juárez-González, V.T.; Arriaga-Canon, C.; Herrera, L.A. Epidrug Repurposing: Discovering New Faces of Old Acquaintances in Cancer Therapy. Front. Oncol. 2020, 10, 2461. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Roboz, G.; Walsh, K.; Kantarjian, H.; Ritchie, E.; Kropf, P.; O’Connell, C.; Tibes, R.; Lunin, S.; Rosenblat, T.; et al. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: Phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol. 2019, 6, e317–e327. [Google Scholar] [CrossRef]
- Graça, I.; Pereira-Silva, E.; Henrique, R.; Packham, G.; Crabb, S.J.; Jerónimo, C. Epigenetic modulators as therapeutic targets in prostate cancer. Clin. Epigenetics 2016, 8, 98. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef]
- Wapenaar, H.; Dekker, F.J. Histone acetyltransferases: Challenges in targeting bi-substrate enzymes. Clin. Epigenetics 2016, 8, 59. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., III; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Stein, E.M.; Garcia-Manero, G.; Rizzieri, D.A.; Tibes, R.; Berdeja, J.G.; Savona, M.R.; Jongen-Lavrenic, M.; Altman, J.K.; Thomson, B.; Blakemore, S.J.; et al. The DOT1L inhibitor pinometostat reduces H3K79 methylation and has modest clinical activity in adult acute leukemia. Blood 2018, 131, 2661–2669. [Google Scholar] [CrossRef]
- Wong, M.; Polly, P.; Liu, T. The histone methyltransferase DOT1L: Regulatory functions and a cancer therapy target. Am. J. Cancer Res. 2015, 5, 2823–2837. [Google Scholar]
- Wojtala, M.; Dąbek, A.; Rybaczek, D.; Śliwińska, A.; Świderska, E.; Słapek, K.; El-Osta, A.; Balcerczyk, A. Silencing Lysine-Specific Histone Demethylase 1 (LSD1) Causes Increased HP1-Positive Chromatin, Stimulation of DNA Repair Processes, and Dysregulation of Proliferation by Chk1 Phosphorylation in Human Endothelial Cells. Cells 2019, 8, 1212. [Google Scholar] [CrossRef]
- Majello, B.; Gorini, F.; Saccà, C.D.; Amente, S. Expanding the Role of the Histone Lysine-Specific Demethylase LSD1 in Cancer. Cancers 2019, 11, 324. [Google Scholar] [CrossRef]
- Alqahtani, A.; Choucair, K.; Ashraf, M.; Hammouda, D.M.; Alloghbi, A.; Khan, T.; Senzer, N.; Nemunaitis, J. Bromodomain and extra-terminal motif inhibitors: A review of preclinical and clinical advances in cancer therapy. Future Sci. OA 2019, 5, Fso372. [Google Scholar] [CrossRef]
- Luan, Y.; Ngo, L.; Han, Z.; Wang, X.; Qu, M.; Zheng, Y.G. Histone acetyltransferases: Enzymes, assays, and inhibitors. In Epigenetic Technological Applications; Elsevier: Amsterdam, The Netherlands, 2015; pp. 291–317. [Google Scholar]
- Meseure, D.; Drak Alsibai, K.; Nicolas, A.; Bieche, I.; Morillon, A. Long Noncoding RNAs as New Architects in Cancer Epigenetics, Prognostic Biomarkers, and Potential Therapeutic Targets. BioMed Res. Int. 2015, 2015, 320214. [Google Scholar] [CrossRef]
- Baumann, V.; Winkler, J. miRNA-based therapies: Strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med. Chem. 2014, 6, 1967–1984. [Google Scholar] [CrossRef]
- Takahashi, R.-U.; Prieto-Vila, M.; Kohama, I.; Ochiya, T. Development of miRNA-based therapeutic approaches for cancer patients. Cancer Sci. 2019, 110, 1140–1147. [Google Scholar] [CrossRef]
- Ma, L.; Chua, M.S.; Andrisani, O.; So, S. Epigenetics in hepatocellular carcinoma: An update and future therapy perspectives. World J. Gastroenterol. 2014, 20, 333–345. [Google Scholar] [CrossRef]
- Falahi, F.; van Kruchten, M.; Martinet, N.; Hospers, G.A.; Rots, M.G. Current and upcoming approaches to exploit the reversibility of epigenetic mutations in breast cancer. Breast Cancer Res. 2014, 16, 412. [Google Scholar] [CrossRef]
- Ari, F.; Napieralski, R.; Ulukaya, E.; Dere, E.; Colling, C.; Honert, K.; Krüger, A.; Kiechle, M.; Schmitt, M. Modulation of protein expression levels and DNA methylation status of breast cancer metastasis genes by anthracycline-based chemotherapy and the demethylating agent decitabine. Cell Biochem. Funct. 2011, 29, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Hurtubise, A.; Momparler, R.L. Evaluation of antineoplastic action of 5-aza-2′-deoxycytidine (Dacogen) and docetaxel (Taxotere) on human breast, lung and prostate carcinoma cell lines. Anticancer Drugs 2004, 15, 161–167. [Google Scholar] [CrossRef]
- Mirza, S.; Sharma, G.; Pandya, P.; Ralhan, R. Demethylating agent 5-aza-2-deoxycytidine enhances susceptibility of breast cancer cells to anticancer agents. Mol. Cell. Biochem. 2010, 342, 101–109. [Google Scholar] [CrossRef]
- Hodges-Gallagher, L.; Valentine, C.D.; Bader, S.E.; Kushner, P.J. Inhibition of histone deacetylase enhances the anti-proliferative action of antiestrogens on breast cancer cells and blocks tamoxifen-induced proliferation of uterine cells. Breast Cancer Res. Treat. 2007, 105, 297–309. [Google Scholar] [CrossRef]
- Basse, C.; Arock, M. The increasing roles of epigenetics in breast cancer: Implications for pathogenicity, biomarkers, prevention and treatment. Int. J. Cancer 2015, 137, 2785–2794. [Google Scholar] [CrossRef]
- Connolly, R.M.; Li, H.; Jankowitz, R.C.; Zhang, Z.; Rudek, M.A.; Jeter, S.C.; Slater, S.A.; Powers, P.; Wolff, A.C.; Fetting, J.H.; et al. Combination Epigenetic Therapy in Advanced Breast Cancer with 5-Azacitidine and Entinostat: A Phase II National Cancer Institute/Stand Up to Cancer Study. Clin. Cancer Res. 2017, 23, 2691–2701. [Google Scholar] [CrossRef]
- Fortunati, N.; Bertino, S.; Costantino, L.; De Bortoli, M.; Compagnone, A.; Bandino, A.; Catalano, M.G.; Boccuzzi, G. Valproic acid restores ER alpha and antiestrogen sensitivity to ER alpha-negative breast cancer cells. Mol. Cell. Endocrinol. 2010, 314, 17–22. [Google Scholar] [CrossRef]
- Jang, E.R.; Lim, S.J.; Lee, E.S.; Jeong, G.; Kim, T.Y.; Bang, Y.J.; Lee, J.S. The histone deacetylase inhibitor trichostatin A sensitizes estrogen receptor alpha-negative breast cancer cells to tamoxifen. Oncogene 2004, 23, 1724–1736. [Google Scholar] [CrossRef]
- Feng, Q.; Zhang, Z.; Shea, M.J.; Creighton, C.J.; Coarfa, C.; Hilsenbeck, S.G.; Lanz, R.; He, B.; Wang, L.; Fu, X.; et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014, 24, 809–819. [Google Scholar] [CrossRef]
- Vázquez, R.; Riveiro, M.E.; Astorgues-Xerri, L.; Odore, E.; Rezai, K.; Erba, E.; Panini, N.; Rinaldi, A.; Kwee, I.; Beltrame, L.; et al. The bromodomain inhibitor OTX015 (MK-8628) exerts anti-tumor activity in triple-negative breast cancer models as single agent and in combination with everolimus. Oncotarget 2017, 8, 7598–7613. [Google Scholar] [CrossRef]
- Min, A.; Im, S.A.; Kim, D.K.; Song, S.H.; Kim, H.J.; Lee, K.H.; Kim, T.Y.; Han, S.W.; Oh, D.Y.; Kim, T.Y.; et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015, 17, 33. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Zhao, F.; Wang, M.; Swaby, R.F.; Sparano, J.A.; Meropol, N.J.; Bhalla, K.N.; Pellegrino, C.M.; Katherine Alpaugh, R.; Falkson, C.I.; et al. A Phase I/II study of suberoylanilide hydroxamic acid (SAHA) in combination with trastuzumab (Herceptin) in patients with advanced metastatic and/or local chest wall recurrent HER2-amplified breast cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1104). Breast Cancer Res. Treat. 2017, 165, 375–382. [Google Scholar] [CrossRef]
- Damaskos, C.; Garmpis, N.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; et al. Histone Deacetylase Inhibitors: An Attractive Therapeutic Strategy Against Breast Cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef]
- Tinkle, S.; McNeil, S.E.; Mühlebach, S.; Bawa, R.; Borchard, G.; Barenholz, Y.C.; Tamarkin, L.; Desai, N. Nanomedicines: Addressing the scientific and regulatory gap. Ann. N. Y. Acad. Sci. 2014, 1313, 35–56. [Google Scholar] [CrossRef]
- EUR-Lex: EU. Available online: lawhttps://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32011H0696&from=EN (accessed on 17 August 2020).
- Schütz, C.A.; Juillerat-Jeanneret, L.; Mueller, H.; Lynch, I.; Riediker, M. Therapeutic nanoparticles in clinics and under clinical evaluation. Nanomedicine 2013, 8, 449–467. [Google Scholar] [CrossRef]
- Boverhof, D.R.; Bramante, C.M.; Butala, J.H.; Clancy, S.F.; Lafranconi, M.; West, J.; Gordon, S.C. Comparative assessment of nanomaterial definitions and safety evaluation considerations. Regul. Toxicol. Pharmacol. 2015, 73, 137–150. [Google Scholar] [CrossRef]
- Nanomaterials definition matters. Nat. Nanotechnol. 2019, 14, 193. [CrossRef]
- Kim, K.; Khang, D. Past, Present, and Future of Anticancer Nanomedicine. Int. J. Nanomed. 2020, 15, 5719. [Google Scholar] [CrossRef]
- Wong, H.L.; Bendayan, R.; Rauth, A.M.; Li, Y.; Wu, X.Y. Chemotherapy with anticancer drugs encapsulated in solid lipid nanoparticles. Adv. Drug Deliv. Rev. 2007, 59, 491–504. [Google Scholar] [CrossRef]
- Martinelli, C.; Pucci, C.; Ciofani, G. Nanostructured carriers as innovative tools for cancer diagnosis and therapy. APL Bioeng. 2019, 3, 011502. [Google Scholar] [CrossRef]
- Smolkova, B.; Dusinska, M.; Gabelova, A. Nanomedicine and epigenome. Possible health risks. Food Chem. Toxicol. 2017, 109, 780–796. [Google Scholar] [CrossRef]
- Takechi-Haraya, Y.; Goda, Y.; Sakai-Kato, K. Control of Liposomal Penetration into Three-Dimensional Multicellular Tumor Spheroids by Modulating Liposomal Membrane Rigidity. Mol. Pharm. 2017, 14, 2158–2165. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. In Nanoscience and Technology: A Collection of Reviews from Nature Journals; World Scientific: Singapore, 2010; pp. 239–250. [Google Scholar]
- Du, M.; Ouyang, Y.; Meng, F.; Ma, Q.; Liu, H.; Zhuang, Y.; Pang, M.; Cai, T.; Cai, Y. Nanotargeted agents: An emerging therapeutic strategy for breast cancer. Nanomedicine 2019, 14, 1771–1786. [Google Scholar] [CrossRef]
- Caster, J.M.; Patel, A.N.; Zhang, T.; Wang, A. Investigational nanomedicines in 2016: A review of nanotherapeutics currently undergoing clinical trials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1416. [Google Scholar] [CrossRef]
- Liyanage, P.Y.; Hettiarachchi, S.D.; Zhou, Y.; Ouhtit, A.; Seven, E.S.; Oztan, C.Y.; Celik, E.; Leblanc, R.M. Nanoparticle-mediated targeted drug delivery for breast cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 419–433. [Google Scholar] [CrossRef]
- Anarjan, F.S. Active targeting drug delivery nanocarriers: Ligands. Nano-Struct. Nano-Objects 2019, 19, 100370. [Google Scholar]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Cole, A.J.; David, A.E.; Wang, J.; Galbán, C.J.; Hill, H.L.; Yang, V.C. Polyethylene glycol modified, cross-linked starch-coated iron oxide nanoparticles for enhanced magnetic tumor targeting. Biomaterials 2011, 32, 2183–2193. [Google Scholar] [CrossRef]
- Saraf, S.; Jain, A.; Tiwari, A.; Verma, A.; Panda, P.K.; Jain, S.K. Advances in liposomal drug delivery to cancer: An overview. J. Drug Deliv. Sci. Technol. 2020, 56, 101549. [Google Scholar] [CrossRef]
- Bottai, G.; Truffi, M.; Corsi, F.; Santarpia, L. Progress in nonviral gene therapy for breast cancer and what comes next? Expert Opin. Biol. Ther. 2017, 17, 595–611. [Google Scholar] [CrossRef]
- Aftab, S.; Shah, A.; Nadhman, A.; Kurbanoglu, S.; Ozkan, S.A.; Dionysiou, D.D.; Shukla, S.S.; Aminabhavi, T.M. Nanomedicine: An effective tool in cancer therapy. Int. J. Pharm. 2018, 540, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zhang, J. Surface engineering of nanomaterials with phospholipid-polyethylene glycol-derived functional conjugates for molecular imaging and targeted therapy. Biomaterials 2020, 230, 119646. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Hong, K.; Kirpotin, D.B.; Colbern, G.; Shalaby, R.; Baselga, J.; Shao, Y.; Nielsen, U.B.; Marks, J.D.; Moore, D. Anti-HER2 immunoliposomes: Enhanced efficacy attributable to targeted delivery. Clin. Cancer Res. 2002, 8, 1172–1181. [Google Scholar] [PubMed]
- Snipstad, S.; Hak, S.; Baghirov, H.; Sulheim, E.; Mørch, Ý.; Lélu, S.; von Haartman, E.; Bäck, M.; Nilsson, K.P.R.; Klymchenko, A.S. Labeling nanoparticles: Dye leakage and altered cellular uptake. Cytom. Part A 2017, 91, 760–766. [Google Scholar] [CrossRef] [PubMed]
- Moumaris, M.; Bretagne, J.-M.; Abuaf, N. Nanomedical Devices and Cancer Theranostics. Open Nanomed. Nanotechnol. J. 2020, 6, 1–11. [Google Scholar] [CrossRef]
- Gurunathan, S.; Kang, M.-H.; Qasim, M.; Kim, J.-H. Nanoparticle-Mediated Combination Therapy: Two-in-One Approach for Cancer. Int. J. Mol. Sci. 2018, 19, 3264. [Google Scholar] [CrossRef]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid-based nanoparticles as pharmaceutical drug carriers: From concepts to clinic. Crit. Rev. Ther. Drug Carr. Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef]
- Monteiro, N.; Martins, A.; Reis, R.L.; Neves, N.M. Liposomes in tissue engineering and regenerative medicine. J. R. Soc. Interface 2014, 11, 20140459. [Google Scholar] [CrossRef]
- Riaz, M.K.; Riaz, M.A.; Zhang, X.; Lin, C.; Wong, K.H.; Chen, X.; Zhang, G.; Lu, A.; Yang, Z. Surface Functionalization and Targeting Strategies of Liposomes in Solid Tumor Therapy: A Review. Int. J. Mol. Sci. 2018, 19, 195. [Google Scholar] [CrossRef]
- Olusanya, T.O.B.; Haj Ahmad, R.R.; Ibegbu, D.M.; Smith, J.R.; Elkordy, A.A. Liposomal Drug Delivery Systems and Anticancer Drugs. Molecules 2018, 23, 907. [Google Scholar] [CrossRef]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.C. Immunological and Toxicological Considerations for the Design of Liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current trends in the use of liposomes for tumor targeting. Nanomedicine 2013, 8, 1509–1528. [Google Scholar] [CrossRef] [PubMed]
- Leo, C.P.; Hentschel, B.; Szucs, T.D.; Leo, C. FDA and EMA Approvals of New Breast Cancer Drugs—A Comparative Regulatory Analysis. Cancers 2020, 12, 437. [Google Scholar] [CrossRef] [PubMed]
- Urbinati, G.; Marsaud, V.; Plassat, V.; Fattal, E.; Lesieur, S.; Renoir, J.M. Liposomes loaded with histone deacetylase inhibitors for breast cancer therapy. Int. J. Pharm. 2010, 397, 184–193. [Google Scholar] [CrossRef]
- Wang, Y.; Tu, S.; Steffen, D.; Xiong, M. Iron complexation to histone deacetylase inhibitors SAHA and LAQ824 in PEGylated liposomes can considerably improve pharmacokinetics in rats. J. Pharm. Pharm. Sci. 2014, 17, 583–602. [Google Scholar] [CrossRef]
- Neupane, Y.R.; Srivastava, M.; Ahmad, N.; Kumar, N.; Bhatnagar, A.; Kohli, K. Lipid based nanocarrier system for the potential oral delivery of decitabine: Formulation design, characterization, ex vivo, and in vivo assessment. Int. J. Pharm. 2014, 477, 601–612. [Google Scholar] [CrossRef]
- Su, X.; Wang, Z.; Li, L.; Zheng, M.; Zheng, C.; Gong, P.; Zhao, P.; Ma, Y.; Tao, Q.; Cai, L. Lipid-polymer nanoparticles encapsulating doxorubicin and 2′-deoxy-5-azacytidine enhance the sensitivity of cancer cells to chemical therapeutics. Mol. Pharm. 2013, 10, 1901–1909. [Google Scholar] [CrossRef]
- Tran, T.H.; Ramasamy, T.; Truong, D.H.; Shin, B.S.; Choi, H.G.; Yong, C.S.; Kim, J.O. Development of vorinostat-loaded solid lipid nanoparticles to enhance pharmacokinetics and efficacy against multidrug-resistant cancer cells. Pharm. Res. 2014, 31, 1978–1988. [Google Scholar] [CrossRef]
- Tran, T.H.; Choi, J.Y.; Ramasamy, T.; Truong, D.H.; Nguyen, C.N.; Choi, H.G.; Yong, C.S.; Kim, J.O. Hyaluronic acid-coated solid lipid nanoparticles for targeted delivery of vorinostat to CD44 overexpressing cancer cells. Carbohydr. Polym. 2014, 114, 407–415. [Google Scholar] [CrossRef]
- Denis, I.; el Bahhaj, F.; Collette, F.; Delatouche, R.; Gueugnon, F.; Pouliquen, D.; Pichavant, L.; Héroguez, V.; Grégoire, M.; Bertrand, P.; et al. Histone deacetylase inhibitor-polymer conjugate nanoparticles for acid-responsive drug delivery. Eur. J. Med. Chem. 2015, 95, 369–376. [Google Scholar] [CrossRef]
- Xu, J.; Sun, J.; Wang, P.; Ma, X.; Li, S. Pendant HDAC inhibitor SAHA derivatised polymer as a novel prodrug micellar carrier for anticancer drugs. J. Drug Target. 2018, 26, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.T.; Hoimes, C.J.; Kaimakliotis, H.Z.; Cheng, C.J.; Zhang, K.; Liu, J.; Wheeler, M.A.; Kelly, W.K.; Tew, G.N.; Saltzman, W.M.; et al. Nanoparticles for urothelium penetration and delivery of the histone deacetylase inhibitor belinostat for treatment of bladder cancer. Nanomedicine 2013, 9, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Sankar, R.; Ravikumar, V. Biocompatibility and biodistribution of suberoylanilide hydroxamic acid loaded poly (DL-lactide-co-glycolide) nanoparticles for targeted drug delivery in cancer. Biomed. Pharmacother. 2014, 68, 865–871. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Cui, Y.; Collins, C.J.; Thompson, D.H.; Irudayaraj, J. PLGA-PEG nano-delivery system for epigenetic therapy. Biomed. Pharmacother. 2017, 90, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Li, S.Y.; Sun, R.; Wang, H.X.; Shen, S.; Liu, Y.; Du, X.J.; Zhu, Y.H.; Jun, W. Combination therapy with epigenetic-targeted and chemotherapeutic drugs delivered by nanoparticles to enhance the chemotherapy response and overcome resistance by breast cancer stem cells. J. Control. Release 2015, 205, 7–14. [Google Scholar] [CrossRef]
- Hong, Y.D.; Zhang, J.; Zhuang, M.; Li, W.; Wu, P.U.; Li, R.T.; Hu, N.; Bian, B.X.; Song, Z.Y.; Wu, F.L. Efficacy of decitabine-loaded gelatinases-stimuli nanoparticles in overcoming cancer drug resistance is mediated via its enhanced demethylating activity to transcription factor AP-2 epsilon. Oncotarget 2017, 8, 114495–114505. [Google Scholar] [CrossRef]
- Wu, F.L.; Li, R.T.; Yang, M.; Yue, G.F.; Wang, H.Y.; Liu, Q.; Cui, F.B.; Wu, P.Y.; Ding, H.; Yu, L.X.; et al. Gelatinases-stimuli nanoparticles encapsulating 5-fluorouridine and 5-aza-2′-deoxycytidine enhance the sensitivity of gastric cancer cells to chemical therapeutics. Cancer Lett. 2015, 363, 7–16. [Google Scholar] [CrossRef]
- Kwak, T.W.; Kim, D.H.; Jeong, Y.I.; Kang, D.H. Antitumor activity of vorinostat-incorporated nanoparticles against human cholangiocarcinoma cells. J. Nanobiotechnol. 2015, 13, 60. [Google Scholar] [CrossRef]
- Wang, E.C.; Min, Y.; Palm, R.C.; Fiordalisi, J.J.; Wagner, K.T.; Hyder, N.; Cox, A.D.; Caster, J.M.; Tian, X.; Wang, A.Z. Nanoparticle formulations of histone deacetylase inhibitors for effective chemoradiotherapy in solid tumors. Biomaterials 2015, 51, 208–215. [Google Scholar] [CrossRef]
- Vijayaraghavalu, S.; Labhasetwar, V. Efficacy of decitabine-loaded nanogels in overcoming cancer drug resistance is mediated via sustained DNA methyltransferase 1 (DNMT1) depletion. Cancer Lett. 2013, 331, 122–129. [Google Scholar] [CrossRef]
- Houshmand, M.; Garello, F.; Circosta, P.; Stefania, R.; Aime, S.; Saglio, G.; Giachino, C. Nanocarriers as Magic Bullets in the Treatment of Leukemia. Nanomaterials 2020, 10, 276. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Dhar, A.; Patel, C.; Khimani, M.; Neogi, S.; Sharma, P.; Kumar, N.S.; Vekariya, R.L. A brief review on solid lipid nanoparticles: Part and parcel of contemporary drug delivery systems. RSC Adv. 2020, 10, 26777–26791. [Google Scholar] [CrossRef]
- Tran, T.H.; Chu, D.T.; Truong, D.H.; Tak, J.W.; Jeong, J.H.; Hoang, V.L.; Yong, C.S.; Kim, J.O. Development of lipid nanoparticles for a histone deacetylases inhibitor as a promising anticancer therapeutic. Drug Deliv. 2016, 23, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Hattori, Y.; Yamada, T.; Uesato, S.; Maitani, Y.; Nagaoka, Y. Histone deacetylase inhibitor prodrugs in nanoparticle vector enhanced gene expression in human cancer cells. Eur. J. Med. Chem. 2009, 44, 4603–4610. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yu, B.; Wu, Y.; Lee, R.J.; Lee, L.J. Efficient down-regulation of CDK4 by novel lipid nanoparticle-mediated siRNA delivery. Anticancer Res. 2011, 31, 1619–1626. [Google Scholar]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef]
- Bobo, D.; Robinson, K.J.; Islam, J.; Thurecht, K.J.; Corrie, S.R. Nanoparticle-Based Medicines: A Review of FDA-Approved Materials and Clinical Trials to Date. Pharm. Res. 2016, 33, 2373–2387. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, B.K.; Park, S.H.; Kim, M.G.; Lee, J.W.; Lee, H.Y.; Lee, H.B.; Kim, J.H.; Kim, M.S. Preparation of biodegradable and elastic poly (ε-caprolactone-co-lactide) copolymers and evaluation as a localized and sustained drug delivery carrier. Int. J. Mol. Sci. 2017, 18, 671. [Google Scholar] [CrossRef]
- Lu, B.; Huang, X.; Mo, J.; Zhao, W. Drug delivery using nanoparticles for cancer stem-like cell targeting. Front. Pharmacol. 2016, 7, 84. [Google Scholar] [CrossRef]
- Nasir, A.; Kausar, A.; Younus, A. A review on preparation, properties and applications of polymeric nanoparticle-based materials. Polym. Plast. Technol. Eng. 2015, 54, 325–341. [Google Scholar] [CrossRef]
- De Souza, C.; Ma, Z.; Lindstrom, A.R.; Chatterji, B.P. Nanomaterials as potential transporters of HDAC inhibitors. Med. Drug Discov. 2020, 6, 100040. [Google Scholar] [CrossRef]
- Kwak, T.W.; Lee, H.L.; Song, Y.H.; Kim, C.; Kim, J.; Seo, S.J.; Jeong, Y.I.; Kang, D.H. Vorinostat-eluting poly(DL-lactide-co-glycolide) nanofiber-coated stent for inhibition of cholangiocarcinoma cells. Int. J. Nanomed. 2017, 12, 7669–7680. [Google Scholar] [CrossRef] [PubMed]
- Alp, E.; Damkaci, F.; Guven, E.; Tenniswood, M. Starch nanoparticles for delivery of the histone deacetylase inhibitor CG-1521 in breast cancer treatment. Int. J. Nanomed. 2019, 14, 1335–1346. [Google Scholar] [CrossRef] [PubMed]
- Honma, K.; Iwao-Koizumi, K.; Takeshita, F.; Yamamoto, Y.; Yoshida, T.; Nishio, K.; Nagahara, S.; Kato, K.; Ochiya, T. RPN2 gene confers docetaxel resistance in breast cancer. Nat. Med. 2008, 14, 939–948. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A.; Caccamo, M.T. Smart Nanoparticles for Drug Delivery Application: Development of Versatile Nanocarrier Platforms in Biotechnology and Nanomedicine. J. Nanomater. 2019, 2019. [Google Scholar] [CrossRef]
- Hanafy, N.A.; El-Kemary, M.; Leporatti, S. Micelles structure development as a strategy to improve smart cancer therapy. Cancers 2018, 10, 238. [Google Scholar] [CrossRef]
- Devulapally, R.; Sekar, N.M.; Sekar, T.V.; Foygel, K.; Massoud, T.F.; Willmann, J.K.; Paulmurugan, R. Polymer nanoparticles mediated codelivery of antimiR-10b and antimiR-21 for achieving triple negative breast cancer therapy. ACS Nano 2015, 9, 2290–2302. [Google Scholar] [CrossRef]
- Mandal, A.K. Dendrimers in targeted drug delivery applications: A review of diseases and cancer. Int. J. Polym. Mater. Polym. Biomater. 2020, 1–11. [Google Scholar] [CrossRef]
- Finlay, J.; Roberts, C.M.; Lowe, G.; Loeza, J.; Rossi, J.J.; Glackin, C.A. RNA-based TWIST1 inhibition via dendrimer complex to reduce breast cancer cell metastasis. Biomed. Res. Int. 2015, 2015, 382745. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, X.H.; Wang, Z.Y.; Meng, M.; Li, X.; Ning, Q. Generation 4 polyamidoamine dendrimers is a novel candidate of nano-carrier for gene delivery agents in breast cancer treatment. Cancer Lett. 2010, 298, 34–49. [Google Scholar] [CrossRef]
- Suhail, M.; Rosenholm, J.M.; Minhas, M.U.; Badshah, S.F.; Naeem, A.; Khan, K.U.; Fahad, M. Nanogels as drug-delivery systems: A comprehensive overview. Ther. Deliv. 2019, 10, 697–717. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.; Reddy, R.; Jiang, Q. Crosslinking biopolymers for biomedical applications. Trends Biotechnol. 2015, 33, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Vijayaraghavalu, S.; Labhasetwar, V. Nanogel-mediated delivery of a cocktail of epigenetic drugs plus doxorubicin overcomes drug resistance in breast cancer cells. Drug Deliv. Transl. Res. 2018, 8, 1289–1299. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Hu, Z.; Nizzero, S.; Zhang, G.; Ramirez, M.R.; Shi, C.; Zhou, J.; Ferrari, M.; Shen, H. Bone-targeting nanoparticle to co-deliver decitabine and arsenic trioxide for effective therapy of myelodysplastic syndrome with low systemic toxicity. J. Control. Release 2017, 268, 92–101. [Google Scholar] [CrossRef]
- Parhizkar, M.; Reardon, P.; Harker, A.; Browning, R.; Stride, E.; Pedley, R.; Knowles, J.; Edirisinghe, M. Enhanced efficacy in drug-resistant cancer cells through synergistic nanoparticle mediated delivery of cisplatin and decitabine. Nanoscale Adv. 2020, 2, 1177–1186. [Google Scholar] [CrossRef]
- Kim, B.; Pena, C.D.; Auguste, D.T. Targeted Lipid Nanoemulsions Encapsulating Epigenetic Drugs Exhibit Selective Cytotoxicity on CDH1(-)/FOXM1(+) Triple Negative Breast Cancer Cells. Mol. Pharm. 2019, 16, 1813–1826. [Google Scholar] [CrossRef]
- Ruttala, H.B.; Ramasamy, T.; Poudal, B.K.; Choi, Y.; Choi, J.Y.; Kim, J.; Kwang Ku, S.; Choi, H.G.; Soon Yong, C.; Oh Kim, J. Molecularly targeted co-delivery of a histone deacetylase inhibitor and paclitaxel by lipid-protein hybrid nanoparticles for synergistic combinational chemotherapy. Oncotarget 2017, 8, 14925–14940. [Google Scholar] [CrossRef]
- Deng, Z.J.; Morton, S.W.; Ben-Akiva, E.; Dreaden, E.C.; Shopsowitz, K.E.; Hammond, P.T. Layer-by-layer nanoparticles for systemic codelivery of an anticancer drug and siRNA for potential triple-negative breast cancer treatment. ACS Nano 2013, 7, 9571–9584. [Google Scholar] [CrossRef]
- Foulkes, R.; Man, E.; Thind, J.; Yeung, S.; Joy, A.; Hoskins, C. The regulation of nanomaterials and nanomedicines for clinical application: Current and future perspectives. Biomater. Sci. 2020, 8, 4653–4664. [Google Scholar] [CrossRef]
- Lungu, I.I.; Grumezescu, A.M.; Volceanov, A.; Andronescu, E. Nanobiomaterials used in cancer therapy: An up-to-date overview. Molecules 2019, 24, 3547. [Google Scholar] [CrossRef]
- Gao, H. Shaping tumor microenvironment for improving nanoparticle delivery. Curr. Drug Metab. 2016, 17, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Liu, W.; Misra, P.; Tanaka, E.; Zimmer, J.P.; Ipe, B.I.; Bawendi, M.G.; Frangioni, J.V. Renal clearance of quantum dots. Nat. Biotechnol. 2007, 25, 1165–1170. [Google Scholar] [CrossRef] [PubMed]
- Longmire, M.; Choyke, P.L.; Kobayashi, H. Clearance properties of nano-sized particles and molecules as imaging agents: Considerations and caveats. Future Med. 2008. [Google Scholar] [CrossRef] [PubMed]
- Hossen, S.; Hossain, M.K.; Basher, M.; Mia, M.; Rahman, M.; Uddin, M.J. Smart nanocarrier-based drug delivery systems for cancer therapy and toxicity studies: A review. J. Adv. Res. 2019, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dusinska, M.; Tulinska, J.; El Yamani, N.; Kuricova, M.; Liskova, A.; Rollerova, E.; Rundén-Pran, E.; Smolkova, B. Immunotoxicity, genotoxicity and epigenetic toxicity of nanomaterials: New strategies for toxicity testing? Food Chem. Toxicol. 2017, 109, 797–811. [Google Scholar] [CrossRef]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical cancer models in tumor biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef]
- Afantitis, A.; Melagraki, G.; Isigonis, P.; Tsoumanis, A.; Varsou, D.D.; Valsami-Jones, E.; Papadiamantis, A.; Ellis, L.-J.A.; Sarimveis, H.; Doganis, P. NanoSolveIT Project: Driving nanoinformatics research to develop innovative and integrated tools for in silico nanosafety assessment. Comput. Struct. Biotechnol. J. 2020, 18, 583–602. [Google Scholar] [CrossRef]
- Collins, A.R.; Annangi, B.; Rubio, L.; Marcos, R.; Dorn, M.; Merker, C.; Estrela-Lopis, I.; Cimpan, M.R.; Ibrahim, M.; Cimpan, E. High throughput toxicity screening and intracellular detection of nanomaterials. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1413. [Google Scholar] [CrossRef]
- Ostermann, M.; Sauter, A.; Xue, Y.; Birkeland, E.; Schoelermann, J.; Holst, B.; Cimpan, M.R. Label-free impedance flow cytometry for nanotoxicity screening. Sci. Rep. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Ruzycka, M.; Cimpan, M.R.; Rios-Mondragon, I.; Grudzinski, I.P. Microfluidics for studying metastatic patterns of lung cancer. J. Nanobiotechnol. 2019, 17, 71. [Google Scholar] [CrossRef]
- Bor, G.; Mat Azmi, I.D.; Yaghmur, A. Nanomedicines for cancer therapy: Current status, challenges and future prospects. Ther. Deliv. 2019, 10, 113–132. [Google Scholar] [CrossRef] [PubMed]
- Dusinska, M.; Mariussen, E.; Rundén-Pran, E.; Hudecova, A.M.; Elje, E.; Kazimirova, A.; El Yamani, N.; Dommershausen, N.; Tharmann, J.; Fieblinger, D.; et al. In Vitro Approaches for Assessing the Genotoxicity of Nanomaterials. Methods Mol. Biol. 2019, 1894, 83–122. [Google Scholar] [CrossRef] [PubMed]
- Jesus, S.; Schmutz, M.; Som, C.; Borchard, G.; Wick, P.; Borges, O. Hazard assessment of polymeric nanobiomaterials for drug delivery: What can we learn from literature so far. Front. Bioeng. Biotechnol. 2019, 7, 261. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, Z.; Raza, M.A.; Manzoor, F.; Riaz, S.; Jabeen, G.; Fatima, S.; Naseem, S. A comparative assessment of nanotoxicity induced by metal (silver, nickel) and metal oxide (cobalt, chromium) nanoparticles in Labeo rohita. Nanomaterials 2019, 9, 309. [Google Scholar] [CrossRef] [PubMed]
- Gatto, F.; Bardi, G. Metallic nanoparticles: General research approaches to immunological characterization. Nanomaterials 2018, 8, 753. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Epi-Drug | Other Interventions | Status | Phase | Trial No. |
|---|---|---|---|---|
| Decitabine | LBH589, Tamoxifen | Terminated | 1, 2 | NCT01194908 |
| Paclitaxel | Unknown | 1b | NCT03282825 | |
| Carboplatin | Recruiting | 2 | NCT03295552 | |
| Doxorubicin and 4 more | Recruiting | 2 | NCT02957968 | |
| Azacitidine | Nab-paclitaxel | Completed | 1, 2 | NCT00748553 |
| Entinostat | Active, not recruiting | 2 | NCT01349959 | |
| Definitive breast | Withdrawn | NA | NCT01292083 | |
| Fulvestrant | Terminated | 2 | NCT02374099 | |
| Durvalumab | Active, not recruiting | 2 | NCT02811497 | |
| Valproic acid | FEC100 | Terminated | 2 | NCT01010854 |
| Hydralazine | Terminated | 2 | NCT00395655 | |
| Bevacizumab, Cetuximab | Recruiting | 1 | NCT01552434 | |
| Entinostat | Capecitabine | Recruiting | 1 | NCT03473639 |
| Exemestane | Recruiting Completed Active, not recruiting Active, not recruiting | 3 2 3 2 | NCT03538171, NCT00676663, NCT02115282, NCT03291886 | |
| Fulvestrant | Withdrawn | 2 | NCT02115594 | |
| Lapatinib Ditosylate, Trastuzumab | Completed | 1 | NCT01434303 | |
| Anastrozole | Terminated | 2 | NCT01234532 | |
| Nivolumab, Ipilimumab | Active, not recruiting | 1 | NCT02453620 | |
| Atezolizumab and 6 more | Recruiting | 1, 2 | NCT03280563 | |
| Romidepsin | Cisplatin, Nivolumab | Suspended | 1, 2 | NCT02393794 |
| Abraxane | Terminated | 1, 2 | NCT01938833 | |
| Alone | Completed | 2 | NCT00098397 | |
| Vorinostat | Olaparib | Not yet recruiting | 1 | NCT03742245 |
| Paclitaxel and 3 more | Completed | 1, 2 | NCT00574587 | |
| Alone | Completed Terminated Completed Withdrawn Completed Terminated | 1 2 2 NA 1 2 | NCT00719875, NCT00132002, NCT00262834, NCT01695057, NCT00788112, NCT00126451 | |
| Tamoxifen | Completed Terminated | 2 2 | NCT00365599, NCT01194427 | |
| Tamoxifen, Pembrolizumab | Terminated Not yet recruiting | 2 2 | NCT02395627, NCT04190056 | |
| Carboplatin, Nab-paclitaxel | Active, not recruiting | 2 | NCT00616967 | |
| Ixabepilone | Completed | 1 | NCT01084057 | |
| Lapatinib | Terminated | 1, 2 | NCT01118975 | |
| Paclitaxel, Bevacizumab | Completed | 1, 2 | NCT00368875 | |
| Anastrozole, Letrozole, Exemestane | Completed Completed | NA NA | NCT01720602, NCT01153672 | |
| Trastuzumab | Completed | 1, 2 | NCT00258349 | |
| Belinostat | Ribociclib | Not yet recruiting | 1 | NCT04315233 |
| Panobinostat | Alone | Completed Terminated Withdrawn | 2 2 1 | NCT00777049, NCT00777335, NCT00993642 |
| Trastuzumab | Terminated | 1, 2 | NCT00567879 | |
| Letrozole | Completed | 1, 2 | NCT01105312 | |
| Trastuzumab, Paclitaxel | Completed | 1 | NCT00788931 | |
| Capecitabine, Lapatinib | Completed | 1 | NCT00632489 |
| Nanocarrier | Loaded Drug | Reference |
|---|---|---|
| PEGylated liposomes | trichostatin A, CG1521, and PXD101 | [189] |
| PEGylated liposomes with Fe complex | VOR and LAQ824 | [190] |
| Hybrid lipid-polymer NPs | DAC | [191,192] |
| Solid lipid NPs | VOR | [193] |
| Solid lipid NPs decorated with hyaluronic acid | VOR | [194] |
| Norbornene polyethylene oxide macromonomer | CI-994 (tacedinaline) | [195] |
| POEG blocks | VOR | [196] |
| PLGA NPs decorated with PGON | belinostat and VOR | [197,198] |
| PLGE-PEG nano-micelles | AZA | [199] |
| PEG-PLA di-block copolymer | DAC | [200] |
| Gelatinases-stimuli di-block copolymers (PEG, PCL) | DAC | [201,202] |
| LGE block copolymer | VOR | [203] |
| Lipid-polymer (DSPE-PEG-COOH-PLGA-lecithin-PEG) core-shell NPs | VOR and quisinostat | [204] |
| Nanogels | DAC | [205] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buocikova, V.; Rios-Mondragon, I.; Pilalis, E.; Chatziioannou, A.; Miklikova, S.; Mego, M.; Pajuste, K.; Rucins, M.; Yamani, N.E.; Longhin, E.M.; et al. Epigenetics in Breast Cancer Therapy—New Strategies and Future Nanomedicine Perspectives. Cancers 2020, 12, 3622. https://doi.org/10.3390/cancers12123622
Buocikova V, Rios-Mondragon I, Pilalis E, Chatziioannou A, Miklikova S, Mego M, Pajuste K, Rucins M, Yamani NE, Longhin EM, et al. Epigenetics in Breast Cancer Therapy—New Strategies and Future Nanomedicine Perspectives. Cancers. 2020; 12(12):3622. https://doi.org/10.3390/cancers12123622
Chicago/Turabian StyleBuocikova, Verona, Ivan Rios-Mondragon, Eleftherios Pilalis, Aristotelis Chatziioannou, Svetlana Miklikova, Michal Mego, Karlis Pajuste, Martins Rucins, Naouale El Yamani, Eleonora Marta Longhin, and et al. 2020. "Epigenetics in Breast Cancer Therapy—New Strategies and Future Nanomedicine Perspectives" Cancers 12, no. 12: 3622. https://doi.org/10.3390/cancers12123622
APA StyleBuocikova, V., Rios-Mondragon, I., Pilalis, E., Chatziioannou, A., Miklikova, S., Mego, M., Pajuste, K., Rucins, M., Yamani, N. E., Longhin, E. M., Sobolev, A., Freixanet, M., Puntes, V., Plotniece, A., Dusinska, M., Cimpan, M. R., Gabelova, A., & Smolkova, B. (2020). Epigenetics in Breast Cancer Therapy—New Strategies and Future Nanomedicine Perspectives. Cancers, 12(12), 3622. https://doi.org/10.3390/cancers12123622