Modelling of Immune Checkpoint Network Explains Synergistic Effects of Combined Immune Checkpoint Inhibitor Therapy and the Impact of Cytokines in Patient Response

 ,
,  and
and
Simple Summary
Abstract
1. Introduction
2. Results
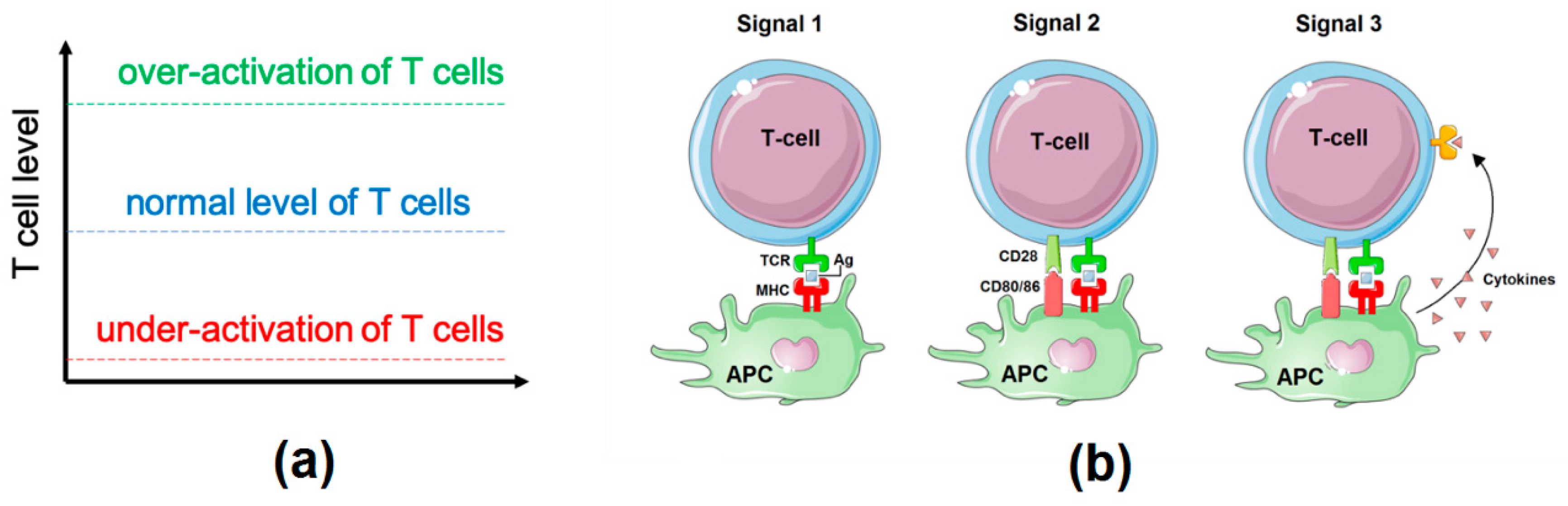
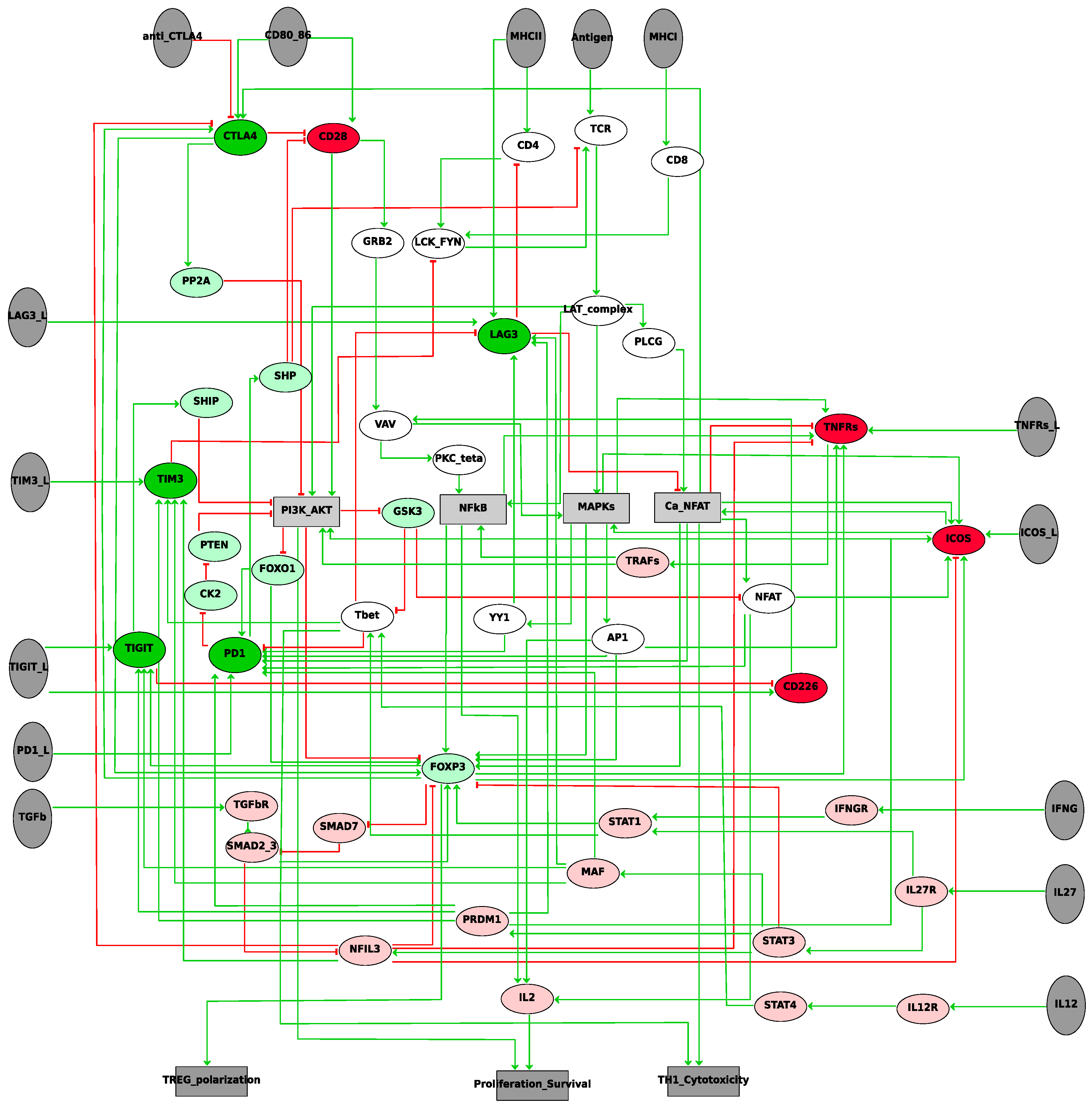
2.1. Influence Network: Proliferation, Survival, and Differentiation of T Cells Downstream of TCR Signalling
2.2. Logical Model of TCR Network
2.3. Model Validation. Simulations of Different Aspects of T Cell Biology
2.4. Role of Individual Inhibiting Immune Checkpoint Receptors in TCR Signalling Modulation
2.5. Two-Step Simulations of T Cell Response in Cancer Treated with PD1 and CTLA4 Inhibitors
2.6. Comparison of the Model Simulations with Experimental Observations
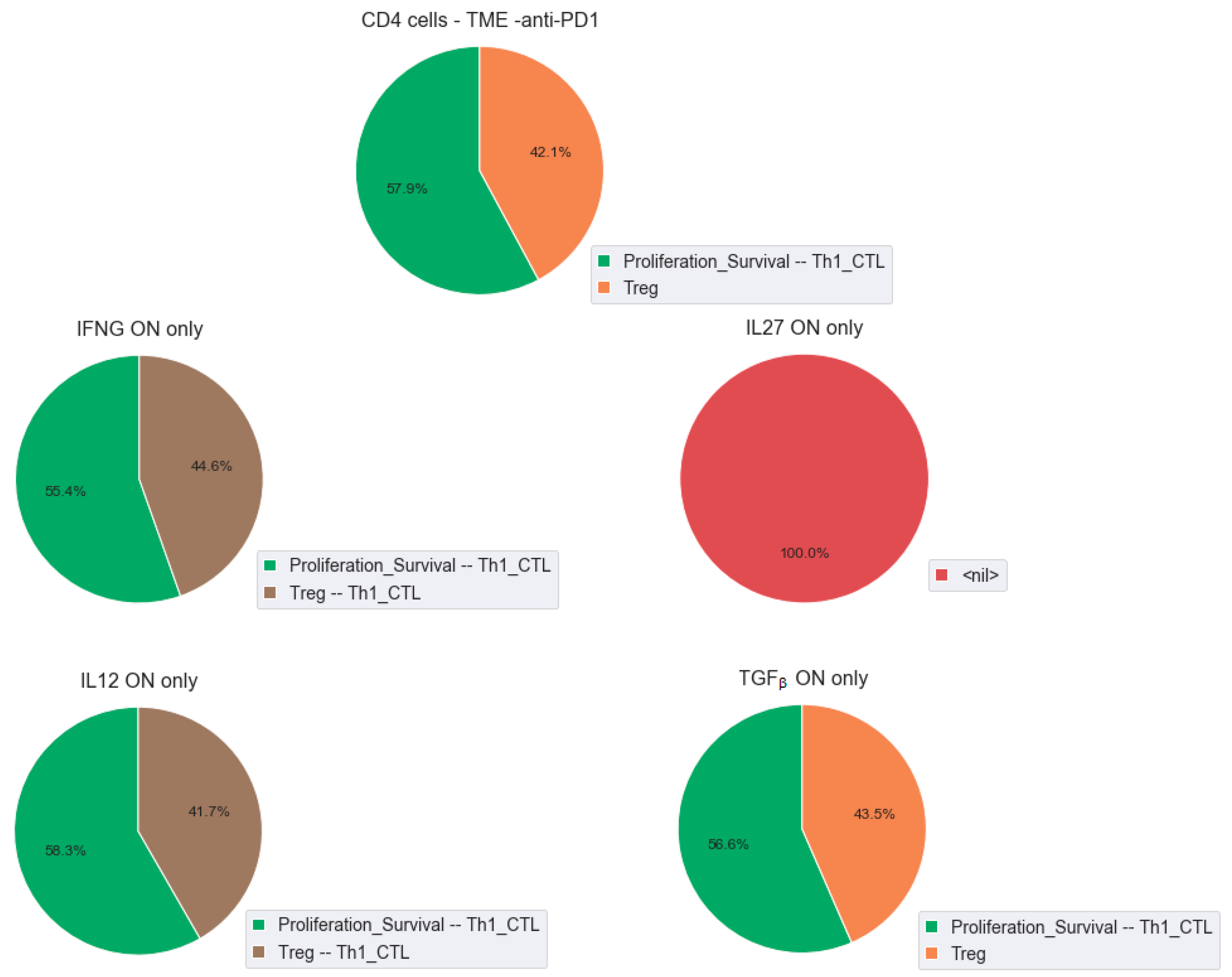
2.7. Role of Cytokines in the Modulation of the Effect of the Checkpoint Therapy
3. Discussion
4. Materials and Methods
4.1. Construction of the Network of the Immune Checkpoint Inhibitor Response
4.2. Logical Modelling and Simulations
4.3. Model Simulation of Immune Checkpoint Inhibiting Treatments
4.4. Model Accessibility and Data Availability
4.5. Data Used for Experimental Validation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef]
- Darvin, P.; Toor, S.M.; Sasidharan Nair, V.; Elkord, E. Immune checkpoint inhibitors: Recent progress and potential biomarkers. Exp. Mol. Med. 2018, 50. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity, NIH Public Access 44, 989–1004.ag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specia. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Quezada, S.A.; Allison, J.P. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS). Curr. Opin. Immunol. 2010, 22, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Fey, D.; Matallanas, D.; Rauch, J.; Rukhlenko, O.S.; Kholodenko, B.N. The complexities and versatility of the RAS-to-ERK signalling system in normal and cancer cells. Semin. Cell Dev. Biol. 2016, 58, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Tyson, J.J.; Tan, M.; Baumann, W.T.; Jin, L.; Xuan, J.; Wang, Y. Systems biology: Perspectives on multiscale modeling in research on endocrine-related cancers. Endocr. Relat. Cancer 2019. [Google Scholar] [CrossRef]
- Hernandez, C.; Thomas-Chollier, M.; Naldi, A.; Thieffry, D. Computational Verification of Large Logical Models—Application to the Prediction of T Cell Response to Checkpoint Inhibitors. Front. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Bolouri, H.; Young, M.; Beilke, J.; Johnson, R.; Fox, B.; Huang, L.; Santini, C.C.; Hill, C.M.; van der Vuurst, V.A.R.; Shannon, P.T.; et al. Integrative network modeling reveals mechanisms underlying T cell exhaustion. Sci. Rep. 2020. [Google Scholar] [CrossRef]
- Le Novère, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015, 16, 146–158. [Google Scholar] [CrossRef]
- Gómez Tejeda Zañudo, J.; Scaltriti, M.; Albert, R. A network modeling approach to elucidate drug resistance mechanisms and predict combinatorial drug treatments in breast cancer. Cancer Converg. 2017, 1, 5. [Google Scholar] [CrossRef] [PubMed]
- Remy, E.; Rebouissou, S.; Chaouiya, C.; Zinovyev, A.; Radvanyi, F.; Calzone, L. A modeling approach to explain mutually exclusive and co-occurring genetic alterations in bladder tumorigenesis. Cancer Res. 2015, 75, 4042–4052. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D.P.A.; Martignetti, L.; Robine, S.; Barillot, E.; Zinovyev, A.; Calzone, L. Mathematical Modelling of Molecular Pathways Enabling Tumour Cell Invasion and Migration. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Fumiã, H.F.; Martins, M.L. Boolean Network Model for Cancer Pathways: Predicting Carcinogenesis and Targeted Therapy Outcomes. PLoS ONE 2013, 8, e69008. [Google Scholar] [CrossRef] [PubMed]
- Grandclaudon, M.; Perrot-Dockès, M.; Trichot, C.; Karpf, L.; Abouzid, O.; Chauvin, C.; Sirven, P.; Abou-Jaoudé, W.; Berger, F.; Hupé, P.; et al. A Quantitative Multivariate Model of Human Dendritic Cell-T Helper Cell Communication. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Abou-Jaoudé, W.; Traynard, P.; Monteiro, P.T.; Saez-Rodriguez, J.; Helikar, T.; Thieffry, D.; Chaouiya, C. Logical modeling and dynamical analysis of cellular networks. Front. Genet. 2016. [Google Scholar] [CrossRef]
- Traynard, P.; Fauré, A.; Fages, F.; Thieffry, D. Logical model specification aided by model-checking techniques: Application to the mammalian cell cycle regulation. Bioinformatics 2016, 32, i772–i780. [Google Scholar] [CrossRef]
- Flobak, Å.; Baudot, A.; Remy, E.; Thommesen, L.; Thieffry, D.; Kuiper, M.; Lægreid, A. Discovery of Drug Synergies in Gastric Cancer Cells Predicted by Logical Modeling. PLoS Comput. Biol. 2015, 11. [Google Scholar] [CrossRef]
- Eduati, F.; Jaaks, P.; Wappler, J.; Cramer, T.; Merten, C.A.; Garnett, M.J.; Saez-Rodriguez, J. Patient-specific logic models of signaling pathways from screenings on cancer biopsies to prioritize personalized combination therapies. Mol. Syst. Biol. 2020, 16. [Google Scholar] [CrossRef]
- Oyeyemi, O.J.; Davies, O.; Robertson, D.L.; Schwartz, J.M. A logical model of HIV-1 interactions with the T-cell activation signalling pathway. Bioinformatics 2015. [Google Scholar] [CrossRef]
- Rodríguez-Jorge, O.; Kempis-Calanis, L.A.; Abou-Jaoudé, W.; Gutiérrez-Reyna, D.Y.; Hernandez, C.; Ramirez-Pliego, O.; Thomas-Chollier, M.; Spicuglia, S.; Santana, M.A.; Thieffry, D. Cooperation between T cell receptor and Toll-like receptor 5 signaling for CD4+ T cell activation. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.J.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O’Gorman, W.E.; Au-Yeung, A.; et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Yost, K.E.; Satpathy, A.T.; Wells, D.K.; Qi, Y.; Wang, C.; Kageyama, R.; McNamara, K.L.; Granja, J.M.; Sarin, K.Y.; Brown, R.A.; et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat. Med. 2019. [Google Scholar] [CrossRef]
- Le Novère, N.; Hucka, M.; Mi, H.; Moodie, S.; Schreiber, F.; Sorokin, A.; Demir, E.; Wegner, K.; Aladjem, M.I.; Wimalaratne, S.M.; et al. The Systems Biology Graphical Notation. Nat. Biotechnol. 2009, 27, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Dustin, M.L. The Cellular Context of T Cell Signaling. Immunity 2009, 30, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Gaud, G.; Lesourne, R.; Love, P.E. Tcell_TCR-sig-regulatory_NatRevImmu2018. Nat. Rev. Immunol. 2018, 1–13. [Google Scholar] [CrossRef]
- Courtney, A.H.; Lo, W.L.; Weiss, A. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem. Sci. 2018, 43, 108–123. [Google Scholar] [CrossRef]
- Takase, K.; Saito, T. T cell activation. Ryumachi 1995, 35, 853–861. [Google Scholar]
- Kim, H.J.; Cantor, H. CD4 T-cell subsets and tumor immunity: The helpful and the not-so-helpful. Cancer Immunol. Res. 2014. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Nishikawa, H. Natural and induced T regulatory cells in cancer. Front. Immunol. 2013, 4, 190. [Google Scholar] [CrossRef] [PubMed]
- Kapp, J.A.; Bucy, R.P. CD8+ suppressor T cells resurrected. Hum. Immunol. 2008, 69, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kryczek, I.; Zou, L.; Daniel, B.; Cheng, P.; Mottram, P.; Curiel, T.; Lange, A.; Zou, W. Plasmacytoid dendritic cells induce CD8+ regulatory T cells in human ovarian carcinoma. Cancer Res. 2005, 65, 5020–5026. [Google Scholar] [CrossRef] [PubMed]
- Kiniwa, Y.; Miyahara, Y.; Wang, H.Y.; Peng, W.; Peng, G.; Wheeler, T.M.; Thompson, T.C.; Old, L.J.; Wang, R.F. CD8+ Foxp3+ regulatory T cells mediate immunosuppression in prostate cancer. Clin. Cancer Res. 2007, 13, 6947–6958. [Google Scholar] [CrossRef]
- Balsari, A.; Merlo, A.; Casalini, P.; Carcangiu, M.L.; Malventano, C.; Triulzi, T.; Menard, S.; Tagliabue, E. FOXP3 expression and overall survival in breast cancer. J. Clin. Oncol. 2009, 27, 1746–1752. [Google Scholar] [CrossRef]
- Wolf, D.; Wolf, A.M.; Rumpold, H.; Fiegl, H.; Zeimet, A.G.; Muller-Holzner, E.; Deibl, M.; Gastl, G.; Gunsilius, E.; Marth, C. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin. Cancer Res. 2005, 11, 8326–8331. [Google Scholar] [CrossRef]
- Hsu, P.; Santner-Nanan, B.; Hu, M.; Skarratt, K.; Lee, C.H.; Stormon, M.; Wong, M.; Fuller, S.J.; Nanan, R. IL-10 Potentiates Differentiation of Human Induced Regulatory T Cells via STAT3 and Foxo1. J. Immunol. 2015, 195, 3665–3674. [Google Scholar] [CrossRef]
- Kerdiles, Y.M.; Stone, E.L.; Beisner, D.L.; McGargill, M.A.; Ch’en, I.L.; Stockmann, C.; Katayama, C.D.; Hedrick, S.M. Foxo Transcription Factors Control Regulatory T Cell Development and Function. Immunity 2010, 33, 890–904. [Google Scholar] [CrossRef]
- Knocke, S.; Fleischmann-Mundt, B.; Saborowski, M.; Manns, M.P.; Kühnel, F.; Wirth, T.C.; Woller, N. Tailored Tumor Immunogenicity Reveals Regulation of CD4 and CD8+ T Cell Responses against Cancer. Cell Rep. 2016. [Google Scholar] [CrossRef]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting Edge: TGF-β Induces a Regulatory Phenotype in CD4 + CD25 − T Cells through Foxp3 Induction and Down-Regulation of Smad7. J. Immunol. 2004. [Google Scholar] [CrossRef]
- Lu, L.; Ma, J.; Wang, X.; Wang, J.; Zhang, F.; Yu, J.; He, G.; Xu, B.; Brand, D.D.; Horwitz, D.A.; et al. Synergistic effect of TGF-β superfamily members on the induction of Foxp3+ Treg. Eur. J. Immunol. 2010. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ochando, J.C.; Bromberg, J.S.; Ding, Y. Identification of a distant T-bet enhancer responsive to IL-12/Stat4 and IFNγ/Stat1 signals. Blood 2007, 110, 2494–2500. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Sakuishi, K.; Xiao, S.; Sun, Z.; Zaghouani, S.; Gu, G.; Wang, C.; Tan, D.J.; Wu, C.; Rangachari, M.; et al. An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef]
- Ouaked, N.; Mantel, P.-Y.; Bassin, C.; Burgler, S.; Siegmund, K.; Akdis, C.A.; Schmidt-Weber, C.B. Regulation of the foxp3 Gene by the Th1 Cytokines: The Role of IL-27-Induced STAT1. J. Immunol. 2009, 182, 1041–1049. [Google Scholar] [CrossRef]
- Huber, M.; Steinwald, V.; Guralnik, A.; Brüstle, A.; Kleemann, P.; Rosenplänter, C.; Decker, T.; Lohoff, M. IL-27 inhibits the development of regulatory T cells via STAT3. Int. Immunol. 2008. [Google Scholar] [CrossRef]
- Wood, J.E.; Schneider, H.; Rudd, C.E. TcR and TcR-CD28 engagement of protein kinase B (PKB/AKT) and glycogen synthase kinase-3 (GSK-3) operates independently of guanine nucleotide exchange factor VAV-1. J. Biol. Chem. 2006, 281, 32385–32394. [Google Scholar] [CrossRef]
- Taylor, A.; Harker, J.A.; Chanthong, K.; Stevenson, P.G.; Zuniga, E.I.; Rudd, C.E. Glycogen Synthase Kinase 3 Inactivation Drives T-bet-Mediated Downregulation of Co-receptor PD-1 to Enhance CD8(+) Cytolytic T Cell Responses. Immunity 2016, 44, 274–286. [Google Scholar] [CrossRef]
- Sauer, S.; Bruno, L.; Hertweck, A.; Finlay, D.; Leleu, M.; Spivakov, M.; Knight, Z.A.; Cobb, B.S.; Cantrell, D.; O’Connor, E.; et al. T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc. Natl. Acad. Sci. USA 2008, 105, 7797–7802. [Google Scholar] [CrossRef]
- Kim, H.S.; Sohn, H.; Jang, S.W.; Lee, G.R. The transcription factor NFIL3 controls regulatory T-cell function and stability. Exp. Mol. Med. 2019, 51, 80. [Google Scholar] [CrossRef]
- Barnes, M.J.; Griseri, T.; Johnson, A.M.F.; Young, W.; Powrie, F.; Izcue, A. CTLA-4 promotes Foxp3 induction and regulatory T cell accumulation in the intestinal lamina propria. Mucosal Immunol. 2013, 6, 324–334. [Google Scholar] [CrossRef]
- Lucca, L.E.; Axisa, P.-P.; Singer, E.R.; Nolan, N.M.; Dominguez-Villar, M.; Hafler, D.A. TIGIT signaling restores suppressor function of Th1 Tregs. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed]
- Chaouiya, C.; Naldi, A.; Thieffry, D. Logical modelling of gene regulatory networks with GINsim. Methods Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Stoll, G.; Caron, B.; Viara, E.; Dugourd, A.; Zinovyev, A.; Naldi, A.; Kroemer, G.; Barillot, E.; Calzone, L. MaBoSS 2.0: An environment for stochastic Boolean modeling. Bioinformatics 2017, 33, 2226–2228. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Lozano, E.; Dominguez-Villar, M.; Kuchroo, V.; Hafler, D.A. The TIGIT/CD226 Axis Regulates Human T Cell Function. J. Immunol. 2012, 188, 3869–3875. [Google Scholar] [CrossRef]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual blockade of PD-1 and CTLA-4 combined with tumor vaccine effectively restores T-cell rejection function in tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef]
- Kroczek, R.A.; Mages, H.W.; Hutloff, A. Emerging paradigms of T-cell co-stimulation. Curr. Opin. Immunol. 2004, 16, 321–327. [Google Scholar] [CrossRef]
- Kinnear, G.; Jones, N.D.; Wood, K.J. Costimulation blockade: Current perspectives and implications for therapy. Transplantation 2013, 95, 527–535. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and anti-CTLA-4 therapies in cancer: Mechanisms of action, efficacy, and limitations. Front. Oncol. 2018. [Google Scholar] [CrossRef]
- Lighvani, A.A.; Frucht, D.M.; Jankovic, D.; Yamane, H.; Aliberti, J.; Hissong, B.D.; Nguyen, B.V.; Gadina, M.; Sher, A.; Paul, W.E.; et al. T-bet is rapidly induced by interferon-γ in lymphoid and myeloid cells. Proc. Natl. Acad. Sci. USA 2001. [Google Scholar] [CrossRef] [PubMed]
- Usui, T.; Preiss, J.C.; Kanno, Y.; Yao, Z.J.; Bream, J.H.; O’Shea, J.J.; Strober, W. T-bet regulates Th1 responses through essential effects on GATA-3 function rather than on IFNG gene acetylation and transcription. J. Exp. Med. 2006. [Google Scholar] [CrossRef] [PubMed]
- Chihara, N.; Madi, A.; Kondo, T.; Zhang, H.; Acharya, N.; Singer, M.; Nyman, J.; Marjanovic, N.D.; Kowalczyk, M.S.; Wang, C.; et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature 2018. [Google Scholar] [CrossRef] [PubMed]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Dual Roles of IL-27 in Cancer Biology and Immunotherapy. Mediat. Inflamm. 2017. [Google Scholar] [CrossRef]
- Gide, T.N.; Quek, C.; Menzies, A.M.; Tasker, A.T.; Shang, P.; Holst, J.; Madore, J.; Lim, S.Y.; Velickovic, R.; Wongchenko, M.; et al. Distinct Immune Cell Populations Define Response to Anti-PD-1 Monotherapy and Anti-PD-1/Anti-CTLA-4 Combined Therapy. Cancer Cell 2019, 35, 238–255.e6. [Google Scholar] [CrossRef]
- Bosisio, D.; Polentarutti, N.; Sironi, M.; Bernasconi, S.; Miyake, K.; Webb, G.R.; Martin, M.U.; Mantovani, A.; Muzio, M. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-γ: A molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 2002, 99, 3427–3431. [Google Scholar] [CrossRef]
- Kondratova, M.; Czerwinska, U.; Sompairac, N.; Amigorena, S.D.; Soumelis, V.; Barillot, E.; Zinovyev, A.; Kuperstein, I. A multiscale signalling network map of innate immune response in cancer reveals cell heterogeneity signatures. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Kondratova, M.; Sompairac, N.; Barillot, E.; Zinovyev, A.; Kuperstein, I. Signalling maps in cancer research: Construction and data analysis. Database 2018. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Conditions | Initial Conditions |
|---|---|
| 1. Role of individual inhibiting immune checkpoint receptors in TCR signalling modulation | CD80/86 (ligand for CTLA4 or CD28) only |
| PD1_L (ligand for PD1) only | |
| LAG3_L (ligand for LAG3) only | |
| TIM3_L (ligand for TIM3) only | |
| TIGIT_L (ligand for TIGIT or CD226) only | |
| 2. Priming in lymph node (LN) vs. secondary tumour microenvironment (TME) activation (two-step process) | LN (immune checkpoints active): CTLA4, ICOS, TNFRs |
| TME (immune checkpoints active): ICOS, TNFRs, PD1, TIGIT, LAG3, TIM3 | |
| 3. CD8+ vs. CD4+ cells | CD8+ (ligands): MHCI |
| CD4+ (ligands): MHCII |
| Cell Population | Type of ICI Therapy | T Cell Proliferation | Th1_Cytotoxicity | Treg | |||
|---|---|---|---|---|---|---|---|
| Model | Experimental Data (Different Methods) | Model | Experimental Data IFNG/pTbet/GZMB (Granzyme B) | Model | Experimental Data ** Treg Ratio/IL10/TGFβ | ||
| All TILs | anti-CTLA4 | NA | ↑ | ↑/NA/NA | ↓ | No effect/No effect/- - | |
| All TILs | anti-PD1 (anti PD1-L) | NA | ↑ | ↑/NA/NA | ↓ | ↓/↓/↓ | |
| All TILs | anti-CTLA4 & anti-PD1 | NA | ↑↑ | ↑↑/NA/NA | ↓↓ | ↓↓/↓/↓↓ | |
| (anti-CTLA4 & anti PD1-L) | |||||||
| CD8+ (total) | Anti-CTLA4 | ↑ (clonal expansion in LN) | ↑ | ↑ | ↑/↑/↑ | NA | |
| CD8+ (total) | anti-PD1 (anti-PD1-L) | ↑ (effect in the TME) | ↑ (↑) | ↑ | ↑/↑/↑ | NA | |
| (↑/↑↑/↑) | |||||||
| CD8+ (total) | anti-CTLA4 & anti-PD1 | ↑↑ | ↑↑ | ↑↑ | ↑↑/↑/↑↑ | NA | |
| (anti PD1-L) | (↑↑) | (↑↑/↑↑/↑↑) | |||||
| CD8+ PD1+CTLA4− | anti-CTLA4 | no effect * | no effect | NA | NA | ||
| CD8+ PD1+CTLA4− | anti-PD1 | ↑ | ↑ | NA | NA | ||
| CD8+ PD1+CTLA4− | anti-CTLA4 & anti-PD1 | ↑ * (same effect as anti-PD1 alone) | ↑ | NA | NA | ||
| CD8+ PD1+CTLA4+ | anti-CTLA4 | ↑ | ↑ | NA | NA | ||
| CD8+ PD1+CTLA4+ | anti-PD1 | ↑ | ↑ | NA | NA | ||
| CD8+ PD1+CTLA4+ | anti-CTLA4 + anti-PD1 | ↑↑ | ↑↑ | NA | NA | ||
| Therapy | LN: Proliferation | LN: Th1_CTL/Treg Ratio | TME: Proliferation | TME: Th1_CTL/Treg Ratio | Clinical Outcome: (Patients Survival) [5] |
|---|---|---|---|---|---|
| anti-CTLA4 | ↑ | ↑ | no effect | no effect | ↑ |
| anti-PD1 | no effect | no effect | ↑ | ↑ | ↑↑ |
| anti-PD1+anti-CTLA4 | ↑ | ↑ | ↑ | ↑ | ↑↑↑ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kondratova, M.; Barillot, E.; Zinovyev, A.; Calzone, L. Modelling of Immune Checkpoint Network Explains Synergistic Effects of Combined Immune Checkpoint Inhibitor Therapy and the Impact of Cytokines in Patient Response. Cancers 2020, 12, 3600. https://doi.org/10.3390/cancers12123600
Kondratova M, Barillot E, Zinovyev A, Calzone L. Modelling of Immune Checkpoint Network Explains Synergistic Effects of Combined Immune Checkpoint Inhibitor Therapy and the Impact of Cytokines in Patient Response. Cancers. 2020; 12(12):3600. https://doi.org/10.3390/cancers12123600
Chicago/Turabian StyleKondratova, Maria, Emmanuel Barillot, Andrei Zinovyev, and Laurence Calzone. 2020. "Modelling of Immune Checkpoint Network Explains Synergistic Effects of Combined Immune Checkpoint Inhibitor Therapy and the Impact of Cytokines in Patient Response" Cancers 12, no. 12: 3600. https://doi.org/10.3390/cancers12123600
APA StyleKondratova, M., Barillot, E., Zinovyev, A., & Calzone, L. (2020). Modelling of Immune Checkpoint Network Explains Synergistic Effects of Combined Immune Checkpoint Inhibitor Therapy and the Impact of Cytokines in Patient Response. Cancers, 12(12), 3600. https://doi.org/10.3390/cancers12123600