A Novel Pipeline for Drug Repurposing for Bladder Cancer Based on Patients’ Omics Signatures

 , , ,
, , ,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
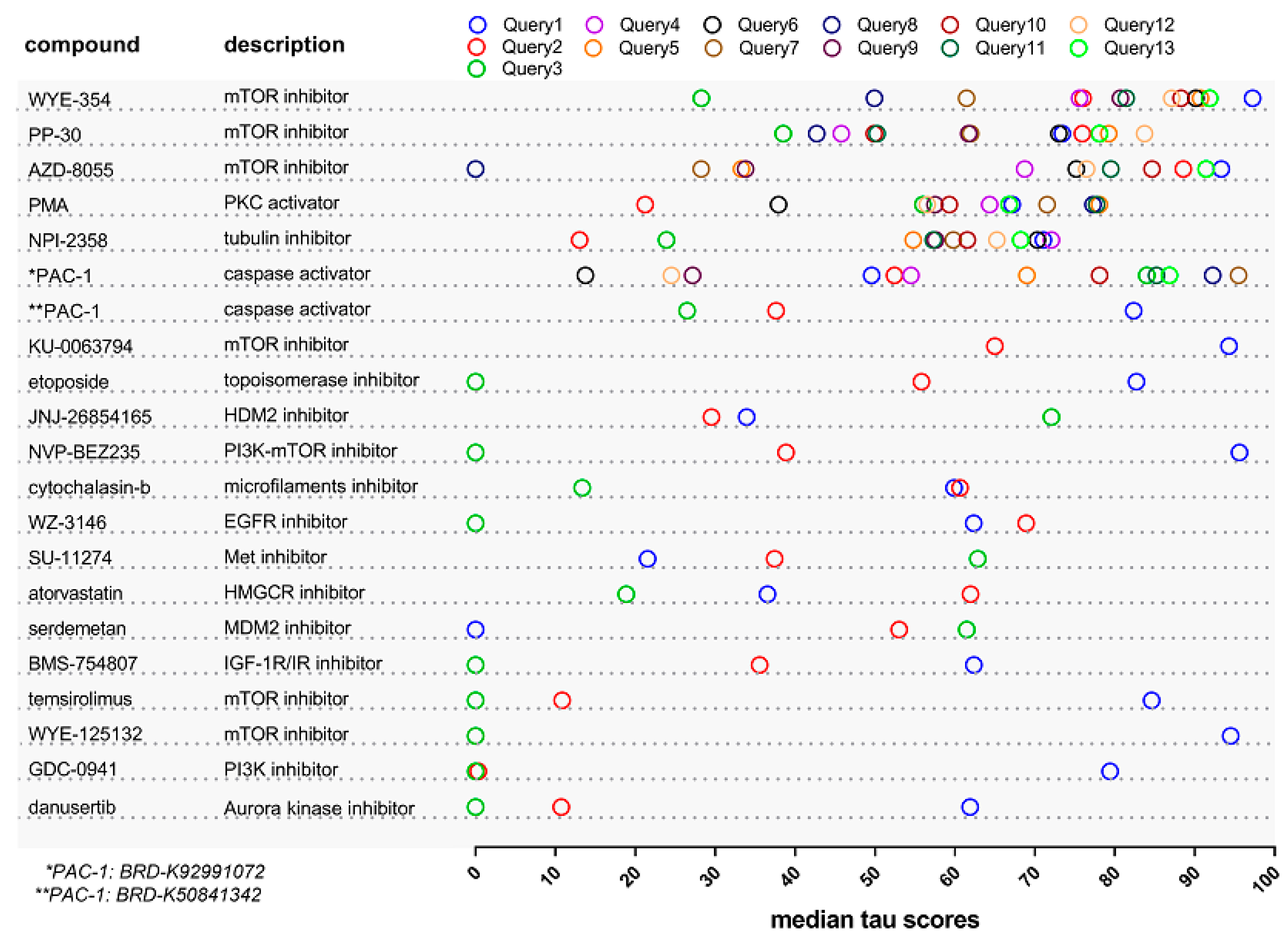
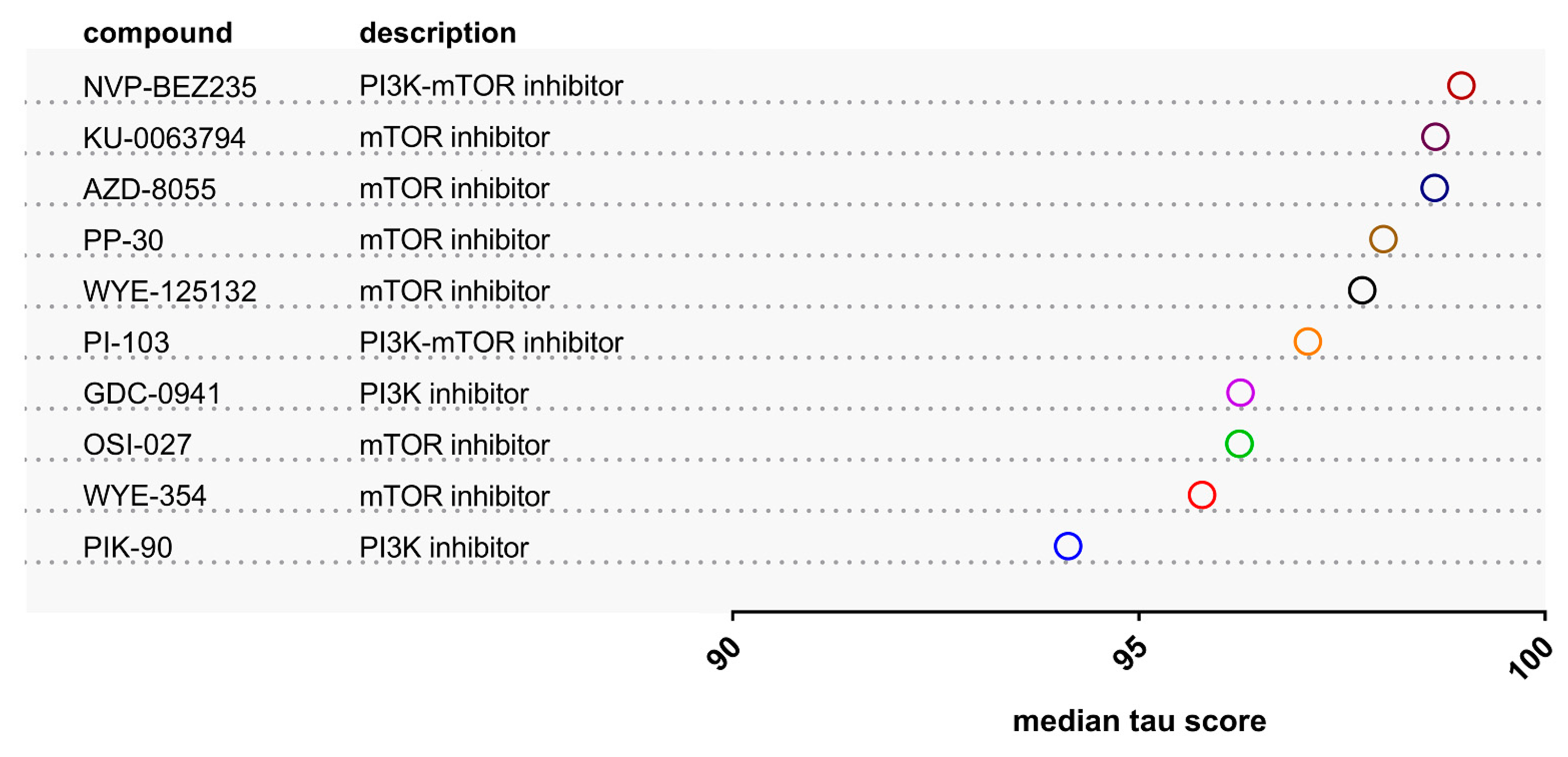
2.1. Identification of Drugs with Reversal Potential for High-Risk NMIBC
2.2. The Effect of the WYE-354 mTOR Inhibitor on BC Cell Lines In Vitro
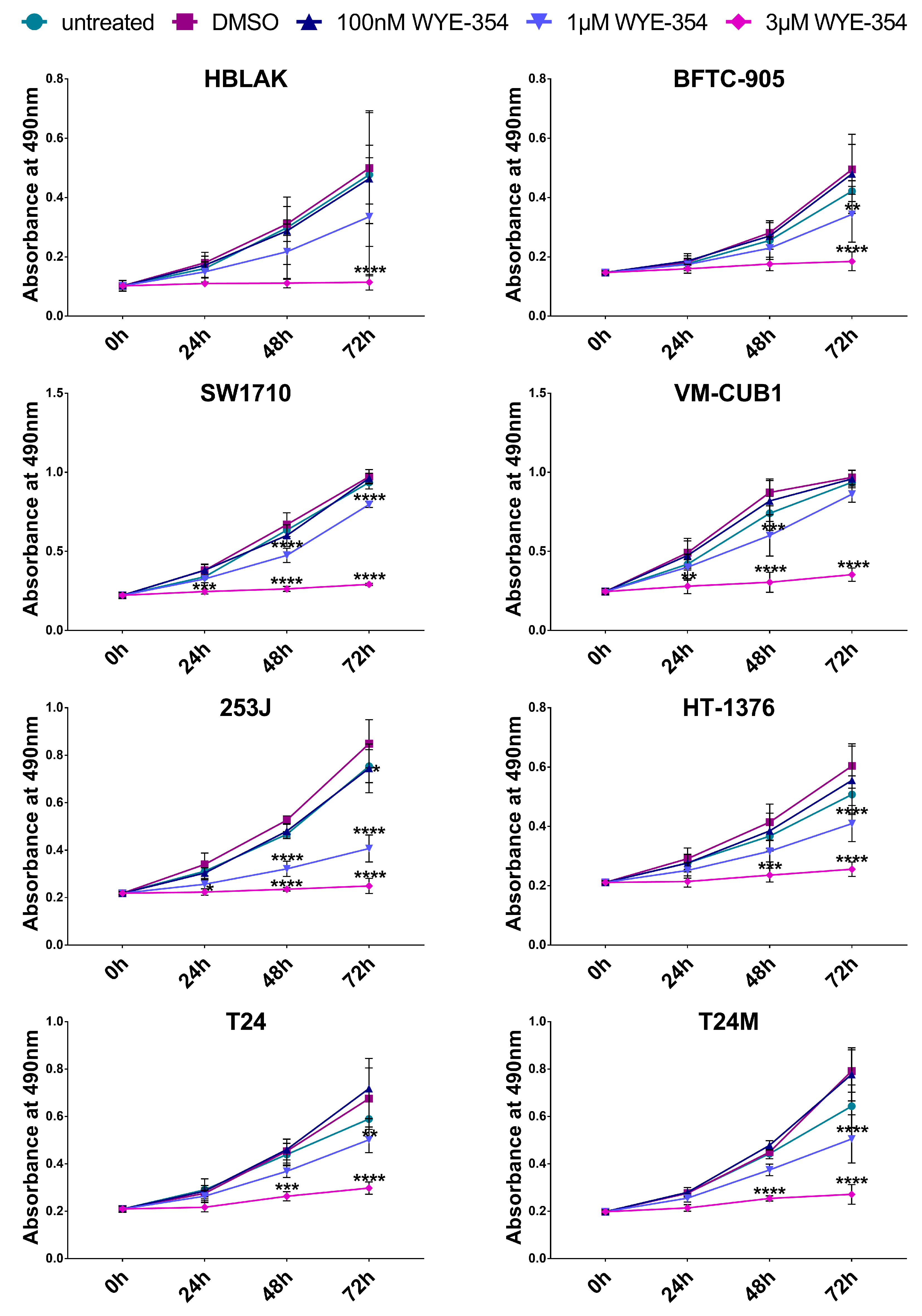
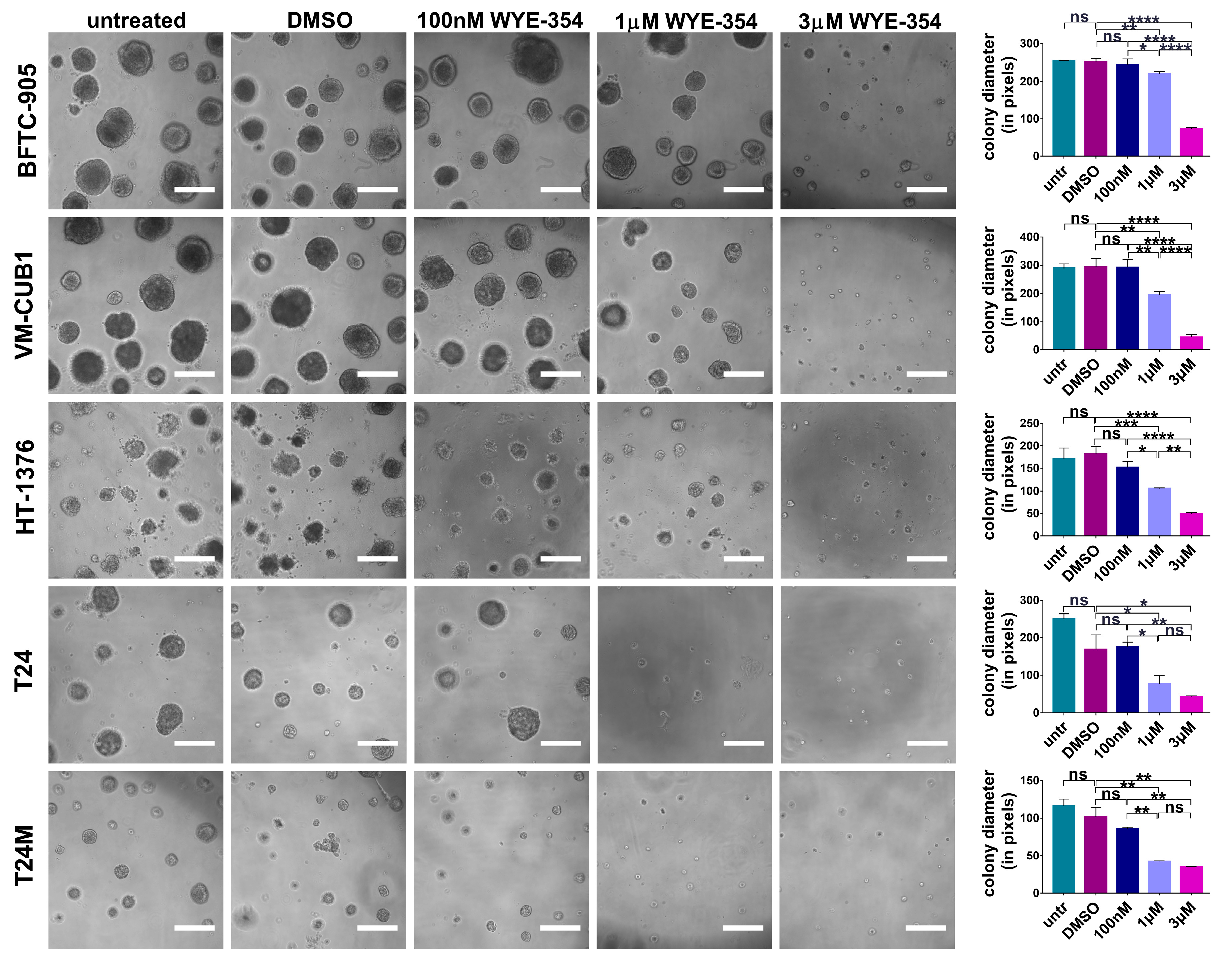
2.2.1. WYE-354 Reduces the Growth of BC Cells
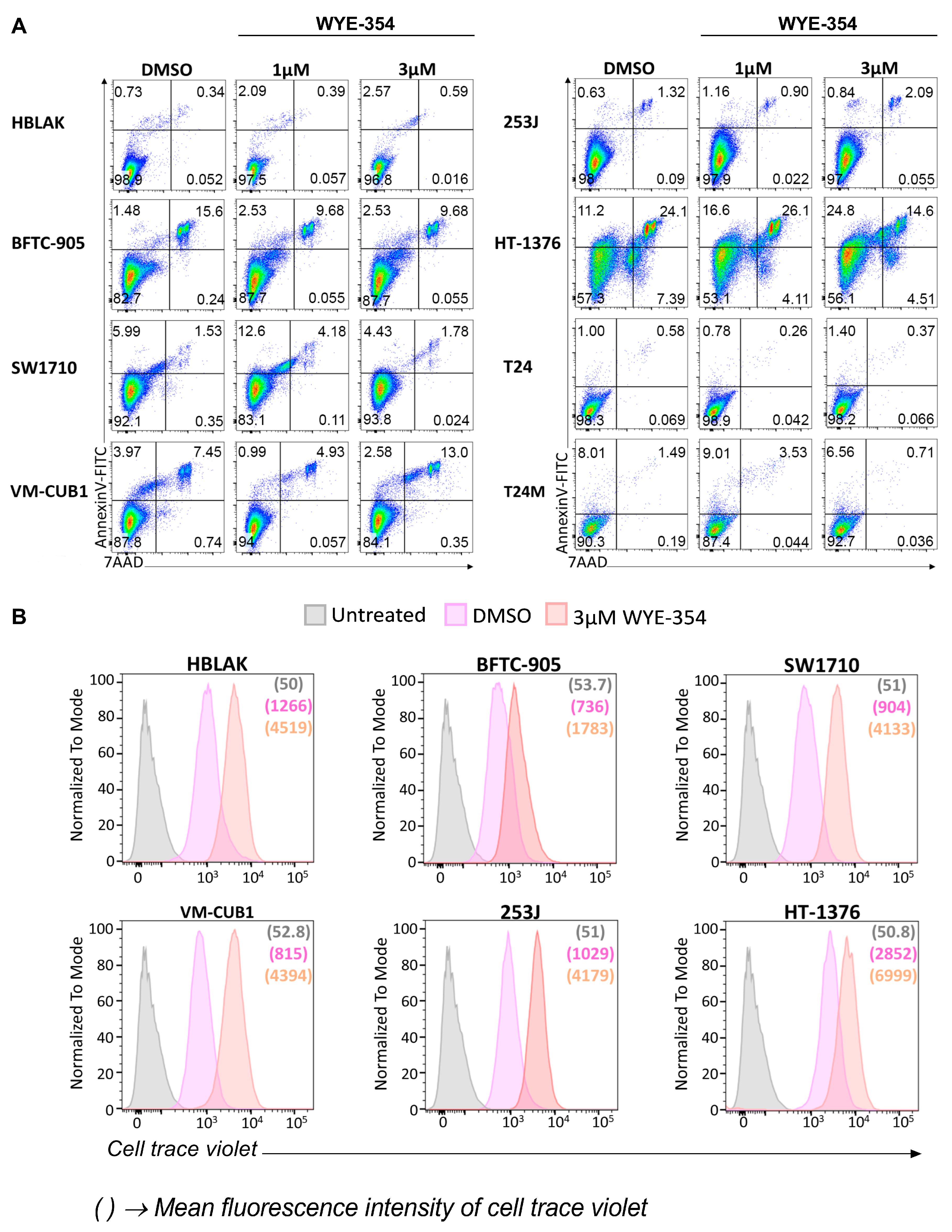
2.2.2. WYE-354 Does Not Affect BC Cell Viability
3. Discussion
4. Materials and Methods
4.1. Connectivity Map Analysis
4.2. Cell Culture
4.3. WYE-354 Inhibitor
4.4. MTS Cell Proliferation Assay
4.5. Colony Formation Assay
4.6. Apoptosis Analysis
4.7. Cell Trace
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.M.; Hahn, N.M.; Efstathiou, J.A.; Lerner, S.P.; Malmstrom, P.U.; Choi, W.; Guo, C.C.; Lotan, Y.; Kassouf, W. Bladder cancer. Lancet 2016, 388, 2796–2810. [Google Scholar] [CrossRef]
- Babjuk, M.; Burger, M.; Compérat, E.M.; Gontero, P.; Mostafid, A.H.; Palou, J.; Van Rhijn, B.W.G.; Rouprêt, M.; Shariat, S.F.; Sylvester, R.; et al. European association of urology guidelines on non-muscle-invasive bladder cancer (TaT1 and Carcinoma in situ)—2019 update. Eur. Urol. 2019, 76, 639–657. [Google Scholar] [CrossRef] [PubMed]
- Kiselyov, A.; Bunimovich-Mendrazitsky, S.; Startsev, V. Key signaling pathways in the muscle-invasive bladder carcinoma: Clinical markers for disease modeling and optimized treatment. Int. J. Cancer 2015, 138, 2562–2569. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.S.; Shipley, W.U.; Feldman, A.S. Bladder cancer. Lancet 2009, 374, 239–249. [Google Scholar] [CrossRef]
- Patel, V.G.; Oh, W.K.; Galsky, M.D. Treatment of muscle-invasive and advanced bladder cancer in 2020. CA Cancer J. Clin. 2020. [Google Scholar] [CrossRef]
- Gopalakrishnan, D.; Koshkin, V.S.; Ornstein, M.C.; Papatsoris, A.; Grivas, P. Immune checkpoint inhibitors in urothelial cancer: Recent updates and future outlook. Ther. Clin. Risk Manag. 2018, 14, 1019–1040. [Google Scholar] [CrossRef]
- Richters, A.; Aben, K.K.H.; Kiemeney, L.A.L.M. The global burden of urinary bladder cancer: An update. World J. Urol. 2020, 38, 1895–1904. [Google Scholar] [CrossRef]
- Leal, J.; Luengo-Fernandez, R.; Sullivan, R.; Witjes, J.A. Economic Burden of Bladder Cancer Across the European Union. Eur. Urol. 2016, 69, 438–447. [Google Scholar] [CrossRef]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Issa, N.T.; Stathias, V.; Schürer, S.; Dakshanamurthy, S. Machine and deep learning approaches for cancer drug repurposing. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 2017, 171, 1437–1452.e17. [Google Scholar] [CrossRef] [PubMed]
- Klein, J.; Caubet, C.; Camus, M.; Makridakis, M.; Denis, C.; Gilet, M.; Feuillet, G.; Rascalou, S.; Neau, E.; Garrigues, L.; et al. Connectivity mapping of glomerular proteins identifies dimethylaminoparthenolide as a new inhibitor of diabetic kidney disease. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Schanstra, J.P.; Luong, T.T.; Makridakis, M.; Van Linthout, S.; Lygirou, V.; Latosisnska, A.; Alesutan, I.; Boehme, B.; Schelski, N.; Von Lewinski, D.; et al. Systems biology identifies cytosolic PLA2 as a target in vascular calcification treatment. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Ganci, F.; Pulito, C.; Valsoni, S.; Sacconi, A.; Turco, C.; Vahabi, M.; Manciocco, V.; Mazza, E.M.C.; Meens, J.; Karamboulas, C.; et al. PI3K inhibitors curtail MYC-dependent mutant p53 gain-of-function in head and neck squamous cell carcinoma. Clin. Cancer Res. 2020, 26, 2956–2971. [Google Scholar] [CrossRef]
- Stroggilos, R.; Mokou, M.; Lotosinska, A.; Makridakis, M.; Lygirou, V.; Mavrogeorgis, E.; Drekolias, D.; Frantzi, M.; Mullen, W.; Fragkoulis, C.; et al. Proteome-based classification of nonmuscle invasive bladder cancer. Int. J. Cancer 2020, 146, 281–294. [Google Scholar]
- Hurst, C.D.; Alder, O.; Platt, F.M.; Droop, A.; Stead, L.F.; Burns, J.E.; Burghel, G.J.; Jain, S.; Klimczak, L.J.; Lindsay, H.; et al. Genomic subtypes of non-invasive bladder cancer with distinct metabolic profile and female gender bias in KDM6A mutation frequency. Cancer Cell 2017, 32, 701–715.e7. [Google Scholar] [CrossRef]
- Sjödahl, G.; Lauss, M.; Lövgren, K.; Chebil, G.; Gudjonsson, S.; Veerla, S.; Patschan, O.; Aine, M.; Fernö, M.; Ringnér, M.; et al. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res. 2012, 18, 3377–3386. [Google Scholar] [CrossRef]
- Frantzi, M.; Latosinska, A.; Mokou, M.; Mischak, H.; Vlahou, A. Drug repurposing in oncology: Better guided strategies in the omics era. Lancet Oncol. 2020, in press. [Google Scholar]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Van Der Horst, G.; Van De Merbel, A.F.; Ruigrok, E.; Van Der Mark, M.H.; Ploeg, E.; Appelman, L.; Tvingsholm, S.; Jäätelä, M.; Van Uhm, J.; De Julio, M.K.; et al. Cationic amphiphilic drugs as potential anticancer therapy for bladder cancer. Mol. Oncol. 2020. [Google Scholar] [CrossRef]
- Kiehl, I.G.A.; Riccetto, E.; Salustiano, A.C.C.; Ossick, M.V.; Ferrari, K.L.; Assalin, H.B.; Ikari, O.; Reis, L.O. Boosting bladder cancer treatment by intravesical nitazoxanide and bacillus calmette-guérin association. World J. Urol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Hamada, A.; Saito, R.; Teramoto, Y.; Tanaka, R.; Takano, K.; Nakayama, K.; Murakami, K.; Matsumoto, K.; Akamatsu, S.; et al. Systematic chemical screening identifies disulfiram as a repurposed drug that enhances sensitivity to cisplatin in bladder cancer: A summary of preclinical studies. Br. J. Cancer 2019, 121, 1027–1038. [Google Scholar] [CrossRef]
- Lee, S.H.; Hu, W.; Matulay, J.T.; Silva, M.V.; Owczarek, T.B.; Kim, K.; Chua, C.W.; Barlow, L.J.; Kandoth, C.; Williams, A.B.; et al. Tumor evolution and drug response in patient-derived organoid models of bladder cancer. Cell 2018, 173, 515–528.e17. [Google Scholar] [CrossRef]
- Liu, S.T.; Hui, G.; Mathis, C.; Chamie, K.; Pantuck, A.J.; Drakaki, A. The current status and future role of the phosphoinositide 3 Kinase/AKT signaling pathway in urothelial cancer: An old pathway in the new immunotherapy era. Clin. Genitourin. Cancer 2018, 16, e269–e276. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Meeks, J.J.; Kuzel, T.M.; Scaranti, M.; Abdulkadir, S.A.; Giles, F.J. Emerging therapeutic targets in bladder cancer. Cancer Treat. Rev. 2015, 41, 170–178. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; Pinheiro, P.D.S.M.; Lima, L.M.; Fraga, C.A.M.; Barreiro, E.J. N-Acylhydrazones as drugs. Bioorganic Med. Chem. Lett. 2018, 28, 2797–2806. [Google Scholar] [CrossRef]
- Kingston, D.G.I. Tubulin-interactive natural products as anticancer agents. J. Nat. Prod. 2009, 72, 507–515. [Google Scholar] [CrossRef]
- Nicholson, B.; Lloyd, G.K.; Miller, B.R.; Palladino, M.A.; Kiso, Y.; Hayashi, Y.; Neuteboom, S.T.C. NPI-2358 is a tubulin-depolymerizing agent: In-vitro evidence for activity as a tumor vascular-disrupting agent. Anti-Cancer Drugs 2006, 17, 25–31. [Google Scholar] [CrossRef]
- Wang, H.; Owens, C.; Chandra, N.; Conaway, M.R.; Brautigan, D.L.; Theodorescu, D. Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res. 2010, 70, 8760–8769. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.; Islam, S.A.; Bommareddy, R.R.; Smalley, T.; Acevedo‑Duncan, M. Simultaneous inhibition of atypical protein kinase‑C and mTOR impedes bladder cancer cell progression. Int. J. Oncol. 2020, 56, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, J. PKCalpha and Netrin-1/UNC5B positive feedback control in relation with chemical therapy in bladder cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 1712–1717. [Google Scholar]
- Dlugosz, A.A.; Mischak, H.; Mushinski, J.F.; Yuspa, S.H. Transcripts encoding protein kinase C-alpha, -delta, -epsilon, -zeta, and -eta are expressed in basal and differentiating mouse keratinocytes in vitro and exhibit quantitative changes in neoplastic cells. Mol. Carcinog. 1992, 5, 286–292. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Newton, A.C.; Brognard, J. Reversing the paradigm: Protein kinase C as a tumor suppressor. Trends Pharmacol. Sci. 2017, 38, 438–447. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 1–19. [Google Scholar] [CrossRef]
- Niegisch, G.; Retz, M.; Thalgott, M.; Balabanov, S.; Honecker, F.; Ohlmann, C.H.; Stöckle, M.; Bögemann, M.; Dorp, F.V.; Gschwend, J.; et al. Second-line treatment of advanced urothelial cancer with paclitaxel and everolimus in a german phase II trial (AUO Trial AB 35/09). Oncology 2015, 89, 70–78. [Google Scholar] [CrossRef]
- Milowsky, M.I.; Iyer, G.; Regazzi, A.M.; Al-Ahmadie, H.; Gerst, S.R.; Ostrovnaya, I.; Gellert, L.L.; Kaplan, R.; Garcia-Grossman, I.R.; Pendse, D.; et al. Phase II study of everolimus in metastatic urothelial cancer. BJU Int. 2013, 112, 462–470. [Google Scholar] [CrossRef]
- Chiarini, F.; Evangelisti, C.; McCubrey, J.A.; Martelli, A.M. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol. Sci. 2015, 36, 124–135. [Google Scholar] [CrossRef]
- Hau, A.M.; Nakasaki, M.; Nakashima, K.; Krish, G.; Hansel, D.E. Differential mTOR pathway profiles in bladder cancer cell line subtypes to predict sensitivity to mTOR inhibition. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009, 69, 6232–6240. [Google Scholar] [CrossRef]
- Callegari, E.; D’Abundo, L.; Guerriero, P.; Simioni, C.; Elamin, B.K.; Russo, M.; Cani, A.; Bassi, C.; Zagatti, B.; Giacomelli, L.; et al. MiR-199a-3p modulates MTOR and PAK4 pathways and inhibits tumor growth in a hepatocellular carcinoma transgenic mouse model. Mol. Ther. Nucleic Acids 2018, 11, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, S.M.; Bakhashab, S.; Ilyas, A.M.; Pushparaj, P.N.; Karim, S.; Khan, J.A.; Abuzenadah, A.M.; Chaudhary, A.G.; Al-Qahtani, M.H.; Ahmed, F. WYE-354 restores Adriamycin sensitivity in multidrug-resistant acute myeloid leukemia cell lines. Oncol. Rep. 2019, 41, 3179–3188. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef]
- Makridakis, M.; Gagos, S.; Petrolekas, A.; Roubelakis, M.G.; Bitsika, V.; Stravodimos, K.; Pavlakis, K.; Anagnou, N.P.; Coleman, J.; Vlahou, A. Chromosomal and proteome analysis of a new T24-based cell line model for aggressive bladder cancer. Proteomics 2009, 9, 287–298. [Google Scholar] [CrossRef]
- Hoffmann, M.J.; Koutsogiannouli, E.; Skowron, M.A.; Pinkerneil, M.; Niegisch, G.; Brandt, A.; Stepanow, S.; Rieder, H.; Schulz, W.A. The new immortalized uroepithelial cell Line HBLAK contains defined genetic aberrations typical of early stage urothelial tumors. Bladder Cancer 2016, 2, 449–463. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mokou, M.; Lygirou, V.; Angelioudaki, I.; Paschalidis, N.; Stroggilos, R.; Frantzi, M.; Latosinska, A.; Bamias, A.; Hoffmann, M.J.; Mischak, H.; et al. A Novel Pipeline for Drug Repurposing for Bladder Cancer Based on Patients’ Omics Signatures. Cancers 2020, 12, 3519. https://doi.org/10.3390/cancers12123519
Mokou M, Lygirou V, Angelioudaki I, Paschalidis N, Stroggilos R, Frantzi M, Latosinska A, Bamias A, Hoffmann MJ, Mischak H, et al. A Novel Pipeline for Drug Repurposing for Bladder Cancer Based on Patients’ Omics Signatures. Cancers. 2020; 12(12):3519. https://doi.org/10.3390/cancers12123519
Chicago/Turabian StyleMokou, Marika, Vasiliki Lygirou, Ioanna Angelioudaki, Nikolaos Paschalidis, Rafael Stroggilos, Maria Frantzi, Agnieszka Latosinska, Aristotelis Bamias, Michèle J. Hoffmann, Harald Mischak, and et al. 2020. "A Novel Pipeline for Drug Repurposing for Bladder Cancer Based on Patients’ Omics Signatures" Cancers 12, no. 12: 3519. https://doi.org/10.3390/cancers12123519
APA StyleMokou, M., Lygirou, V., Angelioudaki, I., Paschalidis, N., Stroggilos, R., Frantzi, M., Latosinska, A., Bamias, A., Hoffmann, M. J., Mischak, H., & Vlahou, A. (2020). A Novel Pipeline for Drug Repurposing for Bladder Cancer Based on Patients’ Omics Signatures. Cancers, 12(12), 3519. https://doi.org/10.3390/cancers12123519