Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes

 , , , ,
, , , ,  ,
,  , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. NK Cells
2.1. PD-1
2.2. KIRs
2.3. NKG2A
2.4. TIM-3
2.5. TIGIT
2.6. LAG-3
3. Helper ILCs
3.1. PD-1
3.2. PD-1 Expression on ILCs in Cancer
3.3. CTLA-4
3.4. TIGIT and CD96, and TGF-β
3.5. KLRG1
3.6. TIM-3
3.7. LAG-3
3.8. NKG2A
3.9. Metabolic Checkpoints
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Klose, C.S.N.; Artis, D. Innate lymphoid cells as regulators of immunity, inflammation and tissue homeostasis. Nat. Immunol. 2016, 17, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; Di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate Lymphoid Cells: 10 Years On. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcenaro, E.; Ferranti, B.; Moretta, A. NK-DC interaction: On the usefulness of auto-aggression. Autoimmun Rev. 2005, 4, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Di Vito, C.; Mikulak, J.; Zaghi, E.; Pesce, S.; Marcenaro, E.; Mavilio, D. NK cells to cure cancer. Semin. Immunol. 2019, 41, 101272. [Google Scholar] [CrossRef]
- Marcenaro, E.; Dondero, A.; Moretta, A. Multi-directional cross-regulation of NK cell function during innate immune responses. Transpl. Immunol. 2006, 17, 16–19. [Google Scholar] [CrossRef]
- Stabile, H.; Fionda, C.; Gismondi, A.; Santoni, A. Role of Distinct Natural Killer Cell Subsets in Anticancer Response. Front. Immun. 2017, 8, 293. [Google Scholar] [CrossRef] [Green Version]
- Lugli, E.; Marcenaro, E.; Mavilio, D. NK Cell Subset Redistribution during the Course of Viral Infections. Front. Immun. 2014, 5, 390. [Google Scholar] [CrossRef] [Green Version]
- Marcenaro, E.; Notarangelo, L.D.; Orange, J.S.; Vivier, E. Editorial: NK Cell Subsets in Health and Disease: New Developments. Front. Immun. 2017, 8, 1363. [Google Scholar] [CrossRef] [Green Version]
- Roberto, A.; Di Vito, C.; Zaghi, E.; Mazza, E.M.C.; Capucetti, A.; Calvi, M.; Tentorio, P.; Zanon, V.; Sarina, B.; Mariotti, J.; et al. The early expansion of anergic NKG2A pos/CD56 dim/CD16 neg natural killer represents a therapeutic target in haploidentical hematopoietic stem cell transplantation. Haematologica 2018, 103, 1390–1402. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial Type 1 Innate Lymphoid Cells Are a Unique Subset of IL-12- and IL-15-Responsive IFN-γ-Producing Cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trabanelli, S.; Gomez-Cadena, A.; Salomé, B.; Michaud, K.; Mavilio, D.; Landis, B.N.; Jandus, P.; Jandus, C. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping: HILC PHENOTYPE. Cytometry 2018, 94, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Salomé, B.; Jandus, C. Innate lymphoid cells in antitumor immunity. J. Leukoc. Biol. 2018, 103, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Herbert, D.B.; Douglas, B.; Zullo, K. Group 2 Innate Lymphoid Cells (ILC2): Type 2 Immunity and Helminth Immunity. IJMS 2019, 20, 2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.W.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin–EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montaldo, E.; Juelke, K.; Romagnani, C. Group 3 innate lymphoid cells (ILC3s): Origin, differentiation, and plasticity in humans and mice: Highlights—(Mini-Review). Eur. J. Immunol. 2015, 45, 2171–2182. [Google Scholar] [CrossRef] [PubMed]
- Vacca, P.; Pesce, S.; Greppi, M.; Fulcheri, E.; Munari, E.; Olive, D.; Mingari, M.C.; Moretta, A.; Moretta, L.; Marcenaro, E. PD-1 is expressed by and regulates human group 3 innate lymphoid cells in human decidua. Mucosal Immunol. 2019, 12, 624–631. [Google Scholar] [CrossRef]
- Ishizuka, I.E.; Chea, S.; Gudjonson, H.; Constantinides, M.G.; Dinner, A.R.; Bendelac, A.; Golub, R. Single-cell analysis defines the divergence between the innate lymphoid cell lineage and lymphoid tissue–inducer cell lineage. Nat. Immunol. 2016, 17, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Zhong, C.; Zheng, M.; Zhu, J. Lymphoid tissue inducer—A divergent member of the ILC family. Cytokine Growth Factor Rev. 2018, 42, 5–12. [Google Scholar] [CrossRef]
- Simoni, Y.; Fehlings, M.; Kløverpris, H.N.; McGovern, N.; Koo, S.-L.; Loh, C.Y.; Lim, S.; Kurioka, A.; Fergusson, J.R.; Tang, C.-L.; et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity 2017, 46, 148–161. [Google Scholar] [CrossRef] [Green Version]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Matsuyama, S.; Miyake, S.; Suga, K.; Nakachi, K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: An 11-year follow-up study of a general population. Lancet 2000, 356, 1795–1799. [Google Scholar] [CrossRef]
- Crinier, A.; Vivier, E.; Bléry, M. Helper-like innate lymphoid cells and cancer immunotherapy. Semin. Immunol. 2019, 41, 101274. [Google Scholar] [CrossRef] [PubMed]
- Salimi, M.; Wang, R.; Yao, X.; Li, X.; Wang, X.; Hu, Y.; Chang, X.; Fan, P.; Dong, T.; Ogg, G. Activated innate lymphoid cell populations accumulate in human tumour tissues. BMC Cancer 2018, 18, 341. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wei, R.; Lin, Y.; Kwok, H.F. Clinical and Recent Patents Applications of PD-1/PD-L1 Targeting Immunotherapy in Cancer Treatment-Current Progress, Strategy, and Future Perspective. Front. Immun. 2020, 11, 1508. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Tabellini, G.; Rampinelli, F.; Parolini, S.; Olive, D.; Moretta, L.; Moretta, A.; Marcenaro, E. Identification of a subset of human natural killer cells expressing high levels of programmed death 1: A phenotypic and functional characterization. J. Allergy Clin. Immunol. 2017, 139, 335–346.e333. [Google Scholar] [CrossRef] [Green Version]
- Beldi-Ferchiou, A.; Lambert, M.; Dogniaux, S.; Vély, F.; Vivier, E.; Olive, D.; Dupuy, S.; Levasseur, F.; Zucman, D.; Lebbé, C.; et al. PD-1 mediates functional exhaustion of activated NK cells in patients with Kaposi sarcoma. Oncotarget 2016, 7, 72961–72977. [Google Scholar] [CrossRef] [Green Version]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef] [Green Version]
- Tumino, N.; Martini, S.; Munari, E.; Scordamaglia, F.; Besi, F.; Mariotti, F.R.; Bogina, G.; Mingari, M.C.; Vacca, P.; Moretta, L. Presence of innate lymphoid cells in pleural effusions of primary and metastatic tumors: Functional analysis and expression of PD-1 receptor. Int. J. Cancer 2019, 145, 1660–1668. [Google Scholar] [CrossRef]
- Niu, C.; Li, M.; Zhu, S.; Chen, Y.; Zhou, L.; Xu, D.; Xu, J.; Li, Z.; Li, W.; Cui, J. PD-1-positive Natural Killer Cells have a weaker antitumor function than that of PD-1-negative Natural Killer Cells in Lung Cancer. Int. J. Med. Sci. 2020, 17, 1964–1973. [Google Scholar] [CrossRef]
- Pesce, S.; Belgrano, V.; Greppi, M.; Carlomagno, S.; Squillario, M.; Barla, A.; Della Chiesa, M.; Di Domenico, S.; Mavilio, D.; Moretta, L.; et al. Different Features of Tumor-Associated NK Cells in Patients With Low-Grade or High-Grade Peritoneal Carcinomatosis. Front. Immun. 2019, 10, 1963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, N.H.; Naidoo, J.; Curigliano, G.; Patel, S.; Sahebjam, S.; Papadopoulos, K.P.; Gordon, M.S.; Wang, D.; Gómez Rueda, A.; Song, X.; et al. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. JCO 2018, 36, 3540. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Qu, Y.; Xia, P.; Chen, Y.; Zhu, X.; Zhang, J.; Wang, G.; Tian, Y.; Ying, J.; Fan, Z. Transdifferentiation of tumor infiltrating innate lymphoid cells during progression of colorectal cancer. Cell Res. 2020, 30, 610–622. [Google Scholar] [CrossRef]
- Vey, N.; Karlin, L.; Sadot-Lebouvier, S.; Broussais, F.; Berton-Rigaud, D.; Rey, J.; Charbonnier, A.; Marie, D.; André, P.; Paturel, C.; et al. A phase 1 study of lirilumab (antibody against killer immunoglobulin-like receptor antibody KIR2D; IPH2102) in patients with solid tumors and hematologic malignancies. Oncotarget 2018, 9, 17675–17688. [Google Scholar] [CrossRef] [Green Version]
- Tinker, A.V.; Hirte, H.W.; Provencher, D.; Butler, M.; Ritter, H.; Tu, D.; Azim, H.A.; Paralejas, P.; Grenier, N.; Hahn, S.-A.; et al. Dose-Ranging and Cohort-Expansion Study of Monalizumab (IPH2201) in Patients with Advanced Gynecologic Malignancies: A Trial of the Canadian Cancer Trials Group (CCTG): IND221. Clin. Cancer Res. 2019, 25, 6052–6060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Bléry, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743.e1713. [Google Scholar] [CrossRef] [Green Version]
- Salomé, B.; Gomez-Cadena, A.; Loyon, R.; Suffiotti, M.; Salvestrini, V.; Wyss, T.; Vanoni, G.; Ruan, D.F.; Rossi, M.; Tozzo, A.; et al. CD56 as a marker of an ILC1-like population with NK cell properties that is functionally impaired in AML. Blood Adv. 2019, 3, 3674–3687. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, J.; Gu, H.; Yuan, Y.; Zhang, B.; Zhu, D.; Zhou, J.; Zhu, Y.; Chen, W. The Clinical Significance of Abnormal Tim-3 Expression on NK Cells from Patients with Gastric Cancer. Immunol. Investig. 2015, 44, 578–589. [Google Scholar] [CrossRef]
- Xu, L.; Huang, Y.; Tan, L.; Yu, W.; Chen, D.; Lu, C.; He, J.; Wu, G.; Liu, X.; Zhang, Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int. Immunopharmacol. 2015, 29, 635–641. [Google Scholar] [CrossRef]
- da Silva, I.P.; Gallois, A.; Jimenez-Baranda, S.; Khan, S.; Anderson, A.C.; Kuchroo, V.K.; Osman, I.; Bhardwaj, N. Reversal of NK-cell exhaustion in advanced melanoma by Tim-3 blockade. Cancer Immunol. Res. 2014, 2, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Gallois, A.; Silva, I.; Osman, I.; Bhardwaj, N. Reversal of natural killer cell exhaustion by TIM-3 blockade. OncoImmunology 2014, 3, e946365. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Kawaguchi, Y.; Sasaki, Y.; Shimada, S.; Murai, S.; Abe, M.; Baba, Y.; Watanuki, M.; Fujiwara, S.; Arai, N.; et al. Monitoring TIGIT/DNAM-1 and PVR/PVRL2 Immune Checkpoint Expression Levels in Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia. Biol. Blood Marrow Transplant. 2019, 25, 861–867. [Google Scholar] [CrossRef]
- Fionda, C.; Stabile, H.; Cerboni, C.; Soriani, A.; Gismondi, A.; Cippitelli, M.; Santoni, A. Hitting More Birds with a Stone: Impact of TGF-β on ILC Activity in Cancer. J. Clin. Med. 2020, 9, 143. [Google Scholar] [CrossRef] [Green Version]
- Sierro, S.; Romero, P.; Speiser, D.E. The CD4-like molecule LAG-3, biology and therapeutic applications. Expert Opin. Ther. Targets 2011, 15, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Magri, G.; Miyajima, M.; Bascones, S.; Mortha, A.; Puga, I.; Cassis, L.; Barra, C.M.; Comerma, L.; Chudnovskiy, A.; Gentile, M.; et al. Innate lymphoid cells integrate stromal and immunological signals to enhance antibody production by splenic marginal zone B cells. Nat. Immunol. 2014, 15, 354–364. [Google Scholar] [CrossRef] [Green Version]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.-C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.J.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, F.R.; Petrini, S.; Ingegnere, T.; Tumino, N.; Besi, F.; Scordamaglia, F.; Munari, E.; Pesce, S.; Marcenaro, E.; Moretta, A.; et al. PD-1 in human NK cells: Evidence of cytoplasmic mRNA and protein expression. OncoImmunology 2019, 8, 1557030. [Google Scholar] [CrossRef] [Green Version]
- Quatrini, L.; Wieduwild, E.; Escaliere, B.; Filtjens, J.; Chasson, L.; Laprie, C.; Vivier, E.; Ugolini, S. Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 2018, 19, 954–962. [Google Scholar] [CrossRef]
- Quatrini, L.; Vacca, P.; Tumino, N.; Besi, F.; Di Pace, A.L.; Scordamaglia, F.; Martini, S.; Munari, E.; Mingari, M.C.; Ugolini, S.; et al. Glucocorticoids and the cytokines IL-12, IL-15, and IL-18 present in the tumor microenvironment induce PD-1 expression on human natural killer cells. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Trefny, M.P.; Kaiser, M.; Stanczak, M.A.; Herzig, P.; Savic, S.; Wiese, M.; Lardinois, D.; Läubli, H.; Uhlenbrock, F.; Zippelius, A. PD-1+ natural killer cells in human non-small cell lung cancer can be activated by PD-1/PD-L1 blockade. Cancer Immunol. Immunother. 2020, 69, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Moretta, A.; Bottino, C.; Pende, D.; Tripodi, G.; Tambussi, G.; Viale, O.; Orengo, A.; Barbaresi, M.; Merli, A.; Ciccone, E. Identification of four subsets of human CD3-CD16+ natural killer (NK) cells by the expression of clonally distributed functional surface molecules: Correlation between subset assignment of NK clones and ability to mediate specific alloantigen recognition. J. Exp. Med. 1990, 172, 1589–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretta, A.; Bottino, C.; Vitale, M.; Pende, D.; Biassoni, R.; Mingari, M.C.; Moretta, L. Receptors for HLA class-I molecules in human natural killer cells. Annu. Rev. Immunol. 1996, 14, 619–648. [Google Scholar] [CrossRef] [PubMed]
- Elliott, J.M.; Yokoyama, W.M. Unifying concepts of MHC-dependent natural killer cell education. Trends Immunol. 2011, 32, 364–372. [Google Scholar] [CrossRef] [Green Version]
- Fauriat, C.; Ivarsson, M.A.; Ljunggren, H.-G.; Malmberg, K.-J.; Michaëlsson, J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood 2010, 115, 1166–1174. [Google Scholar] [CrossRef] [Green Version]
- Marcenaro, E.; Cantoni, C.; Pesce, S.; Prato, C.; Pende, D.; Agaugué, S.; Moretta, L.; Moretta, A. Uptake of CCR7 and acquisition of migratory properties by human KIR+ NK cells interacting with monocyte-derived DC or EBV cell lines: Regulation by KIR/HLA-class I interaction. Blood 2009, 114, 4108–4116. [Google Scholar] [CrossRef]
- Marcenaro, E.; Pesce, S.; Sivori, S.; Carlomagno, S.; Moretta, L.; Moretta, A. KIR2DS1-dependent acquisition of CCR7 and migratory properties by human NK cells interacting with allogeneic HLA-C2+ DCs or T-cell blasts. Blood 2013, 121, 3396–3401. [Google Scholar] [CrossRef] [Green Version]
- Pesce, S.; Moretta, L.; Moretta, A.; Marcenaro, E. Human NK Cell Subsets Redistribution in Pathological Conditions: A Role for CCR7 Receptor. Front. Immun. 2016, 7, 414. [Google Scholar] [CrossRef] [Green Version]
- Pesce, S.; Carlomagno, S.; Moretta, A.; Sivori, S.; Marcenaro, E. Uptake of CCR7 by KIR2DS4⁺ NK cells is induced upon recognition of certain HLA-C alleles. J. Immunol. Res. 2015, 2015, 754373. [Google Scholar] [CrossRef]
- Pesce, S.; Squillario, M.; Greppi, M.; Loiacono, F.; Moretta, L.; Moretta, A.; Sivori, S.; Castagnola, P.; Barla, A.; Candiani, S.; et al. New miRNA Signature Heralds Human NK Cell Subsets at Different Maturation Steps: Involvement of miR-146a-5p in the Regulation of KIR Expression. Front. Immun. 2018, 9, 2360. [Google Scholar] [CrossRef]
- Pesce, S.; Greppi, M.; Ferretti, E.; Obino, V.; Carlomagno, S.; Rutigliani, M.; Thoren, F.B.; Sivori, S.; Castagnola, P.; Candiani, S.; et al. miRNAs in NK Cell-Based Immune Responses and Cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 119. [Google Scholar] [CrossRef] [PubMed]
- Godal, R.; Bachanova, V.; Gleason, M.; McCullar, V.; Yun, G.H.; Cooley, S.; Verneris, M.R.; McGlave, P.B.; Miller, J.S. Natural Killer Cell Killing of Acute Myelogenous Leukemia and Acute Lymphoblastic Leukemia Blasts by Killer Cell Immunoglobulin-Like Receptor–Negative Natural Killer Cells after NKG2A and LIR-1 Blockade. Biol. Blood Marrow Transplant. 2010, 16, 612–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braud, V.M.; Allan, D.S.J.; O’Callaghan, C.A.; Söderström, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; Lopez-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. USA 1998, 95, 5199–5204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulbrecht, M.; Martinozzi, S.; Grzeschik, M.; Hengel, H.; Ellwart, J.W.; Pla, M.; Weiss, E.H. Cutting Edge: The Human Cytomegalovirus UL40 Gene Product Contains a Ligand for HLA-E and Prevents NK Cell-Mediated Lysis. J. Immunol. 2000, 164, 5019–5022. [Google Scholar] [CrossRef] [Green Version]
- Haanen, J.B.; Cerundolo, V. NKG2A, a New Kid on the Immune Checkpoint Block. Cell 2018, 175, 1720–1722. [Google Scholar] [CrossRef] [Green Version]
- Creelan, B.C.; Antonia, S.J. The NKG2A immune checkpoint—A new direction in cancer immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 277–278. [Google Scholar] [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259–1271.e1211. [Google Scholar] [CrossRef]
- Algarra, I.; Garcia-Lora, A.; Cabrera, T.; Ruiz-Cabello, F.; Garrido, F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: Implications for tumor immune escape. Cancer Immunol. Immunother 2004, 53, 904–910. [Google Scholar] [CrossRef]
- Marchesi, M.; Andersson, E.; Villabona, L.; Seliger, B.; Lundqvist, A.; Kiessling, R.; Masucci, G.V. HLA-dependent tumour development: A role for tumour associate macrophages? J. Transl. Med. 2013, 11, 247. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, K.S.; Ginder, G.D. Interferon-γ Induction of the Human Leukocyte Antigen-E Gene Is Mediated through Binding of a Complex Containing STAT1α to a Distinct Interferon-γ-responsive Element. J. Biol. Chem. 1996, 271, 20035–20046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazdi, M.T.; van Riet, S.; van Schadewijk, A.; Fiocco, M.; van Hall, T.; Taube, C.; Hiemstra, P.S.; van der burg, S.H. The positive prognostic effect of stromal CD8+ tumor-infiltrating T cells is restrained by the expression of HLA-E in non-small cell lung carcinoma. Oncotarget 2016, 7, 3477–3488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolpert, F.; Roth, P.; Lamszus, K.; Tabatabai, G.; Weller, M.; Eisele, G. HLA-E contributes to an immune-inhibitory phenotype of glioblastoma stem-like cells. J. Neuroimmunol. 2012, 250, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Kruijf, E.M.; Sajet, A.; van Nes, J.G.H.; Natanov, R.; Putter, H.; Smit, V.T.H.B.M.; Liefers, G.J.; van den Elsen, P.J.; van de Velde, C.J.H.; Kuppen, P.J.K. HLA-E and HLA-G Expression in Classical HLA Class I-Negative Tumors Is of Prognostic Value for Clinical Outcome of Early Breast Cancer Patients. J. Immunol. 2010, 185, 7452–7459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Xu, J.; Huang, Q.; Huang, M.; Wen, H.; Zhang, C.; Wang, J.; Song, J.; Zheng, M.; Sun, H.; et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. OncoImmunology 2017, 6, e1264562. [Google Scholar] [CrossRef]
- Seliger, B.; Jasinski-Bergner, S.; Quandt, D.; Stoehr, C.; Bukur, J.; Wach, S.; Legal, W.; Taubert, H.; Wullich, B.; Hartmann, A. HLA-E expression and its clinical relevance in human renal cell carcinoma. Oncotarget 2016, 7, 67360–67372. [Google Scholar] [CrossRef] [Green Version]
- Gooden, M.; Lampen, M.; Jordanova, E.S.; Leffers, N.; Trimbos, J.B.; van der Burg, S.H.; Nijman, H.; van Hall, T. HLA-E expression by gynecological cancers restrains tumor-infiltrating CD8+ T lymphocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 10656–10661. [Google Scholar] [CrossRef] [Green Version]
- Levy, E.M.; Bianchini, M.; Von Euw, E.M.; Barrio, M.M.; Bravo, A.I.; Furman, D.; Domenichini, E.; Macagno, C.; Pinsky, V.; Zucchini, C.; et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int. J. Oncol. 2008, 32, 633–641. [Google Scholar] [CrossRef] [Green Version]
- Borst, L.; van der Burg, S.H.; van Hall, T. The NKG2A–HLA-E Axis as a Novel Checkpoint in the Tumor Microenvironment. Clin. Cancer. Res. 2020. [Google Scholar] [CrossRef]
- Kamiya, T.; Seow, S.V.; Wong, D.; Robinson, M.; Campana, D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J. Clin. Investig. 2019, 129, 2094–2106. [Google Scholar] [CrossRef] [Green Version]
- Werfel, T.A.; Elion, D.L.; Rahman, B.; Hicks, D.J.; Sanchez, V.; Gonzales-Ericsson, P.I.; Nixon, M.J.; James, J.L.; Balko, J.M.; Scherle, P.A.; et al. Treatment-Induced Tumor Cell Apoptosis and Secondary Necrosis Drive Tumor Progression in the Residual Tumor Microenvironment through MerTK and IDO1. Cancer Res. 2019, 79, 171–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillard-Bocquet, M.; Caer, C.; Cagnard, N.; Crozet, L.; Perez, M.; Fridman, W.H.; Sautès-Fridman, C.; Cremer, I. Lung Tumor Microenvironment Induces Specific Gene Expression Signature in Intratumoral NK Cells. Front. Immun. 2013, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamessier, E.; Sylvain, A.; Thibult, M.-L.; Houvenaeghel, G.; Jacquemier, J.; Castellano, R.; Gonçalves, A.; André, P.; Romagné, F.; Thibault, G.; et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Investig. 2011, 121, 3609–3622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pesce, S.; Tabellini, G.; Cantoni, C.; Patrizi, O.; Coltrini, D.; Rampinelli, F.; Matta, J.; Vivier, E.; Moretta, A.; Parolini, S.; et al. B7-H6-mediated downregulation of NKp30 in NK cells contributes to ovarian carcinoma immune escape. OncoImmunology 2015, 4, e1001224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masilamani, M.; Nguyen, C.; Kabat, J.; Borrego, F.; Coligan, J.E. CD94/NKG2A Inhibits NK Cell Activation by Disrupting the Actin Network at the Immunological Synapse. J. Immunol. 2006, 177, 3590–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringaris, K.; Sekine, T.; Khoder, A.; Alsuliman, A.; Razzaghi, B.; Sargeant, R.; Pavlu, J.; Brisley, G.; de Lavallade, H.; Sarvaria, A.; et al. Leukemia-induced phenotypic and functional defects in natural killer cells predict failure to achieve remission in acute myeloid leukemia. Haematologica 2014, 99, 836–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaghi, E.; Calvi, M.; Marcenaro, E.; Mavilio, D.; Di Vito, C. Targeting NKG2A to elucidate natural killer cell ontogenesis and to develop novel immune-therapeutic strategies in cancer therapy. J. Leukoc. Biol. 2019, 105, 1243–1251. [Google Scholar] [CrossRef]
- Khan, M.; Arooj, S.; Wang, H. NK Cell-Based Immune Checkpoint Inhibition. Front. Immun. 2020, 11, 167. [Google Scholar] [CrossRef]
- Workman, C.J.; Dugger, K.J.; Vignali, D.A.A. Cutting edge: Molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J. Immunol. 2002, 169, 5392–5395. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Cao, J.; Zhao, C.; Li, X.; Zhou, C.; Hirsch, F.R. TIM-3, a promising target for cancer immunotherapy. Onco. Targets 2018, 11, 7005–7009. [Google Scholar] [CrossRef] [Green Version]
- Folgiero, V.; Cifaldi, L.; Li Pira, G.; Goffredo, B.M.; Vinti, L.; Locatelli, F. TIM-3/Gal-9 interaction induces IFNγ-dependent IDO1 expression in acute myeloid leukemia blast cells. J. Hematol. Oncol. 2015, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Wu, K.; Tao, K.; Chen, L.; Zheng, Q.; Lu, X.; Liu, J.; Shi, L.; Liu, C.; Wang, G.; et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 2012, 56, 1342–1351. [Google Scholar] [CrossRef]
- Ji, P.; Chen, D.; Bian, J.; Xia, R.; Song, X.; Wen, W.; Zhang, X.; Zhu, Y. Up-regulation of TIM-3 on CD4+ tumor infiltrating lymphocytes predicts poor prognosis in human non-small-cell lung cancer. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2015, 31, 808–811. [Google Scholar] [PubMed]
- Nebbia, G.; Peppa, D.; Schurich, A.; Khanna, P.; Singh, H.D.; Cheng, Y.; Rosenberg, W.; Dusheiko, G.; Gilson, R.; ChinAleong, J.; et al. Upregulation of the Tim-3/galectin-9 pathway of T cell exhaustion in chronic hepatitis B virus infection. PLoS ONE 2012, 7, e47648. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Akiba, H.; Takeda, K.; Kojima, Y.; Hashiguchi, M.; Azuma, M.; Yagita, H.; Okumura, K. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 2009, 113, 3821–3830. [Google Scholar] [CrossRef] [PubMed]
- DeKruyff, R.H.; Bu, X.; Ballesteros, A.; Santiago, C.; Chim, Y.-L.E.; Lee, H.-H.; Karisola, P.; Pichavant, M.; Kaplan, G.G.; Umetsu, D.T.; et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 2010, 184, 1918–1930. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Zhu, Y.; Li, G.; Huang, H.; Zhang, G.; Wang, F.; Sun, J.; Yang, Q.; Zhang, X.; Lu, B. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS ONE 2012, 7, e30676. [Google Scholar] [CrossRef] [Green Version]
- Golden-Mason, L.; McMahan, R.H.; Strong, M.; Reisdorph, R.; Mahaffey, S.; Palmer, B.E.; Cheng, L.; Kulesza, C.; Hirashima, M.; Niki, T.; et al. Galectin-9 functionally impairs natural killer cells in humans and mice. J. Virol. 2013, 87, 4835–4845. [Google Scholar] [CrossRef] [Green Version]
- Finney, C.A.M.; Ayi, K.; Wasmuth, J.D.; Sheth, P.M.; Kaul, R.; Loutfy, M.; Kain, K.C.; Serghides, L. HIV infection deregulates Tim-3 expression on innate cells: Combination antiretroviral therapy results in partial restoration. J. Acquir. Immune Defic. Syndr. 2013, 63, 161–167. [Google Scholar] [CrossRef]
- Gleason, M.K.; Lenvik, T.R.; McCullar, V.; Felices, M.; O’Brien, M.S.; Cooley, S.A.; Verneris, M.R.; Cichocki, F.; Holman, C.J.; Panoskaltsis-Mortari, A.; et al. Tim-3 is an inducible human natural killer cell receptor that enhances interferon gamma production in response to galectin-9. Blood 2012, 119, 3064–3072. [Google Scholar] [CrossRef] [PubMed]
- Ndhlovu, L.C.; Lopez-Vergès, S.; Barbour, J.D.; Jones, R.B.; Jha, A.R.; Long, B.R.; Schoeffler, E.C.; Fujita, T.; Nixon, D.F.; Lanier, L.L. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood 2012, 119, 3734–3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komita, H.; Koido, S.; Hayashi, K.; Kan, S.; Ito, M.; Kamata, Y.; Suzuki, M.; Homma, S. Expression of immune checkpoint molecules of T cell immunoglobulin and mucin protein 3/galectin-9 for NK cell suppression in human gastrointestinal stromal tumors. Oncol. Rep. 2015, 34, 2099–2105. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.; Hou, N.; Meng, J.; Wang, X.; Zhang, X.; Zhao, D.; Liu, Y.; Zhu, F.; Zhang, L.; Sun, W.; et al. T cell immunoglobulin- and mucin-domain-containing molecule-3 (Tim-3) mediates natural killer cell suppression in chronic hepatitis B. J. Hepatol. 2010, 52, 322–329. [Google Scholar] [CrossRef]
- De Sousa Linhares, A.; Leitner, J.; Grabmeier-Pfistershammer, K.; Steinberger, P. Not All Immune Checkpoints Are Created Equal. Front. Immun. 2018, 9, 1909. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.-T.; Anderson, A.C.; Tan, W.G.; West, E.E.; Ha, S.-J.; Araki, K.; Freeman, G.J.; Kuchroo, V.K.; Ahmed, R. Cooperation of Tim-3 and PD-1 in CD8 T-cell exhaustion during chronic viral infection. Proc. Natl. Acad. Sci. USA 2010, 107, 14733–14738. [Google Scholar] [CrossRef] [Green Version]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 expression is associated with tumor antigen-specific CD8+ T cell dysfunction in melanoma patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef]
- Linedale, R.; Schmidt, C.; King, B.T.; Ganko, A.G.; Simpson, F.; Panizza, B.J.; Leggatt, G.R. Elevated frequencies of CD8 T cells expressing PD-1, CTLA-4 and Tim-3 within tumour from perineural squamous cell carcinoma patients. PLoS ONE 2017, 12, e0175755. [Google Scholar] [CrossRef]
- Liu, F.; Zeng, G.; Zhou, S.; He, X.; Sun, N.; Zhu, X.; Hu, A. Blocking Tim-3 or/and PD-1 reverses dysfunction of tumor-infiltrating lymphocytes in HBV-related hepatocellular carcinoma. Bull Cancer 2018, 105, 493–501. [Google Scholar] [CrossRef]
- Shen, H.; Sheng, H.; Lu, J.J.; Feng, C.; Yao, M.; Pan, H.; Xu, L.S.; Shen, J.F.; Zheng, Y.; Zhou, Y.L. Expression and distribution of programmed death receptor 1 and T cell immunoglobulin mucin 3 in breast cancer microenvironment and its relationship with clinicopathological features. Zhonghua Yi Xue Za Zhi 2018, 98, 1352–1357. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiwada, S.; Sho, M.; Yasuda, S.; Shimada, K.; Yamato, I.; Akahori, T.; Kinoshita, S.; Nagai, M.; Konishi, N.; Nakajima, Y. Clinical significance of CD155 expression in human pancreatic cancer. Anticancer Res. 2015, 35, 2287–2297. [Google Scholar] [PubMed]
- Triebel, F.; Jitsukawa, S.; Baixeras, E.; Roman-Roman, S.; Genevee, C.; Viegas-Pequignot, E.; Hercend, T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 1990, 171, 1393–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sanmamed, M.F.; Datar, I.; Su, T.T.; Ji, L.; Sun, J.; Chen, L.; Chen, Y.; Zhu, G.; Yin, W.; et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell 2019, 176, 334–347.e312. [Google Scholar] [CrossRef] [Green Version]
- Demeure, C.E.; Wolfers, J.; Martin-Garcia, N.; Gaulard, P.; Triebel, F. T Lymphocytes infiltrating various tumour types express the MHC class II ligand lymphocyte activation gene-3 (LAG-3): Role of LAG-3/MHC class II interactions in cell-cell contacts. Eur. J. Cancer 2001, 37, 1709–1718. [Google Scholar] [CrossRef]
- Huang, C.-T.; Workman, C.J.; Flies, D.; Pan, X.; Marson, A.L.; Zhou, G.; Hipkiss, E.L.; Ravi, S.; Kowalski, J.; Levitsky, H.I.; et al. Role of LAG-3 in regulatory T cells. Immunity 2004, 21, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juno, J.A.; Stalker, A.T.; Waruk, J.L.; Oyugi, J.; Kimani, M.; Plummer, F.A.; Kimani, J.; Fowke, K.R. Elevated expression of LAG-3, but not PD-1, is associated with impaired iNKT cytokine production during chronic HIV-1 infection and treatment. Retrovirology 2015, 12, 17. [Google Scholar] [CrossRef] [Green Version]
- Kisielow, M.; Kisielow, J.; Capoferri-Sollami, G.; Karjalainen, K. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 2005, 35, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Andreae, S.; Buisson, S.; Triebel, F. MHC class II signal transduction in human dendritic cells induced by a natural ligand, the LAG-3 protein (CD223). Blood 2003, 102, 2130–2137. [Google Scholar] [CrossRef]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Huard, B.; Prigent, P.; Tournier, M.; Bruniquel, D.; Triebel, F. CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur. J. Immunol. 1995, 25, 2718–2721. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Sun, C.; Xiao, W. Expression regulation of co-inhibitory molecules on human natural killer cells in response to cytokine stimulations. Cytokine 2014, 65, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, J.; Liu, D.; Liu, B.; Wang, M.; Hu, Z.; Du, X.; Tang, L.; He, F. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 2014, 74, 3418–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradshaw, W.E.; Holzapfel, C.M. Interaction between photoperiod, temperature, and chilling in dormant larvae of the tree-hole mosquito, Toxorhynchites rutilus Coq. Biol. Bull. 1977, 152, 147–158. [Google Scholar] [CrossRef]
- Maçon-Lemaître, L.; Triebel, F. The negative regulatory function of the lymphocyte-activation gene-3 co-receptor (CD223) on human T cells. Immunology 2005, 115, 170–178. [Google Scholar] [CrossRef]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [Green Version]
- Camisaschi, C.; Casati, C.; Rini, F.; Perego, M.; De Filippo, A.; Triebel, F.; Parmiani, G.; Belli, F.; Rivoltini, L.; Castelli, C. LAG-3 expression defines a subset of CD4(+)CD25(high)Foxp3(+) regulatory T cells that are expanded at tumor sites. J. Immunol. 2010, 184, 6545–6551. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Lanuza, P.M.; Pesini, C.; Arias, M.A.; Calvo, C.; Ramirez-Labrada, A.; Pardo, J. Recalling the Biological Significance of Immune Checkpoints on NK Cells: A Chance to Overcome LAG3, PD1, and CTLA4 Inhibitory Pathways by Adoptive NK Cell Transfer? Front. Immun. 2020, 10, 3010. [Google Scholar] [CrossRef] [Green Version]
- Merino, A.; Zhang, B.; Dougherty, P.; Luo, X.; Wang, J.; Blazar, B.R.; Miller, J.S.; Cichocki, F. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J. Clin. Invest. 2019, 129, 3770–3785. [Google Scholar] [CrossRef] [Green Version]
- Brignone, C.; Grygar, C.; Marcu, M.; Schäkel, K.; Triebel, F. A soluble form of lymphocyte activation gene-3 (IMP321) induces activation of a large range of human effector cytotoxic cells. J. Immunol. 2007, 179, 4202–4211. [Google Scholar] [CrossRef] [Green Version]
- Brignone, C.; Escudier, B.; Grygar, C.; Marcu, M.; Triebel, F. A phase I pharmacokinetic and biological correlative study of IMP321, a novel MHC class II agonist, in patients with advanced renal cell carcinoma. Clin. Cancer Res. 2009, 15, 6225–6231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brignone, C.; Gutierrez, M.; Mefti, F.; Brain, E.; Jarcau, R.; Cvitkovic, F.; Bousetta, N.; Medioni, J.; Gligorov, J.; Grygar, C.; et al. First-line chemoimmunotherapy in metastatic breast carcinoma: Combination of paclitaxel and IMP321 (LAG-3Ig) enhances immune responses and antitumor activity. J. Transl. Med. 2010, 8, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, H.-Y.; Sciumè, G.; Mikami, Y.; Guo, L.; Sun, H.-W.; Brooks, S.R.; Urban, J.F.; Davis, F.P.; Kanno, Y.; O’Shea, J.J. Developmental Acquisition of Regulomes Underlies Innate Lymphoid Cell Functionality. Cell 2016, 165, 1120–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ercolano, G.; Wyss, T.; Salomé, B.; Romero, P.; Trabanelli, S.; Jandus, C. Distinct and shared gene expression for human innate versus adaptive helper lymphoid cells. J. Leukoc. Biol. 2020, 108, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Morita, H.; Sokolowska, M.; Tan, G.; Boonpiyathad, T.; Opitz, L.; Orimo, K.; Archer, S.K.; Jansen, K.; Tang, M.L.K.; et al. Gene expression signatures of circulating human type 1, 2, and 3 innate lymphoid cells. J. Allergy Clin. Immunol. 2019, 143, 2321–2325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Tsang, J.C.H.; Wang, C.; Clare, S.; Wang, J.; Chen, X.; Brandt, C.; Kane, L.; Campos, L.S.; Lu, L.; et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature 2016, 539, 102–106. [Google Scholar] [CrossRef]
- Seillet, C.; Mielke, L.A.; Amann-Zalcenstein, D.B.; Su, S.; Gao, J.; Almeida, F.F.; Shi, W.; Ritchie, M.E.; Naik, S.H.; Huntington, N.D.; et al. Deciphering the Innate Lymphoid Cell Transcriptional Program. Cell Rep. 2016, 17, 436–447. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Huang, Y.; Mallett, G.; Stathopoulou, C.; Felizardo, T.C.; Sun, M.-A.; Martin, E.L.; Zhu, N.; Woodward, E.L.; Elias, M.S.; et al. PD-1 regulates KLRG1+ group 2 innate lymphoid cells. J. Exp. Med. 2017, 214, 1663–1678. [Google Scholar] [CrossRef] [Green Version]
- Batyrova, B.; Luwaert, F.; Maravelia, P.; Miyabayashi, Y.; Vashist, N.; Stark, J.M.; Soori, S.Y.; Tibbitt, C.A.; Riese, P.; Coquet, J.M.; et al. PD-1 expression affects cytokine production by ILC2 and is influenced by peroxisome proliferator-activated receptor-γ. Immun. Inflamm. Dis. 2020, 8, 8–23. [Google Scholar] [CrossRef] [Green Version]
- Oldenhove, G.; Boucquey, E.; Taquin, A.; Acolty, V.; Bonetti, L.; Ryffel, B.; Le Bert, M.; Englebert, K.; Boon, L.; Moser, M. PD-1 Is Involved in the Dysregulation of Type 2 Innate Lymphoid Cells in a Murine Model of Obesity. Cell Rep. 2018, 25, 2053–2060.e2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helou, D.G.; Shafiei-Jahani, P.; Lo, R.; Howard, E.; Hurrell, B.P.; Galle-Treger, L.; Painter, J.D.; Lewis, G.; Soroosh, P.; Sharpe, A.H.; et al. PD-1 pathway regulates ILC2 metabolism and PD-1 agonist treatment ameliorates airway hyperreactivity. Nat. Commun. 2020, 11, 3998. [Google Scholar] [CrossRef] [PubMed]
- Moral, J.A.; Leung, J.; Rojas, L.A.; Ruan, J.; Zhao, J.; Sethna, Z.; Ramnarain, A.; Gasmi, B.; Gururajan, M.; Redmond, D.; et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 2020, 579, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef]
- Huang, Y.; Guo, L.; Qiu, J.; Chen, X.; Hu-Li, J.; Siebenlist, U.; Williamson, P.R.; Urban, J.F.; Paul, W.E. IL-25-responsive, lineage-negative KLRG1(hi) cells are multipotential ’inflammatory’ type 2 innate lymphoid cells. Nat. Immunol. 2015, 16, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauché, D.; Joyce-Shaikh, B.; Jain, R.; Grein, J.; Ku, K.S.; Blumenschein, W.M.; Ganal-Vonarburg, S.C.; Wilson, D.C.; McClanahan, T.K.; Malefyt, R.D.W.; et al. LAG3+ Regulatory T Cells Restrain Interleukin-23-Producing CX3CR1+ Gut-Resident Macrophages during Group 3 Innate Lymphoid Cell-Driven Colitis. Immunity 2018, 49, 342–352.e345. [Google Scholar] [CrossRef] [Green Version]
- Schuijs, M.J.; Png, S.; Richard, A.C.; Tsyben, A.; Hamm, G.; Stockis, J.; Garcia, C.; Pinaud, S.; Nicholls, A.; Ros, X.R.; et al. ILC2-driven innate immune checkpoint mechanism antagonizes NK cell antimetastatic function in the lung. Nat. Immunol. 2020, 21, 998–1009. [Google Scholar] [CrossRef]
- Pesce, S.; Thoren, F.B.; Cantoni, C.; Prato, C.; Moretta, L.; Moretta, A.; Marcenaro, E. The Innate Immune Cross Talk between NK Cells and Eosinophils Is Regulated by the Interaction of Natural Cytotoxicity Receptors with Eosinophil Surface Ligands. Front. Immun. 2017, 8, 510. [Google Scholar] [CrossRef] [Green Version]
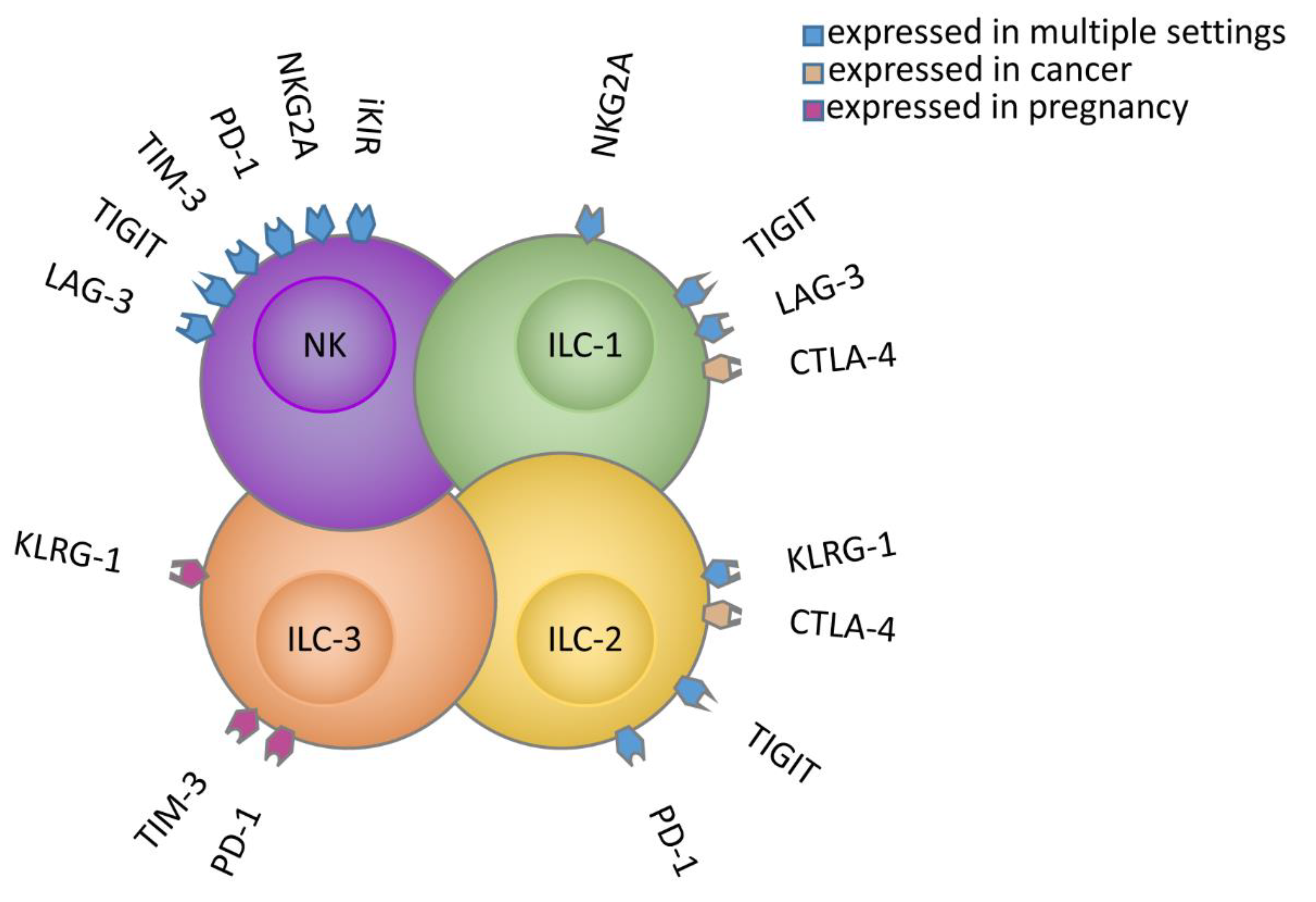

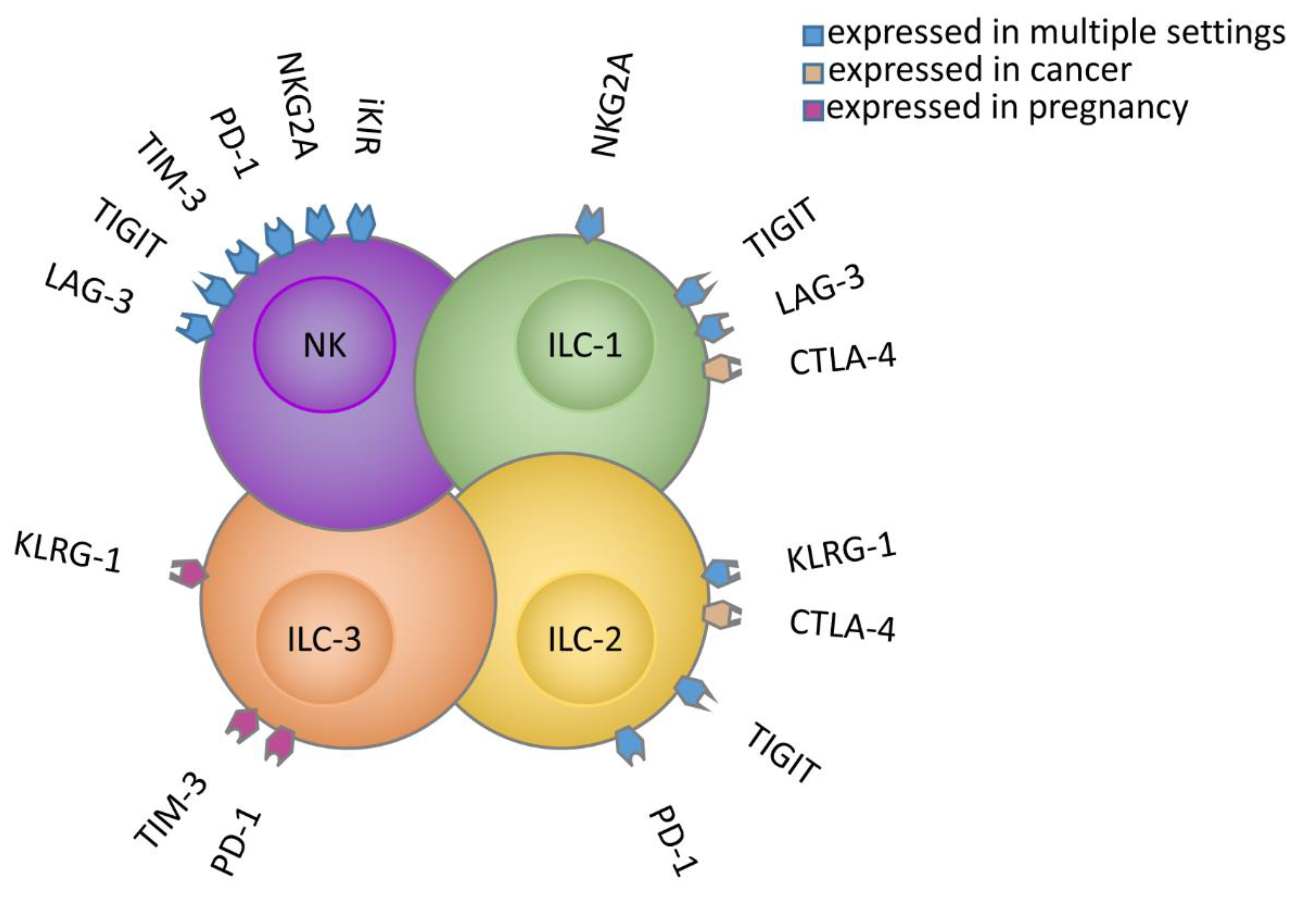{kind=link}
| Receptor | Cell Type | mAbs Used in Clinical Trial | Tissue/Tumor Involvement | Clinical Trial Number/Phase | Recruitment Information |
|---|---|---|---|---|---|
| PD-1 | NK | Several anti-PD-1 mAbs, reviewed in [25] | Solid tumors, including Kaposi sarcoma, PC, ovarian and lung cancers [26,27,28,29,30], and HL [31] | ||
| Use of anti-PD-1 or anti-PD-L1 improves the anti-tumor activity of NK cells against PD-L1/2+ tumor cells [25,26,31,32] | |||||
| ILC | Decidual ILCs during pregnancy [17] | ||||
| Tumor-associated ILC2s (>PD-1 than circulating ILCs) [33] | |||||
| ILC2s and ILC3s in tumors of the gastrointestinal tract (>PD-1 than in the paralesional tissue) | |||||
| ILC3s in human malignant pleural effusion [30] | |||||
| ILC2s in colorectal cancer model [33] | |||||
| KIRs | NK | Monotherapy with IPH2102 (lirilumab) | Solid tumors, hematologic malignancies, all pediatric tumor types [34] | ||
| IPH2102 combined with anti-PD-1 (nivolumab) | Bladder cancer | NCT03532451 phase 1 | 43 participants (active) | ||
| SCCHN | NCT03341936 phase 2 | 58 participants (active/recruiting) | |||
| Advanced solid tumors | NCT03203876 phase 1 | 21 participants (active) | |||
| Leukemia | NCT02599649 phase 2 | 10 participants (terminated/results) | |||
| HL, NHL, MM | NCT01592370 Phase 1/2 | 375 participants (active) | |||
| IPH2102 combined with anti-PD-1 (nivolumab)+ anti-CTLA-4 (ipilimumab) | Advanced solid tumors | NCT01714739 Phase 1/2 | 337 participants (completed) | ||
| Advanced solid tumors | NCT03347123 Phase 1/2 | 11 participants (active) | |||
| IPH2102 combined with anti-CS1 (elotuzumab) | MM | NCT02252263 Phase 1 | 44 participants (completed) | ||
| IPH2102 combined with anti-CD20 (rituximab) | CLL | NCT02481297 phase 2 | 7 participants (completed/results) | ||
| Monotherapy with 1-7F9 | MM | NCT00552396 phase 1 | 32 participants (completed/results) | ||
| IPH2102 combined with azacytidine | AML | NCT02399917 phase 2 | 36 participants (terminated/results) | ||
| MDS | NCT02599649 phase 2 | 10 participants (terminated/results) | |||
| NKG2A | NK | Monotherapy with IPH2201 (monalizumab) [35] | Gynecologic cancers | NCT02459301 phase 1 | 59 participants (completed) |
| IPH2201 combined with anti-PD-L1 (durvalumab) [36] | NSCLC | NCT03822351 phase 2 | 189 participants (active) | ||
| NSCLC | NCT03833440 phase2 | 120 participants (recruiting) | |||
| NSCLC | NCT03794544 phase 2 | 80 participants (active) | |||
| MSS-CRC | NCT04145193 phase 2 | Withdrawn | |||
| Advanced solid tumors | NCT02671435 phase 1/2 | 383 participants (active) | |||
| IPH2201 combined with anti-EGFR (cetuximab) or with anti-EGFR +anti-PD-L1 | SCCHN | NCT02643550 phase 1/2 | 140 participants (recruiting) | ||
| IPH2201 combined with anti-Bruton’s tyrosine kinase (ibrutinib) | CLL | NCT02557516 phase 1/2 | 22 participants (terminated/results) | ||
| ILC | AML [37] | ||||
| TIM-3 | NK | Gastric cancer [38] Lung adenocarcinoma [39] Advanced melanoma [40] Bladder cancer [38,39,40,41] | |||
| Sym023 | Metastatic cancer, solid tumor, lymphomas | NCT03489343 phase 1 | 24 participants (completed) | ||
| Sym023 combined with anti-PD-1 or anti-LAG-3 | NCT03311412 phase 1 | 102 participants (recruiting) | |||
| TSR-022 combine with anti-PD-1 | Advanced solid tumors | NCT02817633 phase 1 | 369 participants (recruiting) | ||
| TSR-022 combine with anti-PD-1 | NCT04139902 phase 2 | 56 participants (recruiting) | |||
| TSR-022 combine with anti-PD-1 | Primary liver cancer | NCT03680508 phase 2 | 42 participants (recruiting) | ||
| BMS-986016 (relatlimab) combined with anti-PD-1 (nivolumab) | Chordoma | NCT03623854 phase 2 | 20 participants (recruiting) | ||
| Metastatic uveal melanoma | NCT04552223 phase 2 | 27 participants (recruiting) | |||
| Melanoma | NCT03743766 phase 2 | 42 participants (recruiting) | |||
| BGB-A425 combined with anti-PD-1 | Advanced or metastatic solid tumors | NCT03744468 phase 1/2 | 162 participants (recruiting) | ||
| Monotherapy with MBG453 | Advanced malignancies | NCT02608268 phase 1/2 | 252 participants (active) | ||
| AML or high risk MDS | NCT03066648 phase 1 | 235 participants (recruiting) | |||
| ILC | TIM-3+ ILC3s were found in the decidua [17] | ||||
| TIGIT | NK | Colon cancer patients and tumor models [42] | |||
| TIGIT blocking was suggested as a potent strategy to increase graft versus leukemia effect in alloSCT in AML [43] | |||||
| Tiragolumab combined with anti-PD-1 (atezolizumab) | Cervical cancer | NCT04300647 phase 2 | 160 participants (recruiting) | ||
| Esophageal squamous cell carcinoma | NCT04543617 phase 3 | 750 participants (recruiting) | |||
| SCLC | NCT04256421 phase 3 | 400 participants (recruiting) | |||
| NSCLC | NCT04513925 phase 3 | 800 participants (recruiting) | |||
| NCT03563716 phase 2 | 135 participants (active) | ||||
| Esophageal Cancer | NCT04540211 phase 3 | 450 participants (recruiting) | |||
| NCT03281369 phase 1/2 | 410 participants (recruiting) | ||||
| Urothelial carcinoma | NCT03869190 phase 1/2 | 385 participants (recruiting) | |||
| Pancreatic adenocarcinoma | NCT03193190 phase 1/2 | 290 participants (recruiting) | |||
| Advanced metastatic tumors | NCT02794571 phase 1 | 400 participants (recruiting) | |||
| Tiragolumab combined with anti-VEGF (bevacizumab) | Advanced liver cancer | NCT04524871 phase 1/2 | 100 participants (recruiting) | ||
| Monotherapy with BMS-986207 or combined with anti-LAG-3 | MM | NCT04150965 phase 1/2 | 104 participants (recruiting) | ||
| BMS-986207 combined with nivolumab | Ovarian cancer, endometrial neoplasmas, solid tumor | NCT04570839 phase 1/2 | 100 participants (recruiting) | ||
| Monotherapy with OMP-313M32 or combined with anti-PD-1 (nivolumab) | Advanced and metastatic cancer | NCT03119428 phase 1 | 33 participants (terminated) | ||
| Monotherapy with AB154 or combined with anti-PD-1 (zimberelimab) | Advanced solid tumors | NCT03628677 phase 1 | 66 participants (recruiting) | ||
| Lung cancer | NCT04262856 phase 2 | 150 participants (recruiting) | |||
| ILC | ILC1 and splenic ILC3 [44] | ||||
| LAG-3 | NNK | Monotherapy with Sym022 | Metastatic cancer, solid tumor, and lymphoma | NCT03489369 phase 1 | 15 participants (completed) |
| Monotherapy with BMS-986016 (relatlimab) or combined with anti-PD-1 | Brain neoplasms, glioblastoma, gliosarcoma | NCT02658981 phase 1 | 63 participants (active) | ||
| Solid tumors | NCT01968109 phase 1/2 | 1500 participants (recruiting) | |||
| NCT03470922 phase 2/3 | 700 participants (recruiting) | ||||
| SCCHN | NCT04080804 phase 2 | 60 participants (recruiting) | |||
| MK-4280 combined with anti-PD-1 | Solid tumors | NCT02720068 phase 1 | 576 participants (recruiting) | ||
| Hematologic malignancies | NCT03598608 phase 1/2 | 134 participants (recruiting) | |||
| Monotherapy with TSR-033 or combined with anti-PD-1 | Advanced solid tumors | NCT03250832 phase 1 | 55 participants (recruiting) | ||
| Monotherapy with LAG525 or combined with anti-PD-1 | Advanced solid tumors | NCT02460224 phase 1/2 | 490 participants (active) | ||
| NCT03499899 phase 2 | 88 participants (active) | ||||
| NCT03484923 phase 2 | 195 participants (recruiting) | ||||
| Solid and hematologic malignancies | NCT03365791 phase 2 | 76 participants (completed) | |||
| Monotherapy with REGN3767 or combined with anti-PD-1 | Advanced malignancies | NCT03005782 phase 1 | 669 participants (recruiting) | ||
| Monotherapy with IMP321 (eftilagimod alpha) or combined with chemotherapy | Melanoma [45] | NCT02676869 phase 1 | 24 participants (completed) | ||
| Metastatic breast cancer | NCT00349934 phase 1 | 33 participants (completed) | |||
| IMP321 (eftilagimod alpha) combined with anti-PD-1 | NSCLC and SCCHN | NCT03625323 phase 2 | 109 participants (recruiting) | ||
| ILC | LAG-3 upregulated upon tumor conversion from NKs to ILC1s [46] | ||||
| KLRG1 | ILC | KLRG1 protein expressed from ILC3s isolated from first trimester decidual tissue [17] and in a subset of ILC2s in vivo expanded with IL-25 [47] | |||
| KLRG1 transcripts identified in ILC2s in lung and colorectal cancer [20] | |||||
| CTLA-4 | ILC | Expression of CTLA-4 in tumor associated ILC1s and ILC2s [33,46] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesce, S.; Trabanelli, S.; Di Vito, C.; Greppi, M.; Obino, V.; Guolo, F.; Minetto, P.; Bozzo, M.; Calvi, M.; Zaghi, E.; et al. Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes. Cancers 2020, 12, 3504. https://doi.org/10.3390/cancers12123504
Pesce S, Trabanelli S, Di Vito C, Greppi M, Obino V, Guolo F, Minetto P, Bozzo M, Calvi M, Zaghi E, et al. Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes. Cancers. 2020; 12(12):3504. https://doi.org/10.3390/cancers12123504
Chicago/Turabian StylePesce, Silvia, Sara Trabanelli, Clara Di Vito, Marco Greppi, Valentina Obino, Fabio Guolo, Paola Minetto, Matteo Bozzo, Michela Calvi, Elisa Zaghi, and et al. 2020. "Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes" Cancers 12, no. 12: 3504. https://doi.org/10.3390/cancers12123504
APA StylePesce, S., Trabanelli, S., Di Vito, C., Greppi, M., Obino, V., Guolo, F., Minetto, P., Bozzo, M., Calvi, M., Zaghi, E., Candiani, S., Lemoli, R. M., Jandus, C., Mavilio, D., & Marcenaro, E. (2020). Cancer Immunotherapy by Blocking Immune Checkpoints on Innate Lymphocytes. Cancers, 12(12), 3504. https://doi.org/10.3390/cancers12123504