Integrated Multi-Tumor Radio-Genomic Marker of Outcomes in Patients with High Serous Ovarian Carcinoma

 , , ,
, , ,
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
2.1. Patient and Tumor Characteristics
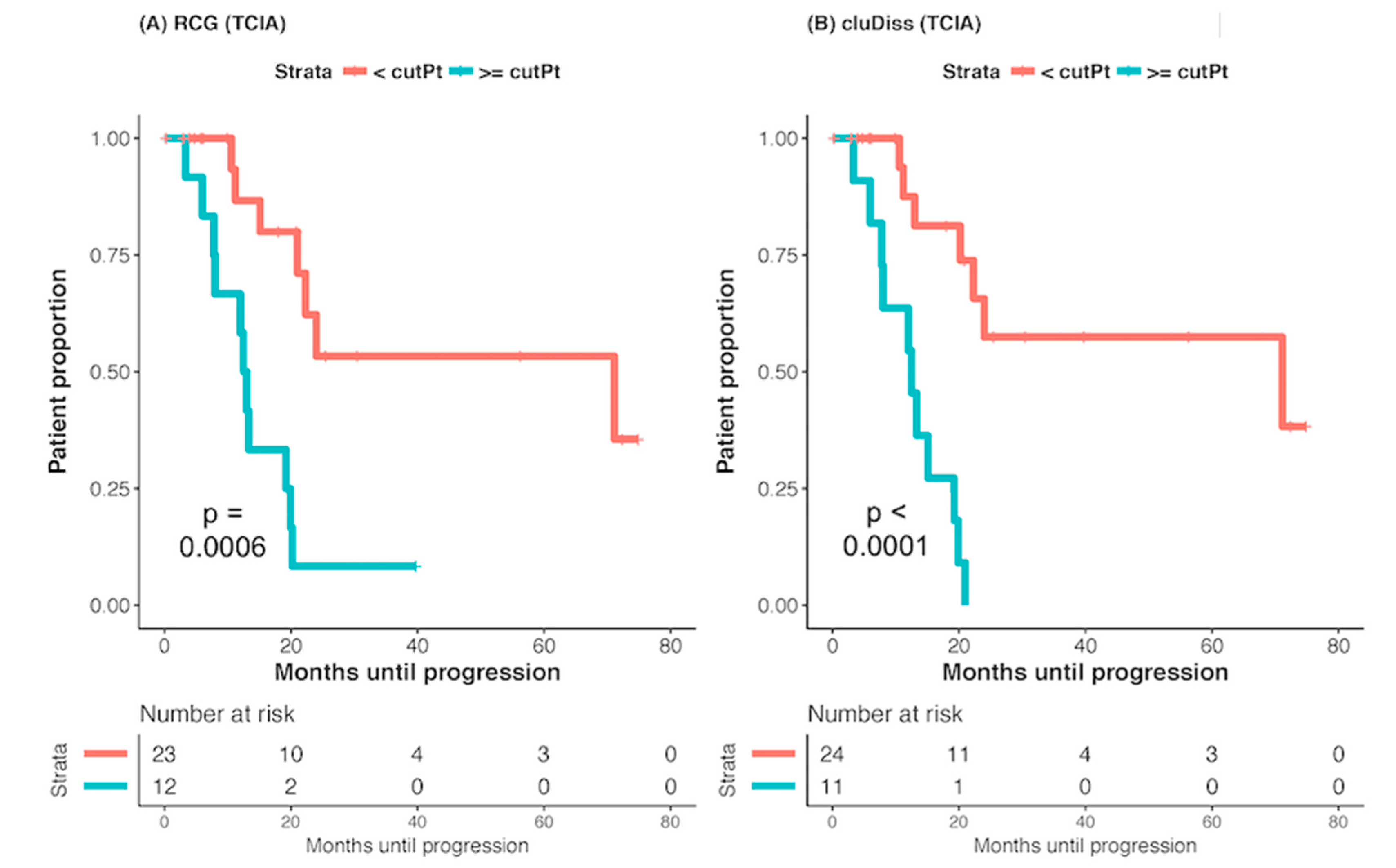
2.2. Association with Survival (PFS)
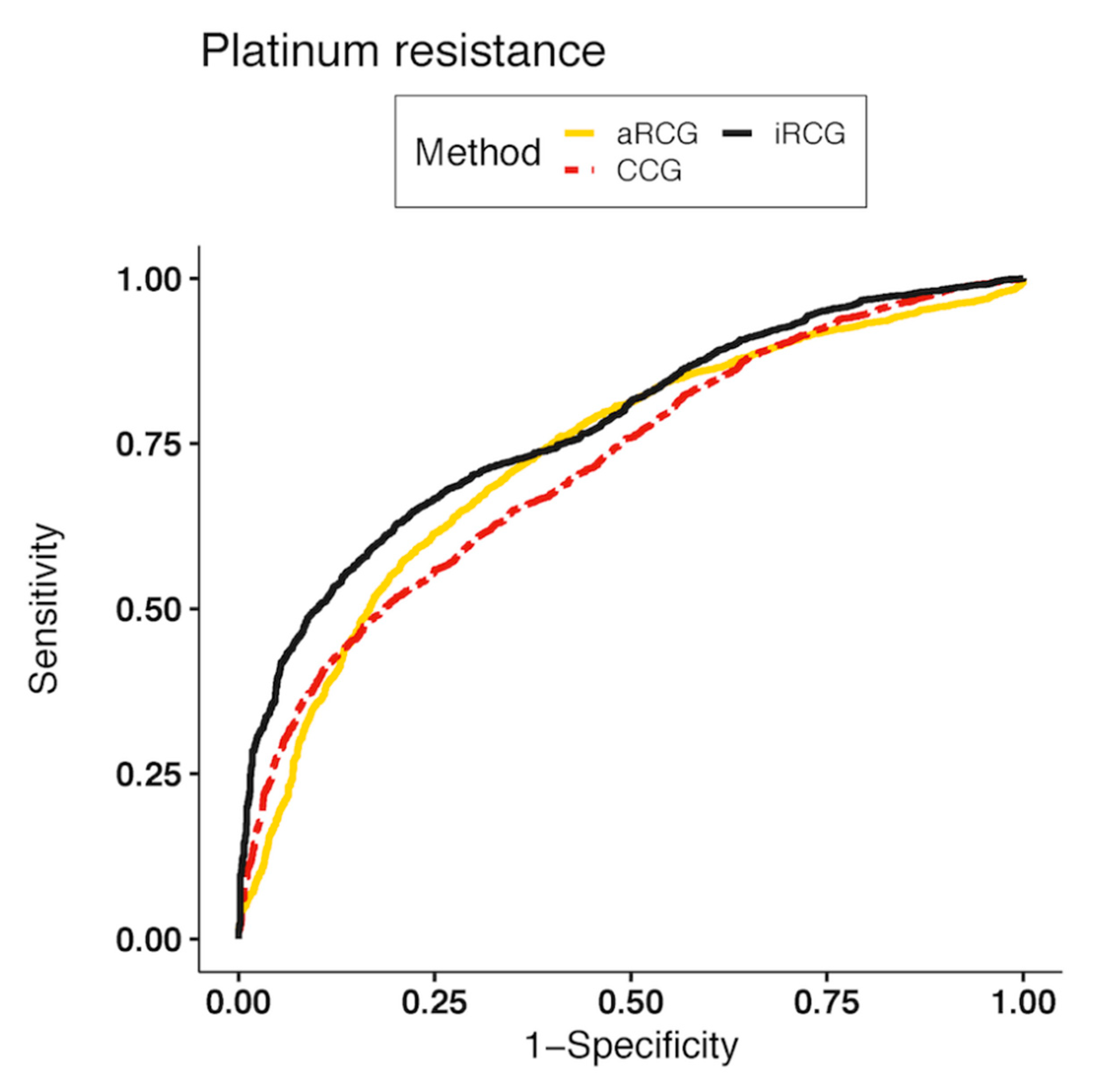
2.3. Classification of Platinum Resistance
2.4. Robustness to the CT Scanner Manufacturer
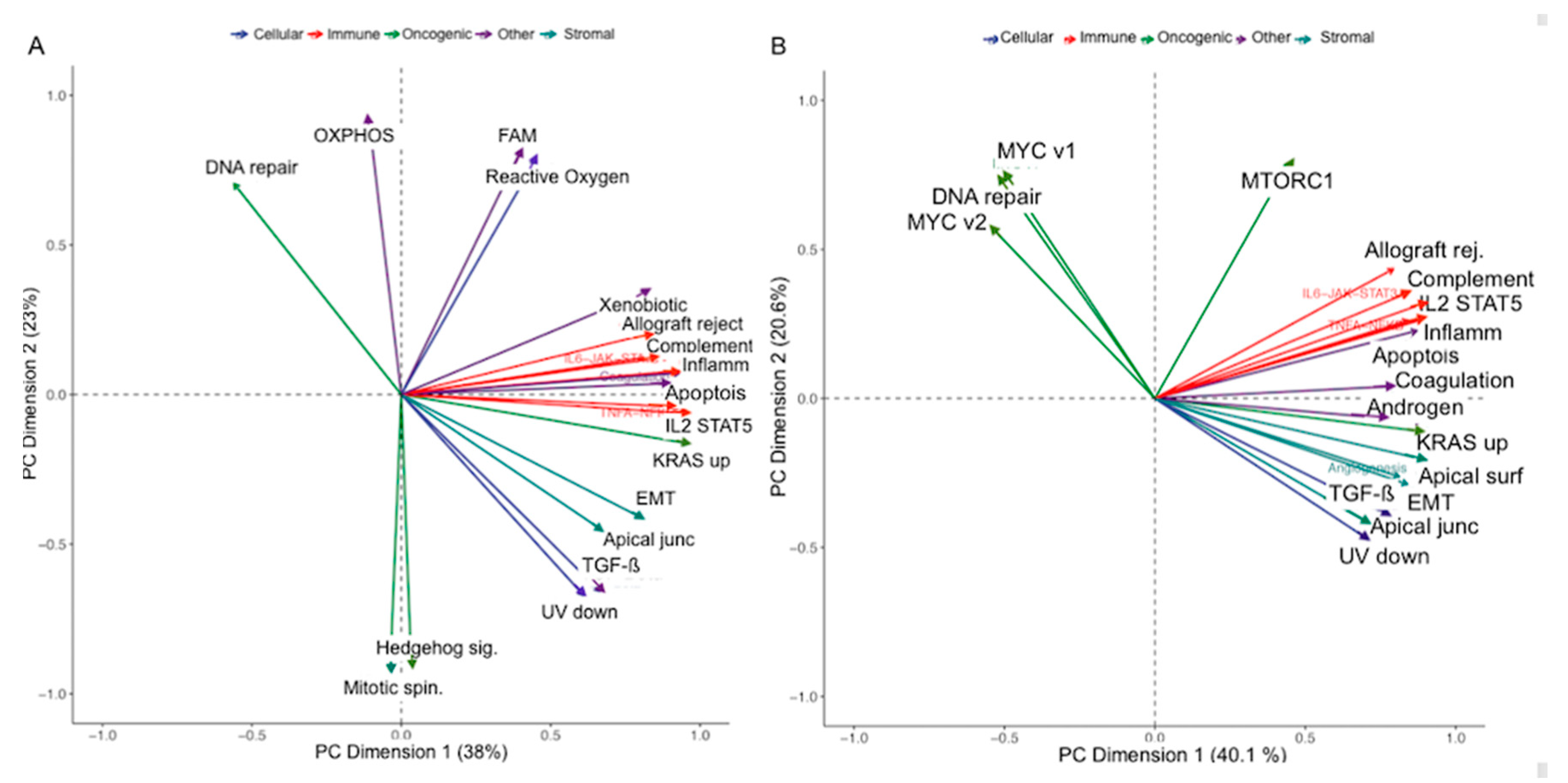
2.5. Correlation of Cludiss to Biological Processes
3. Discussion
4. Materials and Methods
4.1. Ethics and Consent
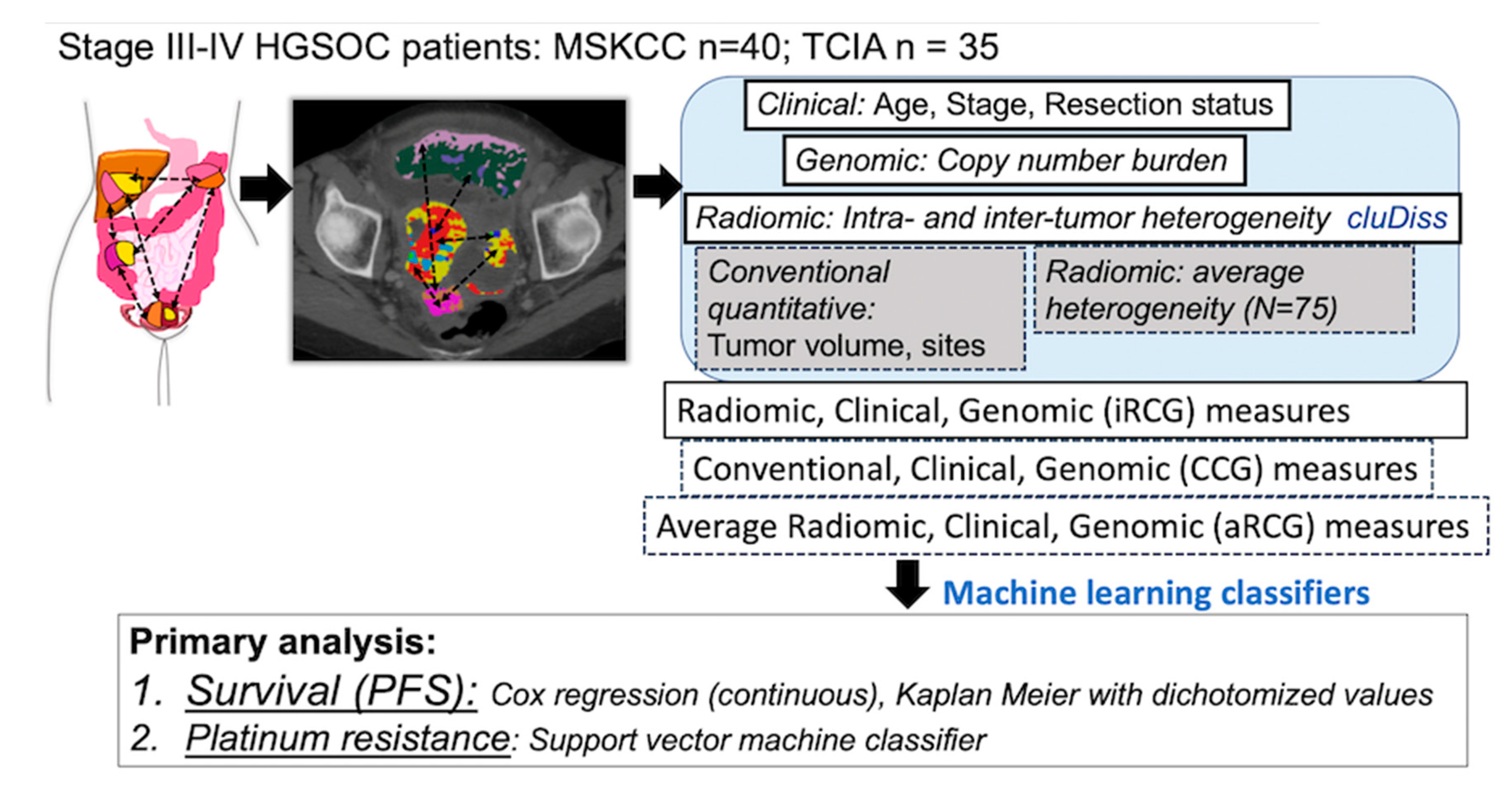
4.2. Study Design and Patients
4.3. Computation of Intra-Site and Inter-Site Tumor Radiomic Heterogeneity
- All suspected primary and metastatic tumors in the abdomen and pelvis (>1 cm) were manually delineated by two oncologic imaging research fellows (4 and 6 years of experience, respectively) using 3DSlicer [43], thereby resulting in multiple volumes of interest (VOI). Two conventional imaging measures, total tumor volume (TTV), estimated as the total number of voxels within each VOI multiplied by the voxel size, and the number of anatomic sites corresponding to the number of radiologist-defined sites of disease on preoperative CT scans were computed.
- CT images were rescaled to 0-255 and discretized into 32 bins. Then, Haralick textures, energy, entropy, homogeneity, and contrast were computed [20] by sliding a fixed sized patch (11 × 11 × 1) centered around every voxel within all VOIs using in-house software implemented in C++ using the Insight ToolKit (ITK) [44].
- Sub-regions of homogeneous texture were extracted from within VOIs by grouping voxels with similar texture values using kernel K-means clustering [45], which exploits the spatial relatedness of voxels to produce a computationally fast clustering. The appropriate number of clusters for each patient was determined using Akaike information criterion from an empirically chosen maximum of five clusters. The mean values of the four individual Haralick texture measures described the sub-regions.
- Sub-region textural differences were quantified using Euclidean distance and summarized into a dissimilarity matrix.
- The group dissimilarity matrix (GDM), which is a 2D histogram that captures the number of sub-region pairs with similar levels of dissimilarity, was computed. The rows of the GDM correspond to the number of sub-regions with a similar dissimilarity and the columns correspond to the dissimilarity level. Ten bins were used to discretize the dissimilarities and the sub-region pairs sizes following min-max normalization.
- The cluDiss measure, which quantifies the peakedness in the distribution of dissimilarities by considering the relatedness between groups of subregions by sharing similar levels of dissimilarity within the GDM is computed as:
4.4. Computation of Average Radiomic Heterogeneity Measures
4.5. Single Sample Gene Set Enrichment Analysis
4.6. Outcomes Classification through Machine Learning Classifiers
4.6.1. Combined Intra-Tumor and Inter-Tumor Site Radiomic, Clinical, and Genomic (iRCG) Score of PFS
4.6.2. Platinum Resistance Classification
4.7. Feature Robustness to CT Manufacturer
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Reid, B.M.; Permuth, J.B.; Sellers, T.A. Epidemiology of ovarian cancer: A review. Cancer Biol. Med. 2017, 14, 9–32. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Piccart, M.J.; Bertelsen, K.; James, K.; Cassidy, J.; Mangioni, C.; Simonsen, E.; Stuart, G.; Kaye, S.; Vergote, I.; Blom, R.; et al. Randomized intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: Three-year results. J. Natl. Cancer Inst. 2000, 92, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Khalique, L.; Ayhan, A.; Weale, M.E.; Jacobs, I.J.; Ramus, S.J.; Gayther, S.A. Genetic intra-tumour heterogeneity in epithelial ovarian cancer and its implications for molecular diagnosis of tumours. J. Pathol. 2007, 211, 286–295. [Google Scholar] [CrossRef] [PubMed]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [PubMed]
- McPherson, A.; Roth, A.; Laks, E.; Masud, T.; Bashashati, A.; Zhang, A.W.; Ha, G.; Biele, J.; Yap, D.; Wan, A.; et al. Divergent modes of clonal spread and intraperitoneal mixing in high-grade serous ovarian cancer. Nat. Genet. 2016, 48, 758–767. [Google Scholar] [CrossRef]
- Zhang, A.W.; McPherson, A.; Milne, K.; Kroeger, D.R.; Hamilton, P.T.; Miranda, A.; Funnell, T.; Little, N.; de Souza, C.P.E.; Laan, S.; et al. Interfaces of Malignant and Immunologic Clonal Dynamics in Ovarian Cancer. Cell 2018, 173, 1755–1769.e22. [Google Scholar] [CrossRef] [PubMed]
- Aerts, H.J.; Velazquez, E.R.; Leijenaar, R.T.; Parmar, C.; Grossmann, P.; Carvalho, S.; Bussink, J.; Monshouwer, R.; Haibe-Kains, B.; Rietveld, D.; et al. Decoding tumour phenotype by noninvasive imaging using a quantitative radiomics approach. Nat. Commun. 2014, 5, 4006. [Google Scholar] [CrossRef]
- Sala, E.; Mema, E.; Himoto, Y.; Veeraraghavan, H.; Brenton, J.D.; Snyder, A.; Weigelt, B.; Vargas, H.A. Unravelling tumour heterogeneity using next-generation imaging: Radiomics, radiogenomics, and habitat imaging. Clin. Radiol. 2017, 72, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Arshad, M.; Thornton, A.; Avesani, G.; Cunnea, P.; Curry, E.; Kanavati, F.; Liang, J.; Nixon, K.; Williams, S.T.; et al. A mathematical-descriptor of tumor-mesoscopic-structure from computed-tomography images annotates prognostic- and molecular-phenotypes of epithelial ovarian cancer. Nat. Commun. 2019, 10, 764. [Google Scholar] [CrossRef] [PubMed]
- Lubner, M.G.; Stabo, N.; Lubner, S.J.; del Rio, A.M.; Song, C.; Halberg, R.B.; Pickhardt, P.J. CT textural analysis of hepatic metastatic colorectal cancer: Pre-treatment tumor heterogeneity correlates with pathology and clinical outcomes. Abdom. Imaging 2015, 40, 2331–2337. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.X.; Lambregts, D.M.; Schnerr, R.S.; Beckers, R.C.; Maas, M.; Albarello, F.; Riedl, R.G.; Dejong, C.H.; Martens, M.H.; Heijnen, L.A.; et al. CT texture analysis in colorectal liver metastases: A better way than size and volume measurements to assess response to chemotherapy? United Eur. Gastroenterol. J. 2016, 4, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Gray, M.R.; del Campo, S.M.; Shlapak, D.; Ganeshan, B.; Zhang, X.; Carson, W.E., 3rd. Predicting Overall Survival in Patients With Metastatic Melanoma on Antiangiogenic Therapy and RECIST Stable Disease on Initial Posttherapy Images Using CT Texture Analysis. Am. J. Roentgenol. 2015, 205, W283–W293. [Google Scholar] [CrossRef]
- Horvat, N.; Veeraraghavan, H.; Khan, M.; Blazic, I.; Zheng, J.; Capanu, M.; Sala, E.; Garcia-Aguilar, J.; Gollub, M.J.; Petkovska, I. MR Imaging of Rectal Cancer: Radiomics Analysis to Assess Treatment Response after Neoadjuvant Therapy. Radiology 2018, 287, 833–843. [Google Scholar] [CrossRef]
- Fehr, D.; Veeraraghavan, H.; Wibmer, A.; Gondo, T.; Matsumoto, K.; Vargas, H.A.; Sala, E.; Hricak, H.; Deasy, J.O. Automatic classification of prostate cancer Gleason scores from multiparametric magnetic resonance images. Proc. Natl. Acad. Sci. USA 2015, 112, E6265–E6273. [Google Scholar] [CrossRef]
- Segal, E.; Sirlin, C.B.; Ooi, C.; Adler, A.S.; Gollub, J.; Chen, X.; Chan, B.K.; Matcuk, G.R.; Barry, C.T.; Chang, H.Y.; et al. Decoding global gene expression programs in liver cancer by noninvasive imaging. Nat. Biotechnol. 2007, 25, 675–680. [Google Scholar] [CrossRef]
- Teruel, J.R.; Heldahl, M.G.; Goa, P.E.; Pickles, M.; Lundgren, S.; Bathen, T.F.; Gibbs, P. Dynamic contrast-enhanced MRI texture analysis for pretreatment prediction of clinical and pathological response to neoadjuvant chemotherapy in patients with locally advanced breast cancer. NMR Biomed. 2014, 27, 887–896. [Google Scholar] [CrossRef]
- Vargas, H.A.; Veeraraghavan, H.; Micco, M.; Nougaret, S.; Lakhman, Y.; Meier, A.A.; Sosa, R.; Soslow, R.A.; Levine, D.A.; Weigelt, B.; et al. A novel representation of inter-site tumour heterogeneity from pre-treatment computed tomography textures classifies ovarian cancers by clinical outcome. Eur. Radiol. 2017, 27, 3991–4001. [Google Scholar] [CrossRef]
- Meier, A.; Veeraraghavan, H.; Nougaret, S.; Lakhman, Y.; Sosa, R.; Soslow, R.A.; Sutton, E.J.; Hricak, H.; Sala, E.; Vargas, H.A. Association between CT-texture-derived tumor heterogeneity, outcomes, and BRCA mutation status in patients with high-grade serous ovarian cancer. Abdom. Radiol. 2019, 44, 2040–2047. [Google Scholar] [CrossRef] [PubMed]
- Himoto, Y.; Veeraraghavan, H.; Zheng, J.; Zamarin, D.; Snyder, A.; Capanu, M.; Nougaret, S.; Vargas, H.A.; Shitano, F.; Callahan, M.; et al. Computed Tomography–Derived Radiomic Metrics Can Identify Responders to Immunotherapy in Ovarian Cancer. JCO Precis. Oncol. 2019, 3, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, A.; Cast, O.; Miller, M.L. Comprehensive Benchmarking and Integration of Tumor Microenvironment Cell Estimation Methods. Cancer Res. 2019, 79, 6238–6246. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Sánchez, A.; Cybulska, P.; Mager, K.L.; Koplev, S.; Cast, O.; Couturier, D.L.; Memon, D.; Selenica, P.; Nikolovski, I.; Mazaheri, Y.; et al. Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Liberzon, A.; Birger, C.; Thorvaldsdottir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, A.; Memon, D.; Pourpe, S.; Veeraraghavan, H.; Li, Y.; Vargas, H.A.; Gill, M.B.; Park, K.J.; Zivanovic, O.; Konner, J.; et al. Heterogeneous Tumor-Immune Microenvironments among Differentially Growing Metastases in an Ovarian Cancer Patient. Cell 2017, 170, 927–938.e20. [Google Scholar] [CrossRef]
- Tixier, F.; Um, H.; Bermudez, D.; Iyer, A.; Apte, A.; Graham, M.S.; Nevel, K.S.; Deasy, J.O.; Young, R.J.; Veeraraghavan, H. Preoperative MRI-radiomics features improve prediction of survival in glioblastoma patients over MGMT methylation status alone. Oncotarget 2019, 10, 660–672. [Google Scholar] [CrossRef]
- Lin, P.; Wen, D.Y.; Chen, L.; Li, X.; Li, S.H.; Yan, H.B.; He, R.Q.; Chen, G.; He, Y.; Yang, H. A radiogenomics signature for predicting the clinical outcome of bladder urothelial carcinoma. Eur. Radiol. 2020, 30, 547–557. [Google Scholar] [CrossRef]
- Vargas, H.A.; Huang, E.P.; Lakhman, Y.; Ippolito, J.E.; Bhosale, P.; Mellnick, V.; Shinagare, A.B.; Anello, M.; Kirby, J.; Fevrier-Sullivan, B.; et al. Radiogenomics of High-Grade Serous Ovarian Cancer: Multireader Multi-Institutional Study from the Cancer Genome Atlas Ovarian Cancer Imaging Research Group. Radiology 2017, 285, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Beig, N.; Bera, K.; Prasanna, P.; Antunes, J.; Correa, R.; Singh, S.; Saeed Bamashmos, A.; Ismail, M.; Braman, N.; Verma, R.; et al. Radiogenomic-Based Survival Risk Stratification of Tumor Habitat on Gd-T1w MRI Is Associated with Biological Processes in Glioblastoma. Clin. Cancer Res. 2020, 26, 1866–1876. [Google Scholar] [CrossRef] [PubMed]
- Jansen, R.W.; van Amstel, P.; Martens, R.M.; Kooi, I.E.; Wesseling, P.; de Langen, A.J.; Menke-Van der Houven van Oordt, C.W.; Jansen, B.H.E.; Moll, A.C.; Dorsman, J.C.; et al. Non-invasive tumor genotyping using radiogenomic biomarkers, a systematic review and oncology-wide pathway analysis. Oncotarget 2018, 9, 20134–20155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Leung, A.; Echegaray, S.; Gentles, A.; Shrager, J.B.; Jensen, K.C.; Berry, G.J.; Plevritis, S.K.; Rubin, D.L.; Napel, S.; et al. Non-Small Cell Lung Cancer Radiogenomics Map Identifies Relationships between Molecular and Imaging Phenotypes with Prognostic Implications. Radiology 2018, 286, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Gevaert, O.; Mitchell, L.A.; Achrol, A.S.; Xu, J.; Echegaray, S.; Steinberg, G.K.; Cheshier, S.H.; Napel, S.; Zaharchuk, G.; Plevritis, S.K. Glioblastoma Multiforme: Exploratory Radiogenomic Analysis by Using Quantitative Image Features. Radiology 2015, 276, 313. [Google Scholar] [CrossRef]
- Horvat, N.; Veeraraghavan, H.; Pelossof, R.A.; Fernandes, M.C.; Arora, A.; Khan, M.; Marco, M.; Cheng, C.T.; Gonen, M.; Golia Pernicka, J.S.; et al. Radiogenomics of rectal adenocarcinoma in the era of precision medicine: A pilot study of associations between qualitative and quantitative MRI imaging features and genetic mutations. Eur. J. Radiol. 2019, 113, 174–181. [Google Scholar] [CrossRef]
- Beer, L.; Sahin, H.; Bateman, N.W.; Blazic, I.; Vargas, H.A.; Veeraraghavan, H.; Kirby, J.; Fevrier-Sullivan, B.; Freymann, J.B.; Jaffe, C.C.; et al. Integration of proteomics with CT-based qualitative and radiomic features in high-grade serous ovarian cancer patients: An exploratory analysis. Eur. Radiol. 2020, 30, 4306–4316. [Google Scholar] [CrossRef]
- Clark, K.; Vendt, B.; Smith, K.; Freymann, J.; Kirby, J.; Koppel, P.; Moore, S.; Phillips, S.; Maffitt, D.; Pringle, M.; et al. The Cancer Imaging Archive (TCIA): Maintaining and operating a public information repository. J. Digit. Imaging 2013, 26, 1045–1057. [Google Scholar] [CrossRef]
- Vargas, H.A.; Micco, M.; Hong, S.I.; Goldman, D.A.; Dao, F.; Weigelt, B.; Soslow, R.A.; Hricak, H.; Levine, D.A.; Sala, E. Association between morphologic CT imaging traits and prognostically relevant gene signatures in women with high-grade serous ovarian cancer: A hypothesis-generating study. Radiology 2015, 274, 742–751. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [CrossRef] [PubMed]
- Friedlander, M.; Trimble, E.; Tinker, A.; Alberts, D.; Avall-Lundqvist, E.; Brady, M.; Harter, P.; Pignata, S.; Pujade-Lauraine, E.; Sehouli, J.; et al. Clinical trials in recurrent ovarian cancer. Int. J. Gynecol. Cancer 2011, 21, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, A.; Beichel, R.; Kalpathy-Cramer, J.; Finet, J.; Fillion-Robin, J.C.; Pujol, S.; Bauer, C.; Jennings, D.; Fennessy, F.; Sonka, M.; et al. 3D Slicer as an image computing platform for the Quantitative Imaging Network. Magn. Reson. Imaging 2012, 30, 1323–1341. [Google Scholar] [CrossRef] [PubMed]
- Yoo, T.S.; Ackerman, M.J.; Lorensen, W.E.; Schroeder, W.; Chalana, V.; Aylward, S.; Metaxas, D.; Whitaker, R. Engineering and algorithm design for an image processing Api: A technical report on ITK—The Insight Toolkit. Stud. Health Technol. Inform. 2002, 85, 586–592. [Google Scholar]
- Dhillon, I.S.; Guan, Y.; Kulis, B. Kernel k-means: Spectral clustering and normalized cuts. In Proceedings of the 10th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, Seattle, WA, USA, 22–25 August 2004; pp. 551–556. [Google Scholar]
- Apte, A.P.; Iyer, A.; Crispin-Ortuzar, M.; Pandya, R.; van Dijk, L.V.; Spezi, E.; Thor, M.; Um, H.; Veeraraghavan, H.; Oh, J.H.; et al. Technical Note: Extension of CERR for computational radiomics: A comprehensive MATLAB platform for reproducible radiomics research. Med. Phys. 2018, 45, 3713–3720. [Google Scholar] [CrossRef]
- Zwanenburg, A.; Vallières, M.; Abdalah, M.A.; Aerts, H.; Andrearczyk, V.; Apte, A.; Ashrafinia, S.; Bakas, S.; Beukinga, R.J.; Boellaard, R.; et al. The Image Biomarker Standardization Initiative: Standardized Quantitative Radiomics for High-Throughput Image-based Phenotyping. Radiology 2020, 295, 328–338. [Google Scholar] [CrossRef]
- Barbie, D.A.; Tamayo, P.; Boehm, J.S.; Kim, S.Y.; Moody, S.E.; Dunn, I.F.; Schinzel, A.C.; Sandy, P.; Meylan, E.; Scholl, C.; et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature 2009, 462, 108–112. [Google Scholar] [CrossRef]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Hearst, M.A. Support Vector Machines. IEEE Intell. Syst. 1998, 13, 18–28. [Google Scholar] [CrossRef]
- Um, H.; Tixier, F.; Bermudez, D.; Deasy, J.O.; Young, R.J.; Veeraraghavan, H. Impact of image preprocessing on the scanner dependence of multi-parametric MRI radiomic features and covariate shift in multi-institutional glioblastoma datasets. Phys. Med. Biol. 2019, 64, 165011. [Google Scholar] [CrossRef]
- DeLong, E.R.; DeLong, D.M.; Clarke-Pearson, D.L. Comparing the areas under two or more correlated receiver operating characteristic curves: A nonparametric approach. Biometrics 1988, 44, 837–845. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | MSKCC (N = 40) | TCIA (N = 35) |
|---|---|---|
| Age (median) (IQR) | 59 (50.8–66) | 61 (52–71) |
| Stage at diagnosis (proportion patients) | ||
| III | 27 (67.5%) | 31 (88.6%) |
| IV | 13 (32.5%) | 4 (11.4%) |
| Surgical debulking outcome (number of patients) | ||
| Complete | 14 | 8 |
| Optimal | 20 | 16 |
| Suboptimal | 6 | 11 |
| Recurrence status * (number of patients) | ||
| Recurring | 38 | 18 |
| Not recurring | 2 | 17 |
| Disease status (number of patients) | ||
| Alive | 17 | 14 |
| Dead | 23 | 21 |
| Follow up * mos (median) (IQR) | 41.9 (22.9–56.3) | 19.3 (6.3–38.6) |
| Survival (median) (IQR) | ||
| PFS + mos | 15.4 (10.5–26.2) | 13.3 (7.0–21.6) |
| OS + mos | 59.0 (43.1–76.4) | 30.0 (14.5–53.1) |
| Platinum status (number of patients) | ||
| Sensitive | 31 | 16 |
| Resistant | 7 | 7 |
| Unknown | 2 § | 12 § |
| Tumor volume (cm3) * (median) (IQR) | 122.0 (65.5–229.0) | 331.0 (158.2–595.0) |
| Tumor sites (median) * (IQR) | 7 (6–9) | 4 (3–5) |
| Copy number alterations (median) (IQR) | 0.546 (0.446–0.653) | 0.584 (0.443–0.654) |
| CT scanners: GE | 40 | 21 |
| Siemens Philips or Toshiba | 0 0 | 12 2 |
| Variable | Univariate Analysis | Multivariable Analysis | ||||||
|---|---|---|---|---|---|---|---|---|
| MSKCC | TCIA | MSKCC | TCIA | |||||
| p-Value | HR (95% CI) | p-Value | HR (95% CI) | p-Value | HR (95% CI) | p-Value | HR (95% CI) | |
| cluDiss | 0.0025 | 1.02 (1.01, 1.03) | 0.002 | 1.03 (1.01, 1.05) | 0.0008 | 1.03 (1.01, 1.04) | 0.004 | 1.04 (1.01, 1.07) |
| Number of sites | 0.049 | 1.13 (1.00, 1.28) | 0.029 | 1.59 (1.05, 2.40) | 0.242 | 1.11 (0.94, 1.31) | 0.009 | 2.00 (1.19, 3.37) |
| TTV | 0.705 | 1.06 (0.78, 1.44) | 0.653 | 0.914 (0.62, 1.35) | 0.813 | 0.953 (0.64, 1.42) | 0.513 | 0.855 (0.54, 1.37) |
| iRCG | 0.0004 | 1.36 (1.15, 1.61) | 0.007 | 1.39 (1.10, 1.76) | 0.001 | 1.38 (1.13, 1.68) | 0.009 | 1.46 (1.10, 1.93) |
| CCG | 0.058 | 1.34 (0.9, 1.81) | 0.515 | 1.13 (0.77, 1.66) | 0.411 | 0.97 (0.91, 1.04) | 0.825 | 1.02 (0.87, 1.19) |
| aRCG | 0.478 | 0.98 (0.92, 1.04) | 0.863 | 0.99 (0.86, 1.13) | 0.539 | 0.82 (0.44, 1.53) | 0.68 | 1.16 (0.56, 2.40) |
| Method | AUROC (95% CI) | Sensitivity (95% CI) | Specificity (95% CI) | p-Value (Method vs. iRCG) |
|---|---|---|---|---|
| iRCG SVM | 0.78 (0.76, 0.79) | 0.75 (0.72, 0.77) | 0.66 (0.65, 0.68) | |
| CCG SVM | 0.72 (0.70, 0.73) | 0.66 (0.64, 0.69) | 0.65 (0.64, 0.67) | <0.001 |
| aRCG SVM * | 0.73 (0.72, 0.75) | 0.68 (0.66, 0.71) | 0.62 (0.60, 0.63) | <0.001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veeraraghavan, H.; Vargas, H.A.; Jimenez-Sanchez, A.; Micco, M.; Mema, E.; Lakhman, Y.; Crispin-Ortuzar, M.; Huang, E.P.; Levine, D.A.; Grisham, R.N.; et al. Integrated Multi-Tumor Radio-Genomic Marker of Outcomes in Patients with High Serous Ovarian Carcinoma. Cancers 2020, 12, 3403. https://doi.org/10.3390/cancers12113403
Veeraraghavan H, Vargas HA, Jimenez-Sanchez A, Micco M, Mema E, Lakhman Y, Crispin-Ortuzar M, Huang EP, Levine DA, Grisham RN, et al. Integrated Multi-Tumor Radio-Genomic Marker of Outcomes in Patients with High Serous Ovarian Carcinoma. Cancers. 2020; 12(11):3403. https://doi.org/10.3390/cancers12113403
Chicago/Turabian StyleVeeraraghavan, Harini, Herbert Alberto Vargas, Alejandro Jimenez-Sanchez, Maura Micco, Eralda Mema, Yulia Lakhman, Mireia Crispin-Ortuzar, Erich P. Huang, Douglas A. Levine, Rachel N. Grisham, and et al. 2020. "Integrated Multi-Tumor Radio-Genomic Marker of Outcomes in Patients with High Serous Ovarian Carcinoma" Cancers 12, no. 11: 3403. https://doi.org/10.3390/cancers12113403
APA StyleVeeraraghavan, H., Vargas, H. A., Jimenez-Sanchez, A., Micco, M., Mema, E., Lakhman, Y., Crispin-Ortuzar, M., Huang, E. P., Levine, D. A., Grisham, R. N., Abu-Rustum, N., Deasy, J. O., Snyder, A., Miller, M. L., Brenton, J. D., & Sala, E. (2020). Integrated Multi-Tumor Radio-Genomic Marker of Outcomes in Patients with High Serous Ovarian Carcinoma. Cancers, 12(11), 3403. https://doi.org/10.3390/cancers12113403